Early-Onset Infantile Facioscapulohumeral Muscular Dystrophy: A Timely Review



Abstract
:
1. Introduction
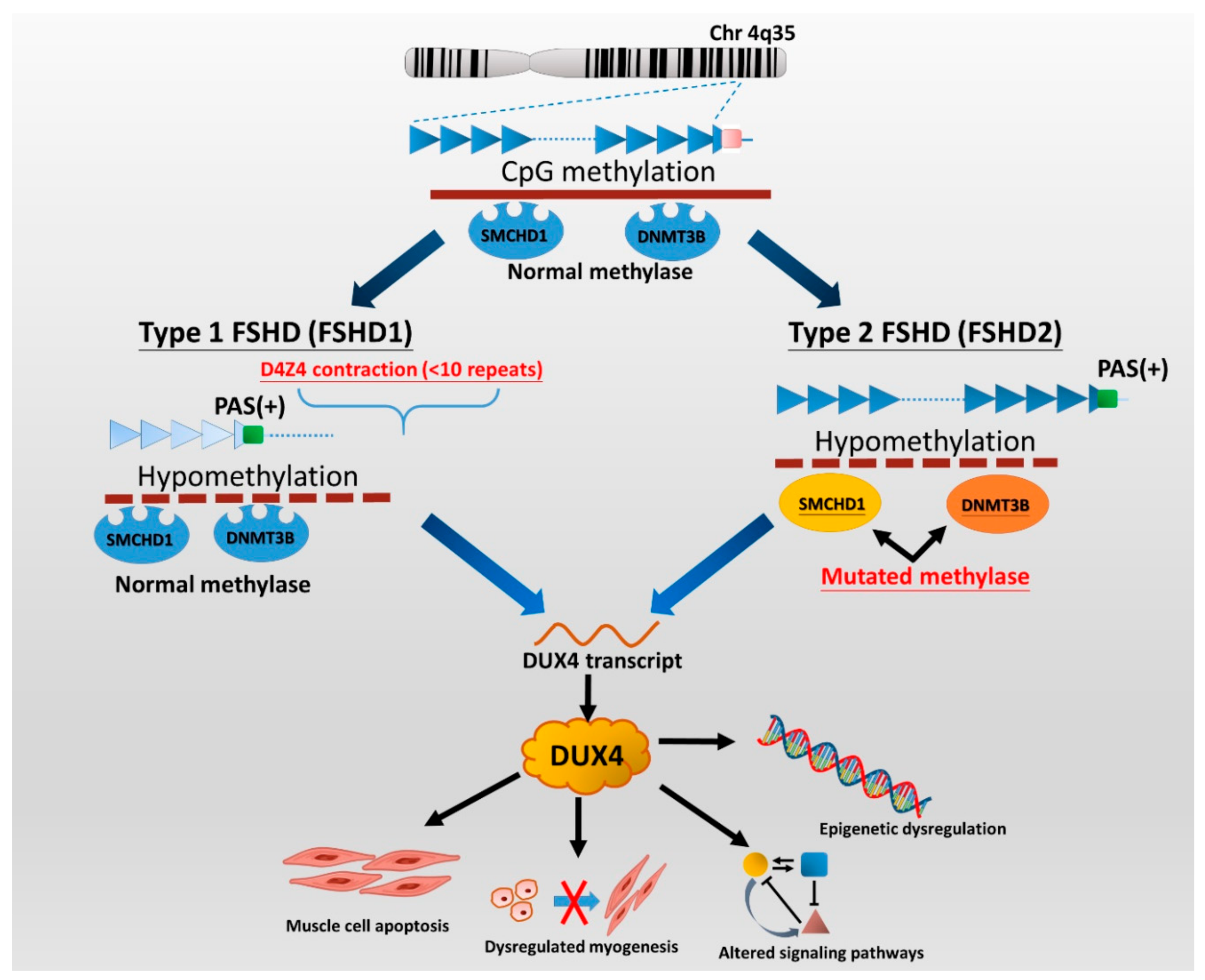
2. Genetics and Pathogenesis of FSHD
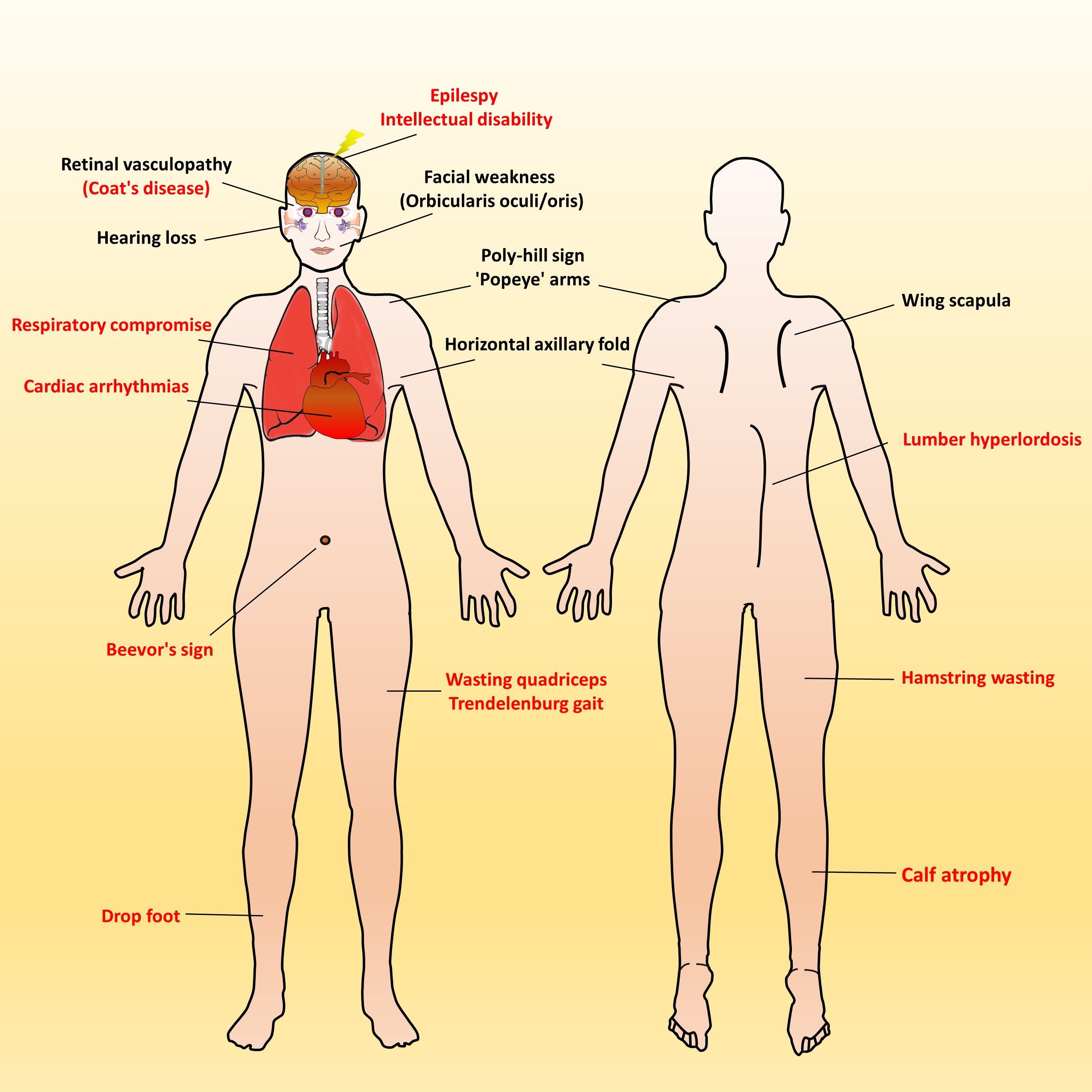
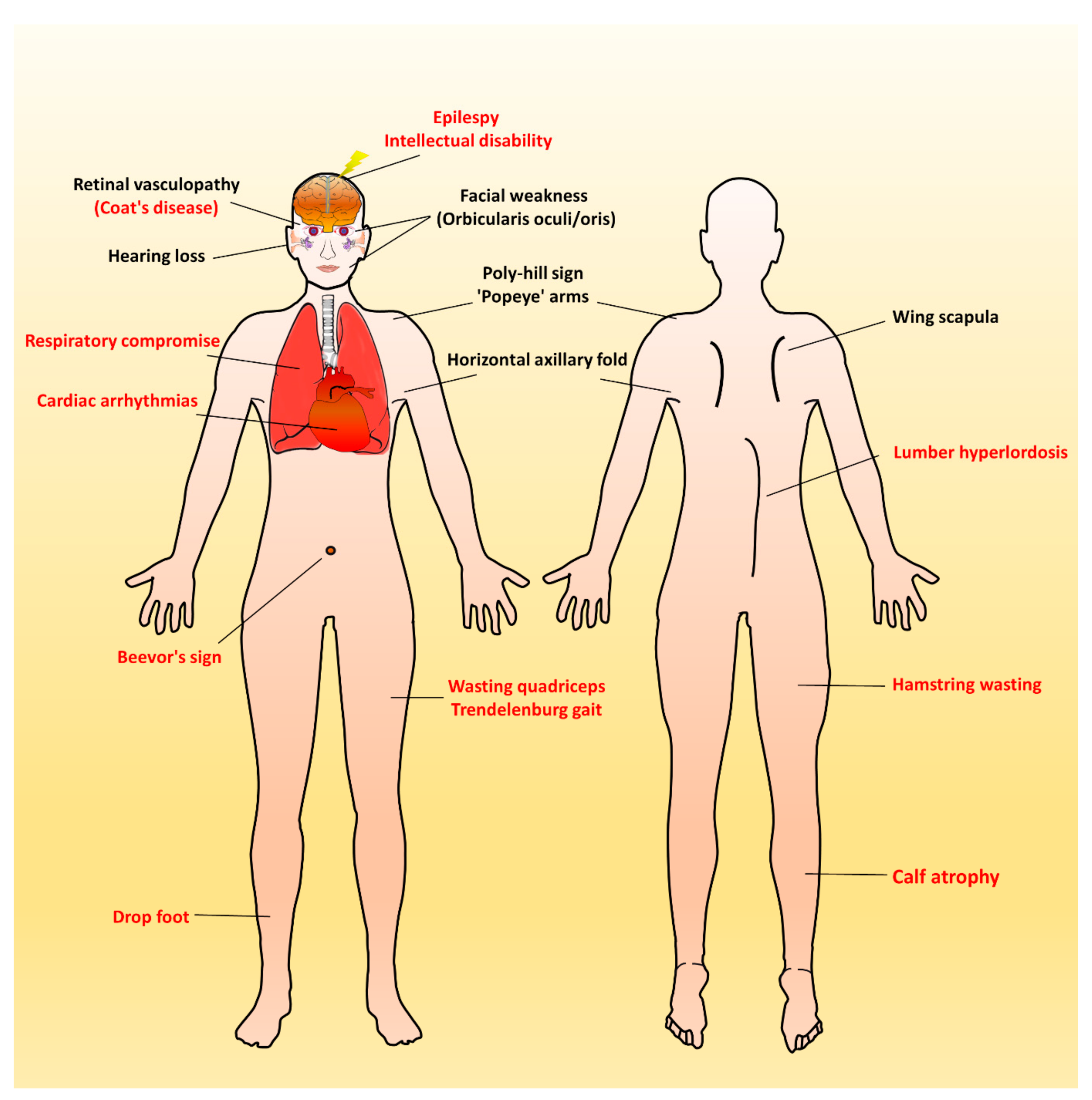
3. Clinical Manifestations of FSHD
4. Early-Onset Infantile FSHD
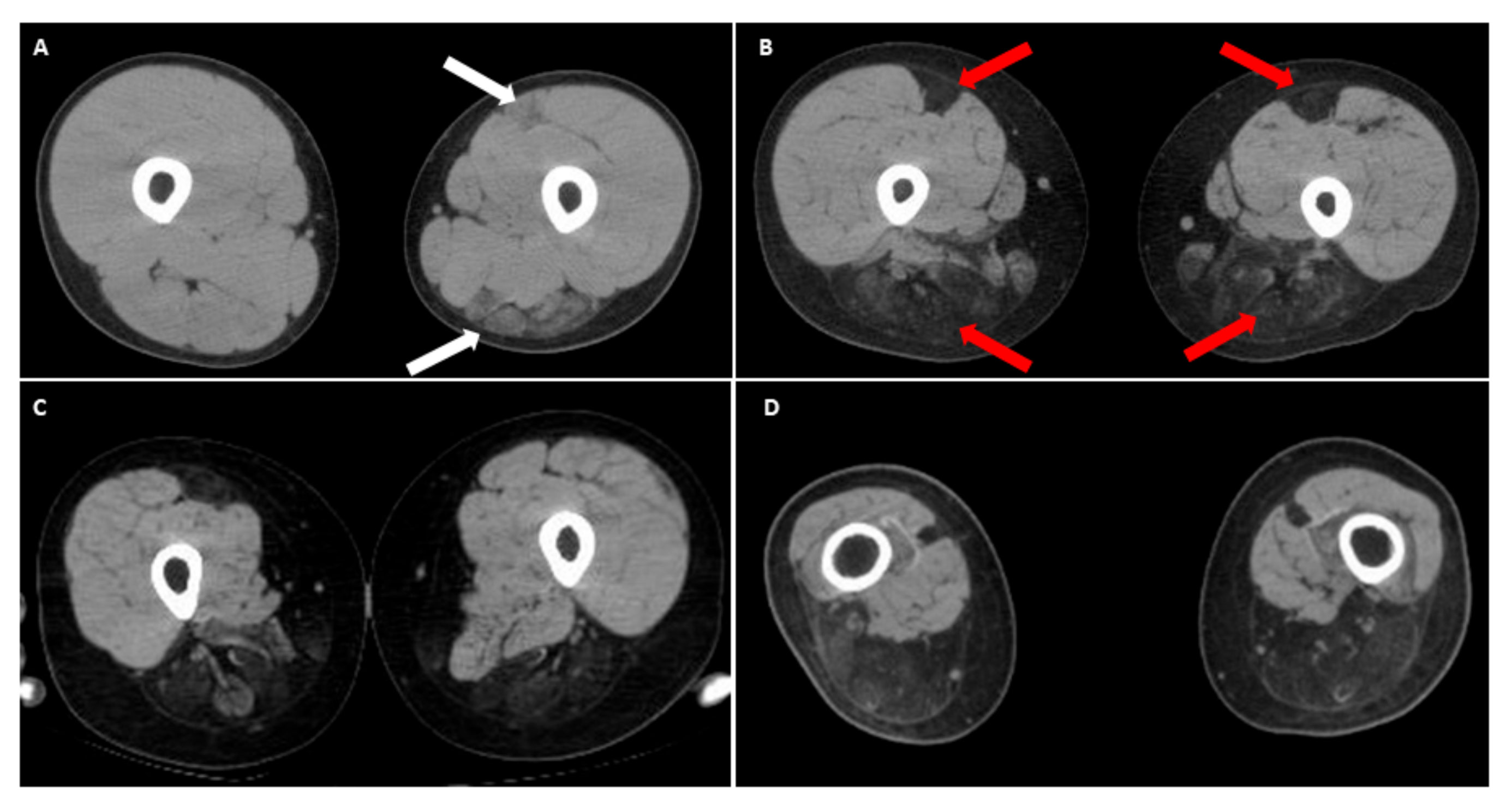
5. Peculiar Pattern of Muscle Involvement in Infantile FSHD
6. Respiratory Involvement in Infantile FSHD
7. Systemic Involvement with Extramuscular Features in Infantile FSHD
7.1. Auditory Impairment
7.2. Retina Vasculopathy
7.3. Brain Dysfunction
7.4. Cardiac Involvement
8. Laboratory Findings and Potential Biomarkers of FSHD
9. Emerging Therapeutic Approaches for FSHD
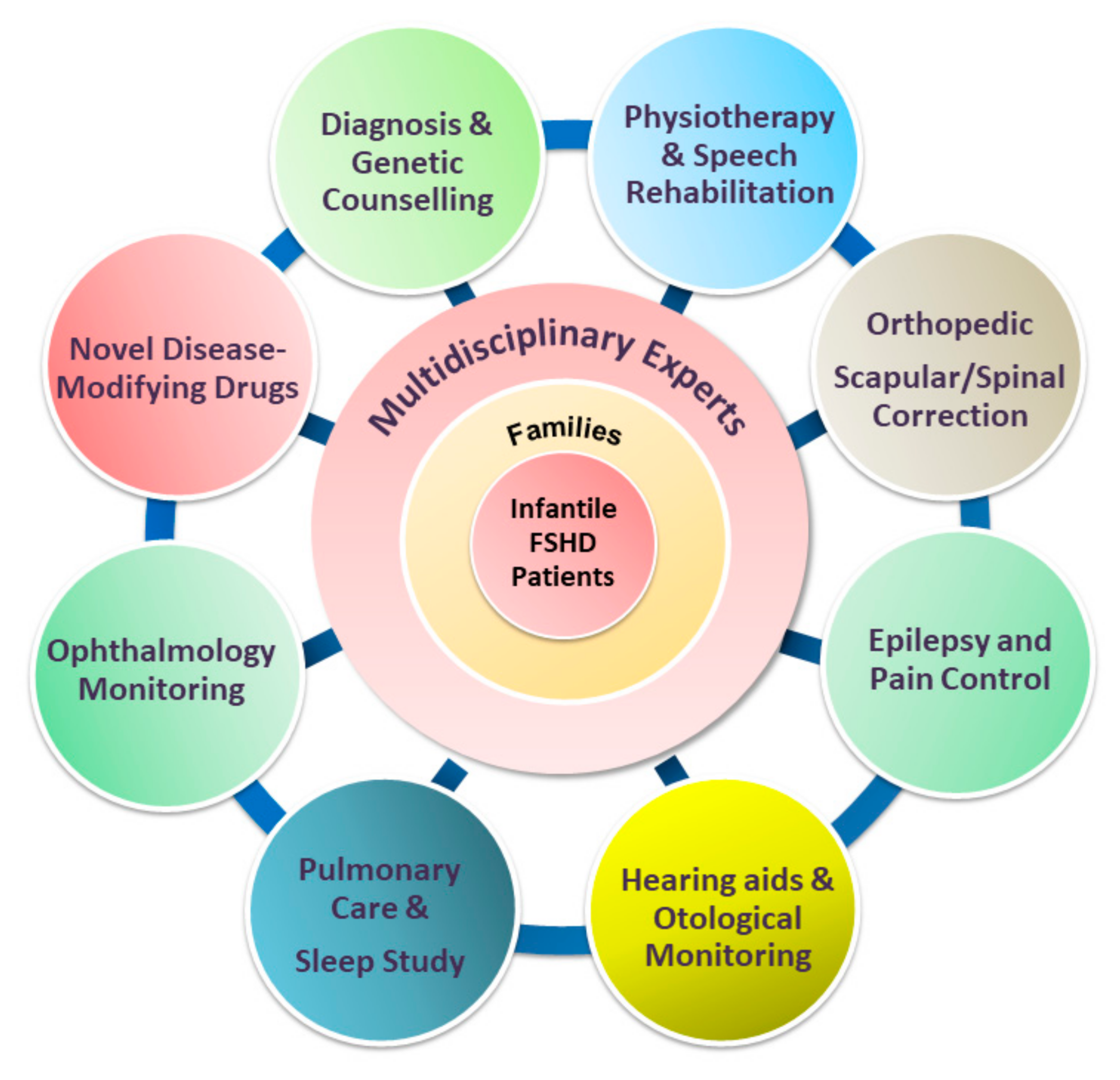
10. Standard Care for Patients with Infantile FSHD
11. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Tawil, R. Facioscapulohumeral muscular dystrophy. Curr. Neurol. Neurosci. Rep. 2004, 4, 51–54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lek, A.; Rahimov, F.; Jones, P.L.; Kunkel, L.M. Emerging preclinical animal models for FSHD. Trends Mol. Med. 2015, 21, 295–306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liew, W.K.M.; van der Maarel, S.M.; Tawil, R. Chapter 32—Facioscapulohumeral Dystrophy. In Neuromuscular Disorders of Infancy, Childhood, and Adolescence, 2nd ed.; Darras, B.T., Jones, H.R., Ryan, M.M., De Vivo, D.C., Eds.; Academic Press: San Diego, CA, USA, 2015; pp. 620–630. [Google Scholar]
- Tawil, R.; Kissel, J.T.; Heatwole, C.; Pandya, S.; Gronseth, G.; Benatar, M. Evidence-based guideline summary: Evaluation, diagnosis, and management of facioscapulohumeral muscular dystrophy: Report of the Guideline Development, Dissemination, and Implementation Subcommittee of the American Academy of Neurology and the Practice Issues Review Panel of the American Association of Neuromuscular & Electrodiagnostic Medicine. Neurology 2015, 85, 357–364. [Google Scholar] [CrossRef] [Green Version]
- Lim, K.R.Q.; Nguyen, Q.; Yokota, T. DUX4 Signalling in the Pathogenesis of Facioscapulohumeral Muscular Dystrophy. Int. J. Mol. Sci. 2020, 21, 729. [Google Scholar] [CrossRef] [Green Version]
- Statland, J.M. Chapter 30—Facioscapulohumeral muscular dystrophy. In Rosenberg’s Molecular and Genetic Basis of Neurological and Psychiatric Disease, 6th ed.; Rosenberg, R.N., Pascual, J.M., Eds.; Academic Press: San Diego, CA, USA, 2020; pp. 511–523. [Google Scholar]
- Daxinger, L.; Tapscott, S.J.; van der Maarel, S.M. Genetic and epigenetic contributors to fshd. Curr. Opin. Genet. Dev. 2015, 33, 56–61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Himeda, C.L.; Jones, P.L. The Genetics and Epigenetics of Facioscapulohumeral Muscular Dystrophy. Ann. Rev. Genom. Hum. Genet. 2019, 20, 265–291. [Google Scholar] [CrossRef] [PubMed]
- Ferreboeuf, M.; Mariot, V.; Bessieres, B.; Vasiljevic, A.; Attie-Bitach, T.; Collardeau, S.; Morere, J.; Roche, S.; Magdinier, F.; Robin-Ducellier, J.; et al. Dux4 and dux4 downstream target genes are expressed in fetal fshd muscles. Hum. Mol. Genet. 2014, 23, 171–181. [Google Scholar] [CrossRef] [Green Version]
- Goselink, R.J.M.; Mul, K.; van Kernebeek, C.R.; Lemmers, R.; van der Maarel, S.M.; Schreuder, T.H.A.; Erasmus, C.E.; Padberg, G.W.; Statland, J.M.; Voermans, N.C.; et al. Early onset as a marker for disease severity in facioscapulohumeral muscular dystrophy. Neurology 2019, 92, e378–e385. [Google Scholar] [CrossRef]
- Mul, K.; Lassche, S.; Voermans, N.C.; Padberg, G.W.; Horlings, C.G.; van Engelen, B.G. What’s in a name? The clinical features of facioscapulohumeral muscular dystrophy. Pr. Neurol. 2016, 16, 201–207. [Google Scholar] [CrossRef]
- Hamel, J.; Tawil, R. Facioscapulohumeral muscular dystrophy: Update on pathogenesis and future treatments. Neurotherapeutics 2018, 15, 863–871. [Google Scholar] [CrossRef] [Green Version]
- Magdinier, F.; Upadhyaya, M. Facioscapulohumeral Muscular Dystrophy: Genetics; eLS John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2018; pp. 1–13. [Google Scholar]
- Trevisan, C.P.; Pastorello, E.; Tomelleri, G.; Vercelli, L.; Bruno, C.; Scapolan, S.; Siciliano, G.; Comacchio, F. Facioscapulohumeral muscular dystrophy: Hearing loss and other atypical features of patients with large 4q35 deletions. Eur. J. Neurol. 2008, 15, 1353–1358. [Google Scholar] [CrossRef] [PubMed]
- Nikolic, A.; Ricci, G.; Sera, F.; Bucci, E.; Govi, M.; Mele, F.; Rossi, M.; Ruggiero, L.; Vercelli, L.; Ravaglia, S.; et al. Clinical expression of facioscapulohumeral muscular dystrophy in carriers of 1–3 d4z4 reduced alleles: Experience of the fshd italian national registry. BMJ Open 2016, 6, e007798. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, T.H.; Lai, Y.H.; Lee, P.L.; Hsu, J.H.; Goto, K.; Hayashi, Y.K.; Nishino, I.; Lin, C.W.; Shih, H.H.; Huang, C.C.; et al. Infantile facioscapulohumeral muscular dystrophy revisited: Expansion of clinical phenotypes in patients with a very short ecori fragment. Neuromuscul. Disord. 2013, 23, 298–305. [Google Scholar] [CrossRef] [PubMed]
- Mah, J.K.; Chen, Y.W. A pediatric review of facioscapulohumeral muscular dystrophy. J. Pediatr. Neurol. 2018, 16, 222–231. [Google Scholar]
- Ruggiero, L.; Mele, F.; Manganelli, F.; Bruzzese, D.; Ricci, G.; Vercelli, L.; Govi, M.; Vallarola, A.; Tripodi, S.; Villa, L.; et al. Phenotypic variability among patients with d4z4 reduced allele facioscapulohumeral muscular dystrophy. JAMA Netw. Open 2020, 3, e204040. [Google Scholar] [CrossRef]
- Steel, D.; Main, M.; Manzur, A.; Muntoni, F.; Munot, P. Clinical features of facioscapulohumeral muscular dystrophy 1 in childhood. Dev. Med. Child Neurol. 2019, 61, 964–971. [Google Scholar] [CrossRef]
- Goselink, R.J.M.; Voermans, N.C.; Okkersen, K.; Brouwer, O.F.; Padberg, G.W.; Nikolic, A.; Tupler, R.; Dorobek, M.; Mah, J.K.; van Engelen, B.G.M.; et al. Early onset facioscapulohumeral dystrophy—A systematic review using individual patient data. Neuromuscul. Disord. 2017, 27, 1077–1083. [Google Scholar] [CrossRef] [Green Version]
- Mah, J.K.; Feng, J.; Jacobs, M.B.; Duong, T.; Carroll, K.; de Valle, K.; Carty, C.L.; Morgenroth, L.P.; Guglieri, M.; Ryan, M.M.; et al. A multinational study on motor function in early-onset fshd. Neurology 2018, 90, e1333–e1338. [Google Scholar] [CrossRef]
- Rijken, N.H.; van der Kooi, E.L.; Hendriks, J.C.; van Asseldonk, R.J.; Padberg, G.W.; Geurts, A.C.; van Engelen, B.G. Skeletal muscle imaging in facioscapulohumeral muscular dystrophy, pattern and asymmetry of individual muscle involvement. Neuromuscul. Disord. 2014, 24, 1087–1096. [Google Scholar] [CrossRef] [Green Version]
- Pastorello, E.; Cao, M.; Trevisan, C.P. Atypical onset in a series of 122 cases with facioscapulohumeral muscular dystrophy. Clin. Neurol. Neurosurg. 2012, 114, 230–234. [Google Scholar] [CrossRef] [Green Version]
- Voet, N.; Bleijenberg, G.; Hendriks, J.; de Groot, I.; Padberg, G.; van Engelen, B.; Geurts, A. Both aerobic exercise and cognitive-behavioral therapy reduce chronic fatigue in fshd: An rct. Neurology 2014, 83, 1914–1922. [Google Scholar] [CrossRef]
- Brooke, M.H. A Clinician’s View of Neuromuscular Diseases; Williams & Wilkins: Baltimore, MD, USA, 1977. [Google Scholar]
- Brouwer, O.F.; Padberg, G.W.; Wijmenga, C.; Frants, R.R. Facioscapulohumeral muscular dystrophy in early childhood. Arch. Neurol. 1994, 51, 387–394. [Google Scholar] [CrossRef]
- Dorobek, M.; van der Maarel, S.M.; Lemmers, R.J.; Ryniewicz, B.; Kabzinska, D.; Frants, R.R.; Gawel, M.; Walecki, J.; Hausmanowa-Petrusewicz, I. Early-onset facioscapulohumeral muscular dystrophy type 1 with some atypical features. J. Child Neurol. 2015, 30, 580–587. [Google Scholar] [CrossRef] [PubMed]
- Brouwer, O.F.; Padberg, G.W.; Bakker, E.; Wijmenga, C.; Frants, R.R. Early onset facioscapulohumeral muscular dystrophy. Muscle Nerve 1995, 2, S67–S72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klinge, L.; Eagle, M.; Haggerty, I.D.; Roberts, C.E.; Straub, V.; Bushby, K.M. Severe phenotype in infantile facioscapulohumeral muscular dystrophy. Neuromuscul. Disord. 2006, 16, 553–558. [Google Scholar] [CrossRef] [PubMed]
- Goselink, R.J.M.; van Kernebeek, C.R.; Mul, K.; Lemmers, R.; van der Maarel, S.M.; Brouwer, O.F.; Voermans, N.; Padberg, G.W.; Erasmus, C.E.; van Engelen, B.G.M. A 22-year follow-up reveals a variable disease severity in early-onset facioscapulohumeral dystrophy. Eur. J. Paediatr. Neurol. 2018, 22, 782–785. [Google Scholar] [CrossRef]
- Goselink, R.J.; Schreuder, T.H.; Mul, K.; Voermans, N.C.; Pelsma, M.; de Groot, I.J.; van Alfen, N.; Franck, B.; Theelen, T.; Lemmers, R.J.; et al. Facioscapulohumeral dystrophy in children: Design of a prospective, observational study on natural history, predictors and clinical impact (ifocus fshd). BMC Neurol. 2016, 16, 138. [Google Scholar] [CrossRef] [Green Version]
- van der Maarel, S.M.; Deidda, G.; Lemmers, R.J.; van Overveld, P.G.; van der Wielen, M.; Hewitt, J.E.; Sandkuijl, L.; Bakker, B.; van Ommen, G.J.; Padberg, G.W.; et al. De novo facioscapulohumeral muscular dystrophy: Frequent somatic mosaicism, sex-dependent phenotype, and the role of mitotic transchromosomal repeat interaction between chromosomes 4 and 10. Am. J. Hum. Genet. 2000, 66, 26–35. [Google Scholar] [CrossRef] [Green Version]
- Lemmers, R.J.; van der Wielen, M.J.; Bakker, E.; Padberg, G.W.; Frants, R.R.; van der Maarel, S.M. Somatic mosaicism in fshd often goes undetected. Ann. Neurol. 2004, 55, 845–850. [Google Scholar] [CrossRef]
- Felice, K.J.; Jones, J.M.; Conway, S.R. Facioscapulohumeral dystrophy presenting as infantile facial diplegia and late-onset limb-girdle myopathy in members of the same family. Muscle Nerve 2005, 32, 368–372. [Google Scholar] [CrossRef]
- Statland, J.M.; Tawil, R. Risk of functional impairment in facioscapulohumeral muscular dystrophy. Muscle Nerve 2014, 49, 520–527. [Google Scholar] [CrossRef] [PubMed]
- Tasca, G.; Monforte, M.; Ottaviani, P.; Pelliccioni, M.; Frusciante, R.; Laschena, F.; Ricci, E. Magnetic resonance imaging in a large cohort of facioscapulohumeral muscular dystrophy patients: Pattern refinement and implications for clinical trials. Ann. Neurol. 2016, 79, 854–864. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.H.; Leung, M.; Liang, W.C.; Hsieh, T.J.; Chen, T.H.; Jong, Y.J. Correlation between muscle involvement, phenotype and d4z4 fragment size in facioscapulohumeral muscular dystrophy. Neuromuscul. Disord. 2012, 22, 331–338. [Google Scholar] [CrossRef] [PubMed]
- Gerevini, S.; Scarlato, M.; Maggi, L.; Cava, M.; Caliendo, G.; Pasanisi, B.; Falini, A.; Previtali, S.C.; Morandi, L. Muscle mri findings in facioscapulohumeral muscular dystrophy. Eur. Radiol. 2016, 26, 693–705. [Google Scholar] [CrossRef] [PubMed]
- Trucco, F.; Pedemonte, M.; Fiorillo, C.; Tacchetti, P.; Brisca, G.; Bruno, C.; Minetti, C. Respiratory pattern in a fshd pediatric population. Respir. Med. 2016, 119, 78–80. [Google Scholar] [CrossRef] [Green Version]
- Wohlgemuth, M.; van der Kooi, E.L.; van Kesteren, R.G.; van der Maarel, S.M.; Padberg, G.W. Ventilatory support in facioscapulohumeral muscular dystrophy. Neurology 2004, 63, 176–178. [Google Scholar] [CrossRef]
- Santos, D.B.; Boussaid, G.; Stojkovic, T.; Orlikowski, D.; Letilly, N.; Behin, A.; Butel, S.; Lofaso, F.; Prigent, H. Respiratory muscle dysfunction in facioscapulohumeral muscular dystrophy. Neuromuscul. Disord. 2015, 25, 632–639. [Google Scholar] [CrossRef]
- Moreira, S.; Wood, L.; Smith, D.; Marini-Bettolo, C.; Guglieri, M.; McMacken, G.; Bailey, G.; Mayhew, A.; Muni-Lofra, R.; Eglon, G.; et al. Respiratory involvement in ambulant and non-ambulant patients with facioscapulohumeral muscular dystrophy. J. Neurol. 2017, 264, 1271–1280. [Google Scholar] [CrossRef]
- Lutz, K.L.; Holte, L.; Kliethermes, S.A.; Stephan, C.; Mathews, K.D. Clinical and genetic features of hearing loss in facioscapulohumeral muscular dystrophy. Neurology 2013, 81, 1374–1377. [Google Scholar] [CrossRef] [Green Version]
- Campbell, A.E.; Shadle, S.C.; Jagannathan, S.; Lim, J.W.; Resnick, R.; Tawil, R.; van der Maarel, S.M.; Tapscott, S.J. Nurd and caf-1-mediated silencing of the d4z4 array is modulated by dux4-induced mbd3l proteins. eLife 2018, 7, e31023. [Google Scholar] [CrossRef]
- Darras, B.T.; Tawil, R. Predicting hearing loss in facioscapulohumeral muscular dystrophy. Neurology 2013, 81, 1370–1371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fitzsimons, R.B. Retinal vascular disease and the pathogenesis of facioscapulohumeral muscular dystrophy. A signalling message from wnt? Neuromuscul. Disord. 2011, 21, 263–271. [Google Scholar] [CrossRef] [PubMed]
- Statland, J.M.; Sacconi, S.; Farmakidis, C.; Donlin-Smith, C.M.; Chung, M.; Tawil, R. Coats syndrome in facioscapulohumeral dystrophy type 1: Frequency and d4z4 contraction size. Neurology 2013, 80, 1247–1250. [Google Scholar] [CrossRef] [Green Version]
- Grosso, S.; Mostardini, R.; Di Bartolo, R.M.; Balestri, P.; Verrotti, A. Epilepsy, speech delay, and mental retardation in facioscapulohumeral muscular dystrophy. Eur. J. Paediatr. Neurol. 2011, 15, 456–460. [Google Scholar] [CrossRef] [PubMed]
- Hobson-Webb, L.D.; Caress, J.B. Facioscapulohumeral muscular dystrophy can be a cause of isolated childhood cognitive dysfunction. J. Child Neurol. 2006, 21, 252–253. [Google Scholar] [PubMed]
- Funakoshi, M.; Goto, K.; Arahata, K. Epilepsy and mental retardation in a subset of early onset 4q35-facioscapulohumeral muscular dystrophy. Neurology 1998, 50, 1791–1794. [Google Scholar] [CrossRef]
- Miura, K.; Kumagai, T.; Matsumoto, A.; Iriyama, E.; Watanabe, K.; Goto, K.; Arahata, K. Two cases of chromosome 4q35-linked early onset facioscapulohumeral muscular dystrophy with mental retardation and epilepsy. Neuropediatrics 1998, 29, 239–241. [Google Scholar] [CrossRef]
- Saito, Y.; Miyashita, S.; Yokoyama, A.; Komaki, H.; Seki, A.; Maegaki, Y.; Ohno, K. Facioscapulohumeral muscular dystrophy with severe mental retardation and epilepsy. Brain Dev. 2007, 29, 231–233. [Google Scholar] [CrossRef]
- Laforet, P.; de Toma, C.; Eymard, B.; Becane, H.M.; Jeanpierre, M.; Fardeau, M.; Duboc, D. Cardiac involvement in genetically confirmed facioscapulohumeral muscular dystrophy. Neurology 1998, 51, 1454–1456. [Google Scholar] [CrossRef]
- Stevenson, W.G.; Perloff, J.K.; Weiss, J.N.; Anderson, T.L. Facioscapulohumeral muscular dystrophy: Evidence for selective, genetic electrophysiologic cardiac involvement. J. Am. Coll. Cardiol. 1990, 15, 292–299. [Google Scholar] [CrossRef]
- Yao, Z.; Snider, L.; Balog, J.; Lemmers, R.J.; Van Der Maarel, S.M.; Tawil, R.; Tapscott, S.J. Dux4-induced gene expression is the major molecular signature in fshd skeletal muscle. Hum. Mol. Genet. 2014, 23, 5342–5352. [Google Scholar] [CrossRef]
- Tawil, R.; Shaw, D.W.; van der Maarel, S.M.; Tapscott, S.J. Clinical trial preparedness in facioscapulohumeral dystrophy: Outcome measures and patient access: 8–9 April 2013, Leiden, The Netherlands. Neuromuscul. Disord. 2014, 24, 79–85. [Google Scholar] [CrossRef]
- Wang, L.H.; Friedman, S.D.; Shaw, D.; Snider, L.; Wong, C.J.; Budech, C.B.; Poliachik, S.L.; Gove, N.E.; Lewis, L.M.; Campbell, A.E.; et al. Mri-informed muscle biopsies correlate mri with pathology and dux4 target gene expression in fshd. Hum. Mol. Genet. 2019, 28, 476–486. [Google Scholar] [CrossRef] [PubMed]
- van den Heuvel, A.; Mahfouz, A.; Kloet, S.L.; Balog, J.; van Engelen, B.G.M.; Tawil, R.; Tapscott, S.J.; van der Maarel, S.M. Single-cell rna sequencing in facioscapulohumeral muscular dystrophy disease etiology and development. Hum. Mol. Genet. 2019, 28, 1064–1075. [Google Scholar] [CrossRef]
- Matsuzaka, Y.; Kishi, S.; Aoki, Y.; Komaki, H.; Oya, Y.; Takeda, S.; Hashido, K. Three novel serum biomarkers, mir-1, mir-133a, and mir-206 for limb-girdle muscular dystrophy, facioscapulohumeral muscular dystrophy, and becker muscular dystrophy. Environ. Health Prev. Med. 2014, 19, 452–458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Statland, J.; Donlin-Smith, C.M.; Tapscott, S.J.; van der Maarel, S.; Tawil, R. Multiplex screen of serum biomarkers in facioscapulohumeral muscular dystrophy. J. Neuromuscul. Dis. 2014, 1, 181–190. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.C.; King, O.D.; Zhang, Y.; Clayton, N.P.; Spencer, C.; Wentworth, B.M.; Emerson, C.P., Jr.; Wagner, K.R. Morpholino-mediated knockdown of dux4 toward facioscapulohumeral muscular dystrophy therapeutics. Mol. Ther. 2016, 24, 1405–1411. [Google Scholar] [CrossRef] [Green Version]
- Pandey, S.N.; Lee, Y.C.; Yokota, T.; Chen, Y.W. Morpholino treatment improves muscle function and pathology of pitx1 transgenic mice. Mol. Ther. 2014, 22, 390–396. [Google Scholar] [CrossRef] [Green Version]
- Wallace, L.M.; Saad, N.Y.; Pyne, N.K.; Fowler, A.M.; Eidahl, J.O.; Domire, J.S.; Griffin, D.A.; Herman, A.C.; Sahenk, Z.; Rodino-Klapac, L.R.; et al. Preclinical safety and off-target studies to support translation of aav-mediated rnai therapy for fshd. Mol. Ther. Methods Clin. Dev. 2018, 8, 121–130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oliva, J.; Galasinski, S.; Richey, A.; Campbell, A.E.; Meyers, M.J.; Modi, N.; Zhong, J.W.; Tawil, R.; Tapscott, S.J.; Sverdrup, F.M. Clinically advanced p38 inhibitors suppress dux4 expression in cellular and animal models of facioscapulohumeral muscular dystrophy. J. Pharmacol. Exp. Ther. 2019, 370, 219–230. [Google Scholar] [CrossRef]
- Mellion, M.; Ronco, L.; Thompson, L.; Hage, M.; Brooks, S.; van Brummelen, E.; Pagan, L.; Badrising, U.; Raines, S.; Tracewell, W.; et al. Phase 1 clinical trial of losmapimod in facioscapulohumeral muscular dystrophy (fshd): Safety, tolerability, and target engagement (1557). Neurology 2020, 94, 1557. [Google Scholar]
- Walker, G.; Butterfield, R.; Mathews, K.; Servais, L.; Day, J.; Gidaro, T.; Shukla, S.; Maggi, L. Results of a phase 1b/2 study of atyr1940 in adolescents and young adults with early onset facioscapulohumeral muscular dystrophy (fshd) (atyr1940-c-003). Neuromuscul. Disord. 2017, 27, S199. [Google Scholar] [CrossRef]
- Gershman, A.; Chiang, K.; Do, M.; Abbink, E.; Harbers, V.; Audebert, C.; Campana-Salort, E.; Monforte, M.; Iyadurai, S.; Carey, L.; et al. A randomized, double-blinded, placebo-controlled, multiple ascending dose study to evaluate the safety, tolerability, pharmacokinetics, immunogenicity, and biological activity of atyr1940 in adult patients with facioscapulohumeral muscular dystrophy (fshd). Neuromuscul. Disord. 2016, 26, S167. [Google Scholar] [CrossRef]
- Wagner, K.R.; Fleckenstein, J.L.; Amato, A.A.; Barohn, R.J.; Bushby, K.; Escolar, D.M.; Flanigan, K.M.; Pestronk, A.; Tawil, R.; Wolfe, G.I.; et al. A phase i/iitrial of myo-029 in adult subjects with muscular dystrophy. Ann. Neurol. 2008, 63, 561–571. [Google Scholar] [CrossRef] [PubMed]
- Glasser, C.E.; Gartner, M.R.; Wilson, D.; Miller, B.; Sherman, M.L.; Attie, K.M. Locally acting ace-083 increases muscle volume in healthy volunteers. Muscle Nerve 2018, 57, 921–926. [Google Scholar] [CrossRef]
- Passerieux, E.; Hayot, M.; Jaussent, A.; Carnac, G.; Gouzi, F.; Pillard, F.; Picot, M.C.; Bocker, K.; Hugon, G.; Pincemail, J.; et al. Effects of vitamin c, vitamin e, zinc gluconate, and selenomethionine supplementation on muscle function and oxidative stress biomarkers in patients with facioscapulohumeral dystrophy: A double-blind randomized controlled clinical trial. Free Radic. Biol. Med. 2015, 81, 158–169. [Google Scholar] [CrossRef]
- Campbell, A.E.; Oliva, J.; Yates, M.P.; Zhong, J.W.; Shadle, S.C.; Snider, L.; Singh, N.; Tai, S.; Hiramuki, Y.; Tawil, R.; et al. Bet bromodomain inhibitors and agonists of the beta-2 adrenergic receptor identified in screens for compounds that inhibit dux4 expression in fshd muscle cells. Skelet Muscle 2017, 7, 16. [Google Scholar] [CrossRef]
- Moyle, L.A.; Blanc, E.; Jaka, O.; Prueller, J.; Banerji, C.R.; Tedesco, F.S.; Harridge, S.D.; Knight, R.D.; Zammit, P.S. Ret function in muscle stem cells points to tyrosine kinase inhibitor therapy for facioscapulohumeral muscular dystrophy. eLife 2016, 5, e11405. [Google Scholar] [CrossRef]
- Sharma, V.; Pandey, S.N.; Khawaja, H.; Brown, K.J.; Hathout, Y.; Chen, Y.W. Parp1 differentially interacts with promoter region of dux4 gene in fshd myoblasts. J. Genet. Syndr. Gene Ther. 2016, 7, 303. [Google Scholar] [CrossRef]
- Tawil, R.; van der Maarel, S.; Padberg, G.W.; van Engelen, B.G. 171st enmc international workshop: Standards of care and management of facioscapulohumeral muscular dystrophy. Neuromuscul. Disord. 2010, 20, 471–475. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Inclusion Criteria |
|
| Exclusion Criteria |
|
| Supplementary Criteria |
|
| Therapeutic Approach (Mechanism of Action) | Compound Agents | Experiment Description/Potential Effectiveness | Trial Phase | Reference |
|---|---|---|---|---|
| Knocking down the aberrantly expressed DUX4 | Antisense oligonucleotides (AON) against DUX4 | Experiments showing morpholinos can either reduce DUX4 expression or inhibit translation of a DUX4-regulated gene, paired-like homeodomain transcription factor 1 (Pitx1) | Preclinical | [61,62] |
| miRNA against DUX4 | Adeno-associated viruses (AAV)-vector miRNA against DUX4 into mouse muscles ectopically expressing DUX4 and was able to reduce DUX4 and improve pathologies induced by it | Preclinical | [63] | |
| Reduce DUX4 expression by suppressing p38, mitogen-activated protein kinases (MAPKs) pathway | Losmapimod (p38α/β inhibitor) |
| Phase 1/2 (NCT04003974, NCT04004000, and NCT04264442), active | [64,65] |
| Immune modulation to reduce FSHD-related inflammatory response in muscles | ATYR1940 |
| Phase 1b/2a (NCT02836418), completed | [66,67] |
| Activating compensatory pathways (Myostatin inhibitors) | MYO-029 |
| Phase 1/2 (NCT00104078), completed | [68] |
| ACE-083 |
|
| [69] | |
| DUX4-related oxidative stress | Antioxidants (vitamin C, vitamin E, zinc gluconate, and selenomethionine) |
| Phase 3 (NCT01596803), completed | [70] |
| Target other molecular pathways related to DUX4 |
|
| All are preclinical. | [71,72,73] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, T.-H.; Wu, Y.-Z.; Tseng, Y.-H. Early-Onset Infantile Facioscapulohumeral Muscular Dystrophy: A Timely Review. Int. J. Mol. Sci. 2020, 21, 7783. https://doi.org/10.3390/ijms21207783
Chen T-H, Wu Y-Z, Tseng Y-H. Early-Onset Infantile Facioscapulohumeral Muscular Dystrophy: A Timely Review. International Journal of Molecular Sciences. 2020; 21(20):7783. https://doi.org/10.3390/ijms21207783
Chicago/Turabian StyleChen, Tai-Heng, Yan-Zhang Wu, and Yung-Hao Tseng. 2020. "Early-Onset Infantile Facioscapulohumeral Muscular Dystrophy: A Timely Review" International Journal of Molecular Sciences 21, no. 20: 7783. https://doi.org/10.3390/ijms21207783
APA StyleChen, T. -H., Wu, Y. -Z., & Tseng, Y. -H. (2020). Early-Onset Infantile Facioscapulohumeral Muscular Dystrophy: A Timely Review. International Journal of Molecular Sciences, 21(20), 7783. https://doi.org/10.3390/ijms21207783