Redox Regulation by Protein S-Glutathionylation: From Molecular Mechanisms to Implications in Health and Disease


Abstract
:1. Introduction
2. The Biochemistry of Protein Cysteine Residues: A Basic Overview
3. Low-Molecular Weight Non-Protein Thiols in Redox Regulation: Focus on Glutathione
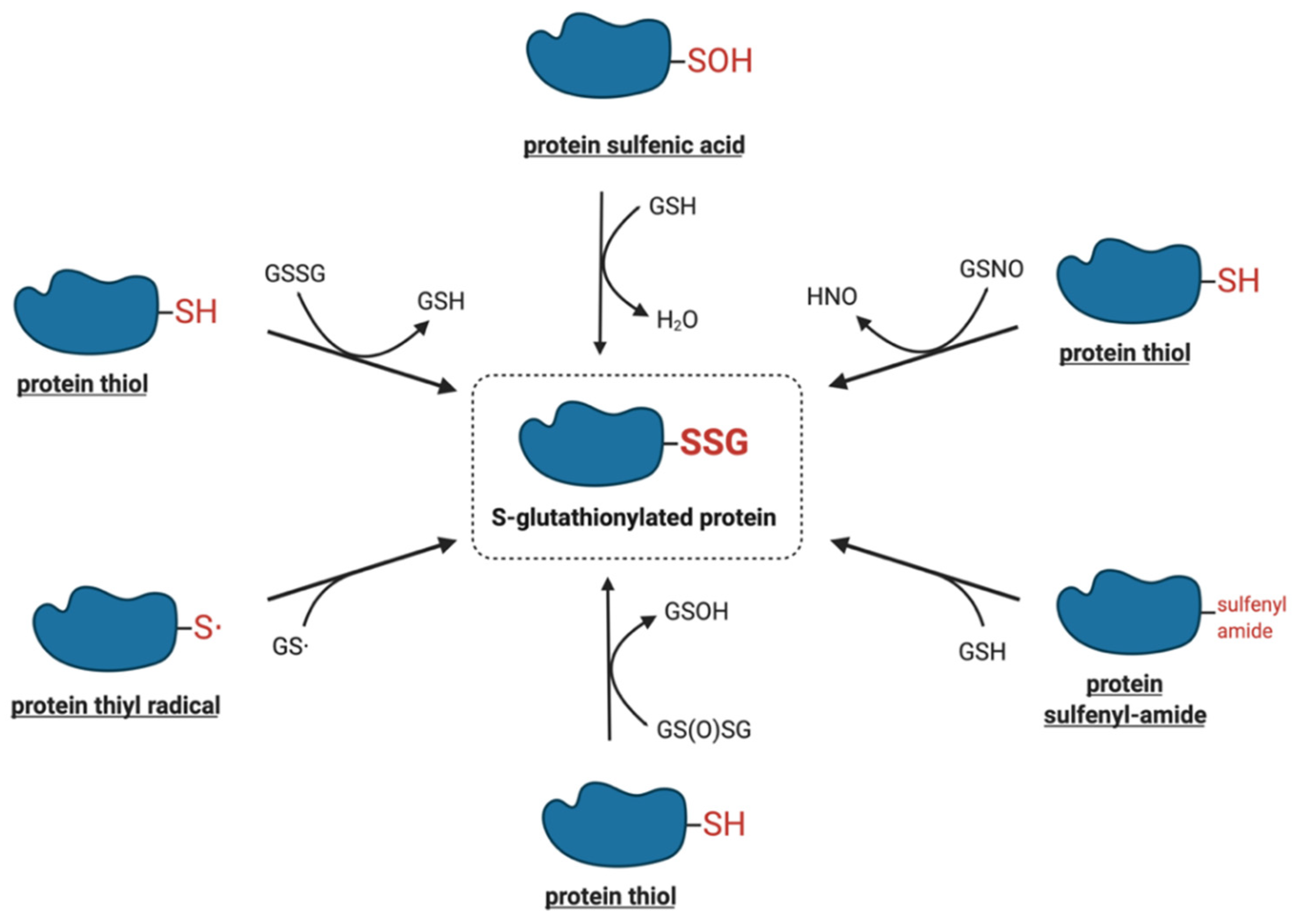
4. Molecular Mechanisms of Protein S-Glutathionylation
4.1. Thiol–Disulfide Exchange Mechanism
4.2. Reactive Thiol Intermediates for S-Glutathionylation
4.2.1. Sulfenic Acids
4.2.2. Sulfenyl-Amides
4.2.3. Thiyl Radicals
4.2.4. S-Nitrosylated Thiols
4.2.5. Thiosulfinates
5. Enzymatic Protein S-Glutathionylation
5.1. Glutathione S-Transferase π
5.2. Other Potential Enzymes
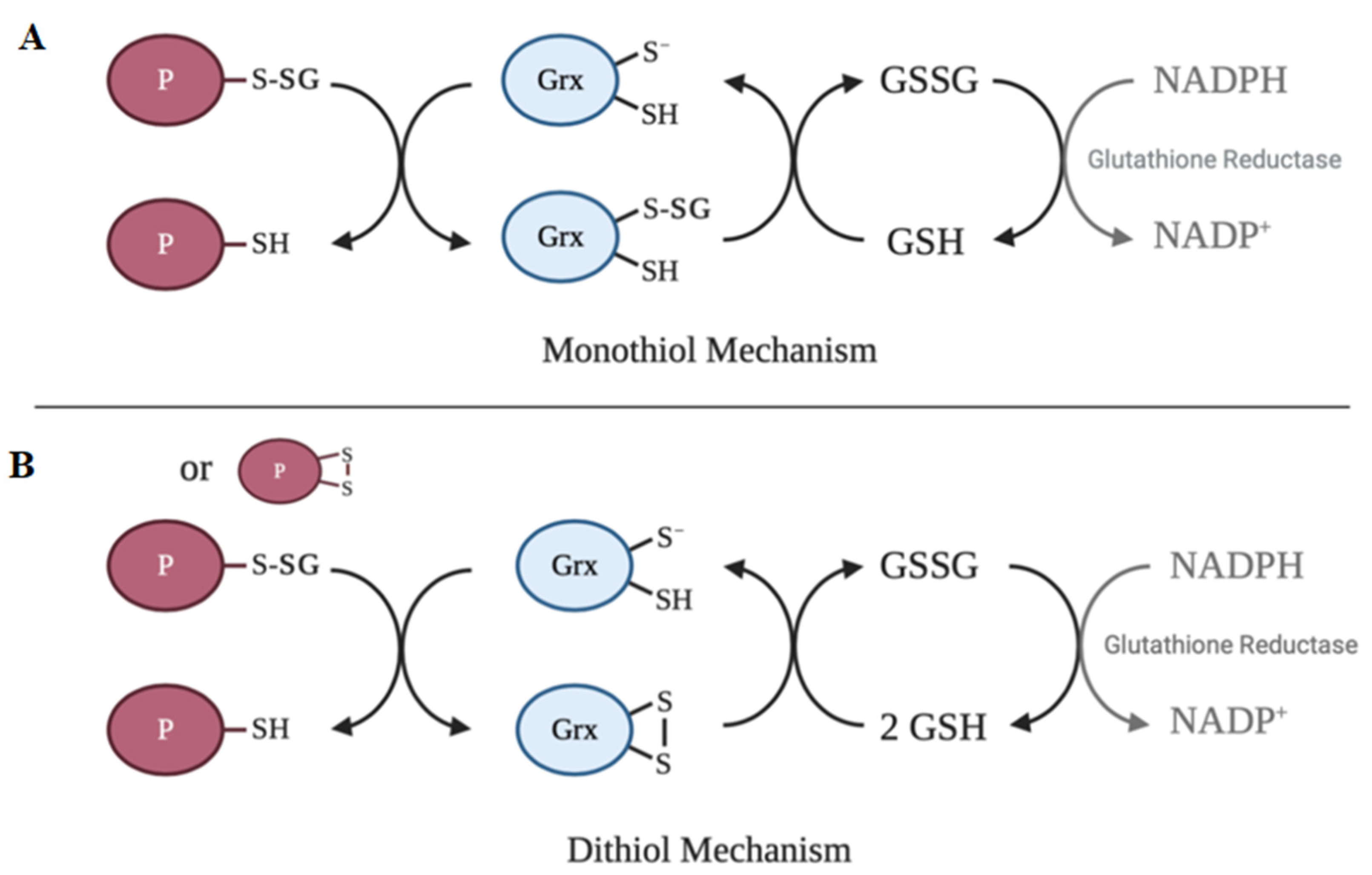
6. Deglutathionylation
7. Structure–Function Relationship of Protein S-Glutathionylation
8. Implications of Protein S-Glutathionylation in Diseases
8.1. Aging and Neurodegeneration
8.2. Cardiovascular Disease
8.2.1. Myocardial Infarction
8.2.2. Cardiac Hypertrophy
8.3. Cancer
8.4. Liver Disease
9. Concluding Remarks
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ROS | reactive oxygen species |
| GSH | reduced glutathione |
| GSSG | oxidized glutathione |
| RSSR’ | disulfide |
| RSOH | sulfenic acid |
| RSO2H | sulfinic acid |
| RSO3H | sulfonic acid |
| RSNHR’ | sulfenyl-amide |
| GGT | γ-glutamyl transpeptidase |
| GST | glutathione S-transferase |
| Grx | glutaredoxin |
References
- Sies, H.; Jones, D.P. Reactive oxygen species (ROS) as pleiotropic physiological signalling agents. Nat. Rev. Mol. Cell Biol. 2020, 21, 363–383. [Google Scholar] [CrossRef] [PubMed]
- Panday, A.; Sahoo, M.K.; Osorio, D.; Batra, S. NADPH oxidases: An overview from structure to innate immunity-associated pathologies. Cell. Mol. Immunol. 2015, 12, 5–23. [Google Scholar] [CrossRef] [Green Version]
- Liguori, I.; Russo, G.; Curcio, F.; Bulli, G.; Aran, L.; Della-Morte, D.; Gargiulo, G.; Testa, G.; Cacciatore, F.; Bonaduce, D.; et al. Oxidative stress, aging, and diseases. Clin. Interv. Aging 2018, 13, 757–772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Finkel, T. Signal transduction by reactive oxygen species. J. Cell Biol. 2011, 194, 7–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sies, H. Hydrogen peroxide as a central redox signaling molecule in physiological oxidative stress: Oxidative eustress. Redox Biol. 2017, 11, 613–619. [Google Scholar] [CrossRef] [PubMed]
- Moran, L.K.; Gutteridge, J.M.; Quinlan, G.J. Thiols in cellular redox signalling and control. Curr. Med. Chem. 2001, 8, 763–772. [Google Scholar] [CrossRef]
- Leonberg, A.K.; Chai, Y.C. The functional role of cysteine residues for c-Abl kinase activity. Mol. Cell. Biochem. 2007, 304, 207–212. [Google Scholar] [CrossRef]
- Paulsen, C.E.; Carroll, K.S. Cysteine-mediated redox signaling: Chemistry, biology, and tools for discovery. Chem. Rev. 2013, 113, 4633–4679. [Google Scholar] [CrossRef]
- Mannaa, A.; Hanisch, F.G. Redox Proteomes in Human Physiology and Disease Mechanisms. J. Proteome Res. 2020, 19, 1–17. [Google Scholar] [CrossRef]
- Poole, L.B. The basics of thiols and cysteines in redox biology and chemistry. Free Radic. Biol. Med. 2015, 80, 148–157. [Google Scholar] [CrossRef] [Green Version]
- Marino, S.M.; Gladyshev, V.N. Analysis and functional prediction of reactive cysteine residues. J. Biol. Chem. 2012, 287, 4419–4425. [Google Scholar] [CrossRef] [Green Version]
- Pace, N.J.; Weerapana, E. A competitive chemical-proteomic platform to identify zinc-binding cysteines. ACS Chem. Biol. 2014, 9, 258–265. [Google Scholar] [CrossRef] [PubMed]
- Reddie, K.G.; Carroll, K.S. Expanding the functional diversity of proteins through cysteine oxidation. Curr. Opin. Chem. Biol. 2008, 12, 746–754. [Google Scholar] [CrossRef] [PubMed]
- Go, Y.M.; Chandler, J.D.; Jones, D.P. The cysteine proteome. Free Radic. Biol. Med. 2015, 84, 227–245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Requejo, R.; Hurd, T.R.; Costa, N.J.; Murphy, M.P. Cysteine residues exposed on protein surfaces are the dominant intramitochondrial thiol and may protect against oxidative damage. FEBS J. 2010, 277, 1465–1480. [Google Scholar] [CrossRef] [Green Version]
- Ferrer-Sueta, G.; Manta, B.; Botti, H.; Radi, R.; Trujillo, M.; Denicola, A. Factors affecting protein thiol reactivity and specificity in peroxide reduction. Chem. Res. Toxicol. 2011, 24, 434–450. [Google Scholar] [CrossRef] [PubMed]
- Trujillo, M.; Alvarez, B.; Radi, R. One-and two-electron oxidation of thiols: Mechanisms, kinetics and biological fates. Free Radic. Res. 2016, 50, 150–171. [Google Scholar] [CrossRef]
- Licht, S.; Gerfen, G.J.; Stubbe, J. Thiyl radicals in ribonucleotide reductases. Science 1996, 271, 477–481. [Google Scholar] [CrossRef] [PubMed]
- Lo Conte, M.; Carroll, K.S. The redox biochemistry of protein sulfenylation and sulfinylation. J. Biol. Chem. 2013, 288, 26480–26488. [Google Scholar] [CrossRef] [Green Version]
- Gupta, V.; Carroll, K.S. Sulfenic acid chemistry, detection and cellular lifetime. Biochim. Biophys. Acta 2014, 1840, 847–875. [Google Scholar] [CrossRef] [Green Version]
- Flohé, L. The impact of thiol peroxidases on redox regulation. Free Radic. Res. 2016, 50, 126–142. [Google Scholar] [CrossRef]
- Ziegler, D.M. Role of reversible oxidation-reduction of enzyme thiols-disulfides in metabolic regulation. Annu. Rev. Biochem. 1985, 54, 305–329. [Google Scholar] [CrossRef]
- Biteau, B.; Labarre, J.; Toledano, M.B. ATP-dependent reduction of cysteine-sulphinic acid by, S. cerevisiae sulphiredoxin. Nature 2003, 425, 980–984. [Google Scholar] [CrossRef]
- Leichert, L.I.; Gehrke, F.; Gudiseva, H.V.; Blackwell, T.; Ilbert, M.; Walker, A.K.; Strahler, J.R.; Andrews, P.C.; Jakob, U. Quantifying changes in the thiol redox proteome upon oxidative stress in vivo. Proc. Natl. Acad. Sci. USA 2008, 105, 8197–8202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le Moan, N.; Clement, G.; Le Maout, S.; Tacnet, F.; Toledano, M.B. The Saccharomyces cerevisiae proteome of oxidized protein thiols: Contrasted functions for the thioredoxin and glutathione pathways. J. Biol. Chem. 2006, 281, 10420–10430. [Google Scholar] [CrossRef] [Green Version]
- Jones, D.P. Radical-free biology of oxidative stress. Am. J. Physiol. Cell Physiol. 2008, 295, C849–C868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Held, J.M.; Gibson, B.W. Regulatory control or oxidative damage? Proteomic approaches to interrogate the role of cysteine oxidation status in biological processes. Mol. Cell. Proteom. 2012, 11, R111.013037. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Winterbourn, C.C.; Metodiewa, D. Reactivity of biologically important thiol compounds with superoxide and hydrogen peroxide. Free Radic. Biol. Med. 1999, 27, 322–328. [Google Scholar] [CrossRef]
- Winterbourn, C.C.; Hampton, M.B. Thiol chemistry and specificity in redox signaling. Free Radic. Biol. Med. 2008, 45, 549–561. [Google Scholar] [CrossRef]
- Hall, A.; Parsonage, D.; Poole, L.B.; Karplus, P.A. Structural evidence that peroxiredoxin catalytic power is based on transition-state stabilization. J. Mol. Biol. 2010, 402, 194–209. [Google Scholar] [CrossRef] [Green Version]
- Allen, E.M.; Mieyal, J.J. Protein-thiol oxidation and cell death: Regulatory role of glutaredoxins. Antioxid. Redox Signal. 2012, 17, 1748–1763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Ye, Z.W.; Singh, S.; Townsend, D.M.; Tew, K.D. An evolving understanding of the S-glutathionylation cycle in pathways of redox regulation. Free Radic. Biol. Med. 2018, 120, 204–216. [Google Scholar] [CrossRef] [PubMed]
- Ulrich, K.; Jakob, U. The role of thiols in antioxidant systems. Free Radic. Biol. Med. 2019, 140, 14–27. [Google Scholar] [CrossRef] [PubMed]
- Van Laer, K.; Hamilton, C.J.; Messens, J. Low-molecular-weight thiols in thiol-disulfide exchange. Antioxid. Redox Signal. 2013, 18, 642–1653. [Google Scholar] [CrossRef] [PubMed]
- Gout, I. Coenzyme A: A protective thiol in bacterial antioxidant defence. Biochem. Soc. Trans. 2019, 47, 469–476. [Google Scholar] [CrossRef]
- Newton, G.L.; Rawat, M.; La Clair, J.J.; Jothivasan, V.K.; Budiarto, T.; Hamilton, C.J.; Claiborne, A.; Helmann, J.D.; Fahey, R.C. Bacillithiol is an antioxidant thiol produced in Bacilli. Nat. Chem. Biol. 2009, 5, 625–627. [Google Scholar] [CrossRef]
- Newton, G.L.; Buchmeier, N.; Fahey, R.C. Biosynthesis and functions of mycothiol, the unique protective thiol of Actinobacteria. Microbiol. Mol. Biol. Rev. 2008, 72, 471–494. [Google Scholar] [CrossRef] [Green Version]
- Manta, B.; Bonilla, M.; Fiestas, L.; Sturlese, M.; Salinas, G.; Bellanda, M.; Comini, M.A. Polyamine-Based Thiols in Trypanosomatids: Evolution, Protein Structural Adaptations, and Biological Functions. Antioxid. Redox Signal. 2018, 28, 463–486. [Google Scholar] [CrossRef]
- Lu, S.C. Glutathione synthesis. Biochim. Biophys. Acta 2013, 1830, 3143–3153. [Google Scholar] [CrossRef] [Green Version]
- Crawford, R.R.; Prescott, E.T.; Sylvester, C.F.; Higdon, A.N.; Shan, J.; Kilberg, M.S.; Mungrue, I.N. Human CHAC1 Protein Degrades Glutathione, and mRNA Induction Is Regulated by the Transcription Factors ATF4 and ATF3 and a Bipartite ATF/CRE Regulatory Element. J. Biol. Chem. 2015, 290, 15878–15891. [Google Scholar] [CrossRef] [Green Version]
- Kumar, A.; Tikoo, S.; Maity, S.; Sengupta, S.; Sengupta, S.; Kaur, A.; Bachhawat, A.K. Mammalian proapoptotic factor ChaC1 and its homologues function as γ-glutamyl cyclotransferases acting specifically on glutathione. EMBO Rep. 2012, 13, 1095–1101. [Google Scholar] [CrossRef] [Green Version]
- Tsunoda, S.; Avezov, E.; Zyryanova, A.; Konno, T.; Mendes-Silva, L.; Melo, E.P.; Ron, D. Intact protein folding in the glutathione-depleted endoplasmic reticulum implicates alternative protein thiol reductants. Elife 2014, 3, e03421. [Google Scholar] [CrossRef]
- Forman, H.J.; Zhang, H.; Rinna, A. Glutathione: Overview of its protective roles, measurement, and biosynthesis. Mol. Asp. Med. 2009, 30, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Hwang, C.; Sinskey, A.J.; Lodish, H.F. Oxidized redox state of glutathione in the endoplasmic reticulum. Science 1992, 257, 1496–1502. [Google Scholar] [CrossRef]
- Pirie, N.W.; Pinhey, K.G. The Titration Curve of Glutathione. J. Biol. Chem. 1929, 84, 321–333. [Google Scholar]
- Vaish, S.; Gupta, D.; Mehrotra, R.; Mehrotra, S.; Basantani, M.K. Glutathione S-transferase: A versatile protein family. 3 Biotech. 2020, 10, 321. [Google Scholar] [CrossRef]
- Molavian, H.; Madani Tonekaboni, A.; Kohandel, M.; Sivaloganathan, S. The Synergetic Coupling among the Cellular Antioxidants Glutathione Peroxidase/Peroxiredoxin and Other Antioxidants and its Effect on the Concentration of H2O2. Sci. Rep. 2015, 5, 13620. [Google Scholar] [CrossRef]
- Matsui, R.; Ferran, B.; Oh, A.; Croteau, D.; Shao, D.; Han, J.; Pimentel, D.R.; Bachschmid, M.M. Redox Regulation via Glutaredoxin-1 and Protein S-Glutathionylation. Antioxid. Redox Signal. 2020, 32, 677–700. [Google Scholar] [CrossRef] [PubMed]
- Mieyal, J.J.; Gallogly, M.M.; Qanungo, S.; Sabens, E.A.; Shelton, M.D. Molecular mechanisms and clinical implications of reversible protein S-glutathionylation. Antioxid. Redox Signal. 2008, 10, 1941–1988. [Google Scholar] [CrossRef] [PubMed]
- Dalle-Donne, I.; Milzani, A.; Gagliano, N.; Colombo, R.; Giustarini, D.; Rossi, R. Molecular mechanisms and potential clinical significance of S-glutathionylation. Antioxid. Redox Signal. 2008, 10, 445–473. [Google Scholar] [CrossRef]
- Chai, Y.C.; Ashraf, S.S.; Rokutan, K.; Johnston, R.B., Jr.; Thomas, J.A. S-thiolation of individual human neutrophil proteins including actin by stimulation of the respiratory burst: Evidence against a role for glutathione disulfide. Arch. Biochem. Biophys. 1994, 310, 273–281. [Google Scholar] [CrossRef] [PubMed]
- Klatt, P.; Molina, E.P.; De Lacoba, M.G.; Padilla, C.A.; Martinez-Galesteo, E.; Barcena, J.A.; Lamas, S. Redox regulation of c-Jun DNA binding by reversible S-glutathiolation. FASEB J. 1999, 13, 1481–1490. [Google Scholar] [CrossRef] [PubMed]
- Zaffagnini, M.; Bedhomme, M.; Marchand, C.H.; Morisse, S.; Trost, P.; Lemaire, S.D. Redox regulation in photosynthetic organisms: Focus on glutathionylation. Antioxid. Redox Signal. 2012, 16, 567–586. [Google Scholar] [CrossRef] [Green Version]
- Gallogly, M.M.; Mieyal, J.J. Mechanisms of reversible protein glutathionylation in redox signaling and oxidative stress. Curr. Opin. Pharmacol. 2007, 7, 381–391. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Sevier, C.S. Formation and Reversibility of BiP Protein Cysteine Oxidation Facilitate Cell Survival during and post Oxidative Stress. J. Biol. Chem. 2016, 291, 7541–7557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.R.; Kwon, K.S.; Kim, S.R.; Rhee, S.G. Reversible inactivation of protein-tyrosine phosphatase 1B in A431 cells stimulated with epidermal growth factor. J. Biol. Chem. 1998, 273, 15366–15372. [Google Scholar] [CrossRef] [Green Version]
- Denu, J.M.; Tanner, K.G. Specific and reversible inactivation of protein tyrosine phosphatases by hydrogen peroxide: Evidence for a sulfenic acid intermediate and implications for redox regulation. Biochemistry 1998, 37, 5633–5642. [Google Scholar] [CrossRef]
- Barrett, W.C.; DeGnore, J.P.; König, S.; Fales, H.M.; Keng, Y.F.; Zhang, Z.Y.; Yim, M.B.; Chock, P.B. Regulation of PTP1B via glutathionylation of the active site cysteine 215. Biochemistry 1999, 38, 6699–6705. [Google Scholar] [CrossRef]
- Heppner, D.E.; Hristova, M.; Dustin, C.M.; Danyal, K.; Habibovic, A.; van der Vliet, A. The NADPH Oxidases DUOX1 and NOX2 Play Distinct Roles in Redox Regulation of Epidermal Growth Factor Receptor Signaling. J. Biol. Chem. 2016, 291, 23282–23293. [Google Scholar] [CrossRef] [Green Version]
- Zaffagnini, M.; Marchand, C.H.; Malferrari, M.; Murail, S.; Bonacchi, S.; Genovese, D.; Montalti, M.; Venturoli, G.; Falini, G.; Baaden, M.; et al. Glutathionylation primes soluble glyceraldehyde-3-phosphate dehydrogenase for late collapse into insoluble aggregates. Proc. Natl. Acad. Sci. USA 2019, 116, 26057–26065. [Google Scholar] [CrossRef]
- Salmeen, A.; Andersen, J.N.; Myers, M.P.; Meng, T.C.; Hinks, J.A.; Tonks, N.K.; Barford, D. Redox regulation of protein tyrosine phosphatase 1B involves a sulphenyl-amide intermediate. Nature 2003, 423, 769–773. [Google Scholar] [CrossRef] [PubMed]
- Stoyanovsky, D.A.; Maeda, A.; Atkins, J.L.; Kagan, V.E. Assessments of thiyl radicals in biosystems: Difficulties and new applications. Anal Chem. 2011, 83, 6432–6438. [Google Scholar] [CrossRef] [PubMed]
- Frey, P.A. Radical mechanisms of enzymatic catalysis. Annu. Rev. Biochem. 2001, 70, 121–148. [Google Scholar] [CrossRef]
- Schöneich, C. Thiyl radicals and induction of protein degradation. Free Radic. Res. 2016, 50, 143–149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Starke, D.W.; Chock, P.B.; Mieyal, J.J. Glutathione-thiyl radical scavenging and transferase properties of human glutaredoxin (thioltransferase). Potential role in redox signal transduction. J. Biol. Chem. 2003, 278, 14607–14613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, P.T.; Zhang, L.; Chen, C.L.; Chen, J.; Green, K.B.; Chen, Y.R. Protein thiyl radical mediates S-glutathionylation of complex, I. Free Radic. Biol. Med. 2012, 53, 962–973. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, P.T.; Chen, C.L.; Chen, Y.R. Increased mitochondrial prooxidant activity mediates up-regulation of Complex I S-glutathionylation via protein thiyl radical in the murine heart of eNOS(-/-). Free Radic. Biol. Med. 2015, 79, 56–68. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.A.; Lin, C.H.; Druhan, L.J.; Wang, T.Y.; Chen, Y.R.; Zweier, J.L. Superoxide induces endothelial nitric-oxide synthase protein thiyl radical formation, a novel mechanism regulating eNOS function and coupling. J. Biol. Chem. 2011, 286, 29098–29107. [Google Scholar] [CrossRef] [Green Version]
- Zweier, J.L.; Chen, C.A.; Druhan, L.J. S-glutathionylation reshapes our understanding of endothelial nitric oxide synthase uncoupling and nitric oxide/reactive oxygen species-mediated signaling. Antioxid. Redox Signal. 2011, 14, 1769–1775. [Google Scholar] [CrossRef] [Green Version]
- Foster, M.W.; Hess, D.T.; Stamler, J.S. Protein S-nitrosylation in health and disease: A current perspective. Trends Mol. Med. 2009, 15, 391–404. [Google Scholar] [CrossRef] [Green Version]
- Ehrenfeld, P.; Cordova, F.; Duran, W.N.; Sanchez, F.A. S-nitrosylation and its role in breast cancer angiogenesis and metastasis. Nitric Oxide 2019, 87, 52–59. [Google Scholar] [CrossRef]
- Stomberski, C.T.; Hess, D.T.; Stamler, J.S. Protein S-Nitrosylation: Determinants of Specificity and Enzymatic Regulation of S-Nitrosothiol-Based Signaling. Antioxid. Redox Signal. 2019, 30, 1331–1351. [Google Scholar] [CrossRef] [PubMed]
- Williams, D.L.H. The Chemistry of S-Nitrosothiols. Acc. Chem. Res. 1999, 32, 689–876. [Google Scholar] [CrossRef]
- Grek, C.L.; Zhang, J.; Manevich, Y.; Townsend, D.M.; Tew, K.D. Causes and consequences of cysteine S-glutathionylation. J. Biol. Chem. 2013, 288, 26497–26504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belcastro, E.; Gaucher, C.; Corti, A.; Leroy, P.; Lartaud, I.; Pompella, A. Regulation of protein function by S-nitrosation and S-glutathionylation: Processes and targets in cardiovascular pathophysiology. Biol. Chem. 2017, 398, 1267–1293. [Google Scholar] [CrossRef]
- Klatt, P.; Molina, E.P.; Lamas, S. Nitric oxide inhibits c-Jun DNA binding by specifically targeted S-glutathionylation. J. Biol. Chem. 1999, 274, 15857–15864. [Google Scholar] [CrossRef] [Green Version]
- Giustarini, D.; Milzani, A.; Aldini, G.; Carini, M.; Rossi, R.; Dalle-Donne, I. S-nitrosation versus S-glutathionylation of protein sulfhydryl groups by S-nitrosoglutathione. Antioxid. Redox Signal. 2005, 7, 930–939. [Google Scholar] [CrossRef]
- Mohr, S.; Hallak, H.; de Boitte, A.; Lapetina, E.G.; Brüne, B. Nitric oxide-induced S-glutathionylation and inactivation of glyceraldehyde-3-phosphate dehydrogenase. J. Biol. Chem. 1999, 274, 9427–9430. [Google Scholar] [CrossRef] [Green Version]
- Dutka, T.L.; Mollica, J.P.; Lamboley, C.R.; Weerakkody, V.C.; Greening, D.W.; Posterino, G.S.; Murphy, R.M.; Lamb, G.D. S-nitrosylation and S-glutathionylation of Cys134 on troponin I have opposing competitive actions on Ca2+ sensitivity in rat fast-twitch muscle fibers. Am. J. Physiol. Cell Physiol. 2017, 312, C316–C327. [Google Scholar] [CrossRef]
- Huang, K.P.; Huang, F.L. Glutathionylation of proteins by glutathione disulfide S-oxide. Biochem. Pharmacol. 2002, 64, 1049–1056. [Google Scholar] [CrossRef]
- Li, J.; Huang, F.L.; Huang, K.P. Glutathiolation of proteins by glutathione disulfide S-oxide derived from S-nitrosoglutathione. Modifications of rat brain neurogranin/RC3 and neuromodulin/GAP-43. J. Biol. Chem. 2001, 276, 3098–3105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okamoto, T.; Akaike, T.; Sawa, T.; Miyamoto, Y.; van der Vliet, A.; Maeda, H. Activation of matrix metalloproteinases by peroxynitrite-induced protein S-glutathiolation via disulfide S-oxide formation. J. Biol. Chem. 2001, 276, 29596–29602. [Google Scholar] [CrossRef] [Green Version]
- Sadidi, M.; Geddes, T.J.; Kuhn, D.M. S-thiolation of tyrosine hydroxylase by reactive nitrogen species in the presence of cysteine or glutathione. Antioxid. Redox Signal. 2005, 7, 863–869. [Google Scholar] [CrossRef] [PubMed]
- Boyland, E.; Chasseaud, L.F. Enzyme-catalysed conjugations of glutathione with unsaturated compounds. Biochem. J. 1967, 104, 95–102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Townsend, D.M.; Manevich, Y.; He, L.; Hutchens, S.; Pazoles, C.J.; Tew, K.D. Novel role for glutathione S-transferase pi. Regulator of protein S-Glutathionylation following oxidative and nitrosative stress. J. Biol. Chem. 2009, 284, 436–445. [Google Scholar] [CrossRef] [Green Version]
- Manevich, Y.; Feinstein, S.I.; Fisher, A.B. Activation of the antioxidant enzyme 1-CYS peroxiredoxin requires glutathionylation mediated by heterodimerization with pi GST. Proc. Natl. Acad. Sci. USA 2004, 101, 3780–3785. [Google Scholar] [CrossRef] [Green Version]
- Bartolini, D.; Torquato, P.; Piroddi, M.; Galli, F. Targeting glutathione S-transferase P and its interactome with selenium compounds in cancer therapy. Biochim. Biophys. Acta Gen. Subj. 2019, 1863, 130–143. [Google Scholar] [CrossRef]
- Townsend, D.M.; He, L.; Hutchens, S.; Garrett, T.E.; Pazoles, C.J.; Tew, K.D. NOV-002, a glutathione disulfide mimetic, as a modulator of cellular redox balance. Cancer Res. 2008, 68, 2870–2877. [Google Scholar] [CrossRef] [Green Version]
- Ko, K.Y.; Lee, J.H.; Jang, J.K.; Jin, Y.; Kang, H.; Kim, I.Y. S-Glutathionylation of mouse selenoprotein W prevents oxidative stress-induced cell death by blocking the formation of an intramolecular disulfide bond. Free Radic. Biol. Med. 2019, 141, 362–371. [Google Scholar] [CrossRef]
- Ye, Z.W.; Zhang, J.; Ancrum, T.; Manevich, Y.; Townsend, D.M.; Tew, K.D. Glutathione S-Transferase P-Mediated Protein S-Glutathionylation of Resident Endoplasmic Reticulum Proteins Influences Sensitivity to Drug-Induced Unfolded Protein Response. Antioxid. Redox Signal. 2017, 26, 247–261. [Google Scholar] [CrossRef] [Green Version]
- Carvalho, A.N.; Marques, C.; Guedes, R.C.; Castro-Caldas, M.; Rodrigues, E.; van Horssen, J.; Gama, M.J. S-Glutathionylation of Keap1: A new role for glutathione S-transferase pi in neuronal protection. FEBS Lett. 2016, 590, 1455–1466. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Ye, Z.W.; Chen, W.; Manevich, Y.; Mehrotra, S.; Ball, L.; Janssen-Heininger, Y.; Tew, K.D.; Townsend, D.M. S-Glutathionylation of estrogen receptor α affects dendritic cell function. J. Biol. Chem. 2018, 293, 4366–4380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uemura, T.; Tsaprailis, G.; Gerner, E.W. GSTΠ stimulates caveolin-1-regulated polyamine uptake via actin remodeling. Oncotarget 2019, 10, 5713–5723. [Google Scholar] [CrossRef] [PubMed]
- Klaus, A.; Zorman, S.; Berthier, A.; Polge, C.; Ramirez, S.; Michelland, S.; Sève, M.; Vertommen, D.; Rider, M.; Lentze, N.; et al. Glutathione S-transferases interact with AMP-activated protein kinase: Evidence for S-glutathionylation and activation in vitro. PLoS ONE 2013, 8, e62497. [Google Scholar] [CrossRef]
- Tew, K.D.; Townsend, D.M. Regulatory functions of glutathione S-transferase P1-1 unrelated to detoxification. Drug Metab. Rev. 2011, 43, 179–193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujitani, N.; Yoneda, A.; Takahashi, M.; Takasawa, A.; Aoyama, T.; Miyazaki, T. Silencing of Glutathione S-Transferase Pi Inhibits Cancer Cell Growth via Oxidative Stress Induced by Mitochondria Dysfunction. Sci. Rep. 2019, 9, 14764. [Google Scholar] [CrossRef]
- Janssen-Heininger, Y.M.; Nolin, J.D.; Hoffman, S.M.; van der Velden, J.L.; Tully, J.E.; Lahue, K.G.; Abdalla, S.T.; Chapman, D.G.; Reynaert, N.L.; van der Vliet, A.; et al. Emerging mechanisms of glutathione-dependent chemistry in biology and disease. J. Cell Biochem. 2013, 114, 1962–1968. [Google Scholar] [CrossRef] [Green Version]
- Ercolani, L.; Scirè, A.; Galeazzi, R.; Massaccesi, L.; Cianfruglia, L.; Amici, A.; Piva, F.; Urbanelli, L.; Emiliani, C.; Principato, G.; et al. A possible S-glutathionylation of specific proteins by glyoxalase II: An in vitro and in silico study. Cell Biochem. Funct. 2016, 34, 620–627. [Google Scholar] [CrossRef]
- Galeazzi, R.; Laudadio, E.; Falconi, E.; Massaccesi, L.; Ercolani, L.; Mobbili, G.; Minnelli, C.; Scirè, A.; Cianfruglia, L.; Armeni, T. Protein-protein interactions of human glyoxalase II: Findings of a reliable docking protocol. Org. Biomol. Chem. 2018, 16, 5167–5177. [Google Scholar] [CrossRef]
- Brings, S.; Fleming, T.; Freichel, M.; Muckenthaler, M.U.; Herzig, S.; Nawroth, P.P. Dicarbonyls and Advanced Glycation End-Products in the Development of Diabetic Complications and Targets for Intervention. Int. J. Mol. Sci. 2017, 18, 984. [Google Scholar] [CrossRef] [Green Version]
- Harmel, R.; Fiedler, D. Features and regulation of non-enzymatic post-translational modifications. Nat. Chem. Biol. 2018, 14, 244–252. [Google Scholar] [CrossRef] [PubMed]
- Ren, G.; Stephan, D.; Xu, Z.; Zheng, Y.; Tang, D.; Harrison, R.S.; Kurz, M.; Jarrott, R.; Shouldice, S.R.; Hiniker, A. Properties of the thioredoxin fold superfamily are modulated by a single amino acid residue. J. Biol. Chem. 2009, 284, 10150–10159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zimmermann, J.; Oestreicher, J.; Hess, S.; Herrmann, J.M.; Deponte, M.; Morgan, B. One cysteine is enough: A monothiol Grx can functionally replace all cytosolic Trx and dithiol Grx. Redox Biol. 2020, 36, 101598. [Google Scholar] [CrossRef]
- Chrestensen, C.A.; Starke, D.W.; Mieyal, J.J. Acute cadmium exposure inactivates thioltransferase (Glutaredoxin), inhibits intracellular reduction of protein-glutathionyl-mixed disulfides, and initiates apoptosis. J. Biol. Chem. 2000, 275, 26556–26565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jung, C.H.; Thomas, J.A. S-glutathiolated hepatocyte proteins and insulin disulfides as substrates for reduction by glutaredoxin, thioredoxin, protein disulfide isomerase, and glutathione. Arch. Biochem. Biophys. 1996, 335, 61–72. [Google Scholar] [CrossRef] [PubMed]
- Herrero, E.; de la Torre-Ruiz, M.A. Monothiol glutaredoxins: A common domain for multiple functions. Cell. Mol. Life Sci. 2007, 64, 1518–1530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Couturier, J.; Przybyla-Toscano, J.; Roret, T.; Didierjean, C.; Rouhier, N. The roles of glutaredoxins ligating Fe-S clusters: Sensing, transfer or repair functions? Biochim. Biophys. Acta 2015, 1853, 1513–1527. [Google Scholar] [CrossRef]
- Berndt, C.; Lillig, C.H.; Holmgren, A. Thioredoxins and glutaredoxins as facilitators of protein folding. Biochim. Biophys. Acta 2008, 1783, 641–650. [Google Scholar] [CrossRef] [Green Version]
- Park, J.W.; Mieyal, J.J.; Rhee, S.G.; Chock, P.B. Deglutathionylation of 2-Cys peroxiredoxin is specifically catalyzed by sulfiredoxin. J. Biol. Chem. 2009, 284, 23364–23374. [Google Scholar] [CrossRef] [Green Version]
- Findlay, V.J.; Tapiero, H.; Townsend, D.M. Sulfiredoxin: A potential therapeutic agent? Biomed. Pharmacother. 2005, 59, 374–379. [Google Scholar] [CrossRef]
- Findlay, V.J.; Townsend, D.M.; Morris, T.E.; Fraser, J.P.; He, L.; Tew, K.D. A novel role for human sulfiredoxin in the reversal of glutathionylation. Cancer Res. 2006, 66, 6800–6806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shao, D.; Fry, J.L.; Han, J.; Hou, X.; Pimentel, D.R.; Matsui, R.; Cohen, R.A.; Bachschmid, M.M. A redox-resistant sirtuin-1 mutant protects against hepatic metabolic and oxidant stress. J. Biol. Chem. 2014, 289, 7293–7306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mandato, A.; Chai, Y.C. Regulation of antigen 85C activity by reversible S-glutathionylation. IUBMB Life 2018, 70, 1111–1114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gandhirajan, R.K.; Jain, M.; Walla, B.; Johnsen, M.; Bartram, M.P.; Huynh Anh, M.; Rinschen, M.M.; Benzing, T.; Schermer, B. Cysteine S-Glutathionylation Promotes Stability and Activation of the Hippo Downstream Effector Transcriptional Co-activator with PDZ-binding Motif (TAZ). J. Biol. Chem. 2016, 291, 11596–11607. [Google Scholar] [CrossRef] [Green Version]
- Nagarkoti, S.; Dubey, M.; Sadaf, S.; Awasthi, D.; Chandra, T.; Jagavelu, K.; Kumar, S.; Dikshit, M. Catalase S-Glutathionylation by NOX2 and Mitochondrial-Derived ROS Adversely Affects Mice and Human Neutrophil Survival. Inflammation 2019, 42, 2286–2296. [Google Scholar] [CrossRef]
- Sánchez, G.; Pedrozo, Z.; Domenech, R.J.; Hidalgo, C.; Donoso, P. Tachycardia increases NADPH oxidase activity and RyR2 S-glutathionylation in ventricular muscle. J. Mol. Cell. Cardiol. 2005, 39, 982–991. [Google Scholar] [CrossRef]
- Yang, J.; Zhang, H.; Gong, W.; Liu, Z.; Wu, H.; Hu, W.; Chen, X.; Wang, L.; Wu, S.; Chen, C.; et al. S-Glutathionylation of human inducible Hsp70 reveals a regulatory mechanism involving the C-terminal α-helical lid. J. Biol. Chem. 2020, 295, 8302–8324. [Google Scholar] [CrossRef] [Green Version]
- Porter, C.M.; Truman, A.W.; Truttmann, M.C. Post-translational modifications of Hsp70 family proteins: Expanding the chaperone code. J. Biol. Chem. 2020, 295, 10689–10708. [Google Scholar] [CrossRef]
- Zhang, J.; Ye, Z.W.; Chen, W.; Culpepper, J.; Jiang, H.; Ball, L.E.; Mehrotra, S.; Blumental-Perry, A.; Tew, K.D.; Townsend, D.M. Altered Redox Regulation and S-Glutathionylation of BiP Contribute to Bortezomib Resistance in Multiple Myeloma Free Radic. Biol. Med. 2020, 160, 755–767. [Google Scholar] [CrossRef]
- Gething, M.J. Role and regulation of the ER chaperone BiP. Semin. Cell Dev. Biol. 1999, 10, 465–472. [Google Scholar] [CrossRef]
- Pizzino, G.; Irrera, N.; Cucinotta, M.; Pallio, G.; Mannino, F.; Arcoraci, V.; Squadrito, F.; Altavilla, D.; Bitto, A. Oxidative Stress: Harms and Benefits for Human Health. Oxid. Med. Cell. Longev. 2017, 8416763. [Google Scholar] [CrossRef]
- Zhou, L.; Chan, J.C.Y.; Chupin, S.; Gueguen, N.; Desquiret-Dumas, V.; Koh, S.K.; Li, J.; Gao, Y.; Deng, L.; Verma, C.; et al. Increased Protein S-Glutathionylation in Leber’s Hereditary Optic Neuropathy (LHON). Int. J. Mol. Sci. 2020, 21, 3027. [Google Scholar] [CrossRef] [PubMed]
- Jeon, D.; Park, H.J.; Kim, H.S. Protein S-glutathionylation induced by hypoxia increases hypoxia-inducible factor-1α in human colon cancer cells. Biochem. Biophys. Res. Commun. 2018, 495, 212–216. [Google Scholar] [CrossRef] [PubMed]
- Anathy, V.; Lahue, K.G.; Chapman, D.G.; Chia, S.B.; Casey, D.T.; Aboushousha, R.; van der Velden, J.; Elko, E.; Hoffman, S.M.; McMillan, D.H.; et al. Reducing protein oxidation reverses lung fibrosis. Nat. Med. 2018, 24, 1128–1135. [Google Scholar] [CrossRef]
- Tamma, G.; Valenti, G. Evaluating the Oxidative Stress in Renal Diseases: What Is the Role for S-Glutathionylation? Antioxid. Redox Signal. 2016, 25, 147–164. [Google Scholar] [CrossRef] [PubMed]
- Maki, K.; Nagai, K.; Suzuki, M.; Inomata, T.; Yoshida, T.; Nishimura, M. Temporal changes in glutaredoxin 1 and protein s-glutathionylation in allergic airway inflammation. PLoS ONE 2015, 10, e0122986. [Google Scholar] [CrossRef] [PubMed]
- Hong, C.; Seo, H.; Kwak, M.; Jeon, J.; Jang, J.; Jeong, E.M.; Myeong, J.; Hwang, Y.J.; Ha, K.; Kang, M.J.; et al. Increased TRPC5 glutathionylation contributes to striatal neuron loss in Huntington’s disease. Brain 2015, 138, 3030–3047. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sánchez-Gómez, F.J.; Espinosa-Díez, C.; Dubey, M.; Dikshit, M.; Lamas, S. S-glutathionylation: Relevance in diabetes and potential role as a biomarker. Biol. Chem. 2013, 394, 1263–1280. [Google Scholar] [CrossRef] [Green Version]
- Nonaka, K.; Kume, N.; Urata, Y.; Seto, S.; Kohno, T.; Honda, S.; Ikeda, S.; Muroya, T.; Ikeda, Y.; Ihara, Y.; et al. Serum levels of S-glutathionylated proteins as a risk-marker for arteriosclerosis obliterans. Circ. J. 2007, 71, 100–105. [Google Scholar] [CrossRef] [Green Version]
- Grek, C.L.; Reyes, L.; Townsend, D.M.; Tew, K.D. S-glutathionylation of buccal cell proteins as biomarkers of exposure to hydrogen peroxide. BBA Clin. 2014, 2, 31–39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cui, H.; Kong, Y.; Zhang, H. Oxidative stress, mitochondrial dysfunction, and aging. J. Signal Transduct. 2012, 2012, 646354. [Google Scholar] [CrossRef] [Green Version]
- Harman, D. Aging: A theory based on free radical and radiation chemistry. J. Gerontol. 1956, 11, 298–300. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Y.; Carvey, P.M.; Ling, Z. Age-related changes in glutathione and glutathione-related enzymes in rat brain. Brain Res. 2006, 1090, 35–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cha, S.J.; Kim, H.; Choi, H.J.; Lee, S.; Kim, K. Protein Glutathionylation in the Pathogenesis of Neurodegenerative Diseases. Oxid. Med. Cell. Longev. 2017, 2818565. [Google Scholar] [CrossRef]
- Hodson, R. Alzheimer’s disease. Nature 2018, 559, 7715. [Google Scholar] [CrossRef] [Green Version]
- Huang, W.J.; Zhang, X.; Chen, W.W. Role of oxidative stress in Alzheimer’s disease. Biomed. Rep. 2016, 4, 519–522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Newman, S.F.; Sultana, R.; Perluigi, M.; Coccia, R.; Cai, J.; Pierce, W.M.; Klein, J.B.; Turner, D.M.; Butterfield, D.A. An increase in S-glutathionylated proteins in the Alzheimer’s disease inferior parietal lobule, a proteomics approach. J. Neurosci. Res. 2007, 85, 1506–1514. [Google Scholar] [CrossRef]
- Di Domenico, F.; Cenini, G.; Sultana, R.; Perluigi, M.; Uberti, D.; Memo, M.; Butterfield, D.A. Glutathionylation of the pro-apoptotic protein p53 in Alzheimer’s disease brain: Implications for AD pathogenesis. Neurochem. Res. 2009, 34, 727–733. [Google Scholar] [CrossRef] [PubMed]
- Rani, P.; Krishnan, S.; Rani Cathrine, C. Study on Analysis of Peripheral Biomarkers for Alzheimer’s Disease Diagnosis. Front. Neurol. 2017, 8, 328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonora, M.; Wieckowski, M.R.; Sinclair, D.A.; Kroemer, G.; Pinton, P.; Galluzzi, L. Targeting mitochondria for cardiovascular disorders: Therapeutic potential and obstacles. Nat. Rev. Cardiol. 2019, 16, 33–55. [Google Scholar] [CrossRef]
- Tahrir, F.G.; Langford, D.; Amini, S.; Mohseni Ahooyi, T.; Khalili, K. Mitochondrial quality control in cardiac cells: Mechanisms and role in cardiac cell injury and disease. J. Cell. Physiol. 2019, 234, 8122–8133. [Google Scholar] [CrossRef] [PubMed]
- Pastore, A.; Piemonte, F. Protein glutathionylation in cardiovascular diseases. Int. J. Mol. Sci. 2013, 14, 20845–20876. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, L.; Liu, M.; Sun, R.; Zheng, Y.; Zhang, P. Myocardial Infarction: Symptoms and Treatments. Cell Biochem. Biophys. 2015, 72, 865–867. [Google Scholar] [CrossRef]
- Eaton, P.; Wright, N.; Hearse, D.J.; Shattock, M.J. Glyceraldehyde phosphate dehydrogenase oxidation during cardiac ischemia and reperfusion. J. Mol. Cell. Cardiol. 2002, 34, 1549–1560. [Google Scholar] [CrossRef]
- Chen, F.C.; Ogut, O. Decline of contractility during ischemia-reperfusion injury: Actin glutathionylation and its effect on allosteric interaction with tropomyosin. Am. J. Physiol. Cell Physiol. 2006, 290, C719–C727. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.R.; Chen, C.L.; Pfeiffer, D.R.; Zweier, J.L. Mitochondrial complex II in the post-ischemic heart: Oxidative injury and the role of protein S-glutathionylation. J. Biol. Chem. 2007, 282, 32640–32654. [Google Scholar] [CrossRef] [Green Version]
- Avner, B.S.; Shioura, K.M.; Scruggs, S.B.; Grachoff, M.; Geenen, D.L.; Helseth, D.L., Jr.; Farjah, M.; Goldspink, P.H.; Solaro, R.J. Myocardial infarction in mice alters sarcomeric function via post-translational protein modification. Mol. Cell. Biochem. 2012, 363, 203–215. [Google Scholar] [CrossRef] [Green Version]
- Alegre-Cebollada, J.; Kosuri, P.; Giganti, D.; Eckels, E.; Rivas-Pardo, J.A.; Hamdani, N.; Warren, C.M.; Solaro, R.J.; Linke, W.A.; Fernández, J.M. S-glutathionylation of cryptic cysteines enhances titin elasticity by blocking protein folding. Cell 2014, 156, 1235–1246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chakouri, N.; Reboul, C.; Boulghobra, D.; Kleindienst, A.; Nottin, S.; Gayrard, S.; Roubille, F.; Matecki, S.; Lacampagne, A.; Cazorla, O. Stress-induced protein S-glutathionylation and phosphorylation crosstalk in cardiac sarcomeric proteins—Impact on heart function. Int. J. Cardiol. 2018, 258, 207–216. [Google Scholar] [CrossRef]
- Murry, C.E.; Jennings, R.B.; Reimer, K.A. Preconditioning with ischemia: A delay of lethal cell injury in ischemic myocardium. Circulation 1986, 74, 1124–1136. [Google Scholar] [CrossRef] [Green Version]
- Domenech, R.J.; Sánchez, G.; Donoso, P.; Parra, V.; Macho, P. Effect of tachycardia on myocardial sarcoplasmic reticulum and Ca2+ dynamics: A mechanism for preconditioning? J. Mol. Cell. Cardiol. 2003, 35, 1429–1437. [Google Scholar] [CrossRef] [PubMed]
- Nikolaienko, R.; Bovo, E.; Zima, A.V. Redox Dependent Modifications of Ryanodine Receptor: Basic Mechanisms and Implications in Heart Diseases. Front. Physiol. 2018, 9, 1775. [Google Scholar] [CrossRef] [Green Version]
- Sánchez, G.; Escobar, M.; Pedrozo, Z.; Macho, P.; Domenech, R.; Härtel, S.; Hidalgo, C.; Donoso, P. Exercise and tachycardia increase NADPH oxidase and ryanodine receptor-2 activity: Possible role in cardioprotection. Cardiovasc. Res. 2008, 77, 380–386. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, I.; Minamino, T. Physiological and pathological cardiac hypertrophy. J. Mol. Cell. Cardiol. 2016, 97, 245–262. [Google Scholar] [CrossRef]
- Nakamura, M.; Sadoshima, J. Mechanisms of physiological and pathological cardiac hypertrophy. Nat. Rev. Cardiol. 2018, 15, 387–407. [Google Scholar] [CrossRef]
- Bueno, O.F.; De Windt, L.J.; Tymitz, K.M.; Witt, S.A.; Kimball, T.R.; Klevitsky, R.; Hewett, T.E.; Jones, S.P.; Lefer, D.J.; Peng, C.F. The MEK1-ERK1/2 signaling pathway promotes compensated cardiac hypertrophy in transgenic mice. EMBO J. 2000, 19, 6341–6350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gallo, S.; Vitacolonna, A.; Bonzano, A.; Comoglio, P.; Crepaldi, T. ERK: A Key Player in the Pathophysiology of Cardiac Hypertrophy. Int. J. Mol. Sci. 2019, 20, 2164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pimentel, D.R.; Adachi, T.; Ido, Y.; Heibeck, T.; Jiang, B.; Lee, Y.; Melendez, J.A.; Cohen, R.A.; Colucci, W.S. Strain-stimulated hypertrophy in cardiac myocytes is mediated by reactive oxygen species-dependent Ras S-glutathiolation. J. Mol. Cell. Cardiol. 2006, 41, 613–622. [Google Scholar] [CrossRef]
- Adachi, T.; Pimentel, D.R.; Heibeck, T.; Hou, X.; Lee, Y.J.; Jiang, B.; Ido, Y.; Cohen, R.A. S-glutathiolation of Ras mediates redox-sensitive signaling by angiotensin II in vascular smooth muscle cells. J. Biol. Chem. 2004, 279, 29857–29862. [Google Scholar] [CrossRef] [Green Version]
- Panieri, E.; Santoro, M.M. ROS homeostasis and metabolism: A dangerous liason in cancer cells. Cell Death Dis. 2016, 7, e2253. [Google Scholar] [CrossRef]
- Gorrini, C.; Harris, I.S.; Mak, T.W. Modulation of oxidative stress as an anticancer strategy. Nat. Rev. Drug Discov. 2013, 12, 931–947. [Google Scholar] [CrossRef]
- Garg, R.; Benedetti, L.G.; Abera, M.B.; Wang, H.; Abba, M.; Kazanietz, M.G. Protein kinase C and cancer: What we know and what we do not. Oncogene 2014, 33, 5225–5237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steinberg, S.F. Mechanisms for redox-regulation of protein kinase C. Front. Pharmacol. 2015, 6, 128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chu, F.; Ward, N.E.; O’Brian, C.A. PKC isozyme S-cysteinylation by cystine stimulates the pro-apoptotic isozyme PKC delta and inactivates the oncogenic isozyme PKC epsilon. Carcinogenesis 2003, 24, 317–325. [Google Scholar] [CrossRef] [Green Version]
- Humphries, K.M.; Juliano, C.; Taylor, S.S. Regulation of cAMP-dependent protein kinase activity by glutathionylation. J. Biol. Chem. 2002, 277, 43505–43511. [Google Scholar] [CrossRef] [Green Version]
- Humphries, K.M.; Deal, M.S.; Taylor, S.S. Enhanced dephosphorylation of cAMP-dependent protein kinase by oxidation and thiol modification. J. Biol. Chem. 2005, 280, 2750–2758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cruz, C.M.; Rinna, A.; Forman, H.J.; Ventura, A.L.; Persechini, P.M.; Ojcius, D.M. ATP activates a reactive oxygen species-dependent oxidative stress response and secretion of proinflammatory cytokines in macrophages. J. Biol. Chem. 2007, 282, 2871–2879. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rao, R.K.; Clayton, L.W. Regulation of protein phosphatase 2A by hydrogen peroxide and glutathionylation. Biochem. Biophys. Res. Commun. 2002, 293, 610–616. [Google Scholar] [CrossRef]
- Velu, C.S.; Niture, S.K.; Doneanu, C.E.; Pattabiraman, N.; Srivenugopal, K.S. Human p53 is inhibited by glutathionylation of cysteines present in the proximal DNA-binding domain during oxidative stress. Biochemistry 2007, 46, 7765–7780. [Google Scholar] [CrossRef] [Green Version]
- Pineda-Molina, E.; Klatt, P.; Vázquez, J.; Marina, A.; García de Lacoba, M.; Pérez-Sala, D.; Lamas, S. Glutathionylation of the p50 subunit of NF-kappaB: A mechanism for redox-induced inhibition of DNA binding. Biochemistry 2001, 40, 14134–14142. [Google Scholar] [CrossRef]
- Qanungo, S.; Starke, D.W.; Pai, H.V.; Mieyal, J.J.; Nieminen, A.L. Glutathione supplementation potentiates hypoxic apoptosis by S-glutathionylation of p65-NFkappaB. J. Biol. Chem. 2007, 282, 18427–18436. [Google Scholar] [CrossRef] [Green Version]
- Butturini, E.; Darra, E.; Chiavegato, G.; Cellini, B.; Cozzolino, F.; Monti, M.; Pucci, P.; Dell’Orco, D.; Mariotto, S. S-Glutathionylation at Cys328 and Cys542 impairs STAT3 phosphorylation. ACS Chem. Biol. 2014, 9, 1885–1893. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hensley, P.; Mishra, M.; Kyprianou, N. Targeting caspases in cancer therapeutics. Biol. Chem. 2013, 394, 831–843. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zamaraev, A.V.; Kopeina, G.S.; Prokhorova, E.A.; Zhivotovsky, B.; Lavrik, I.N. Post-translational Modification of Caspases: The Other Side of Apoptosis Regulation. Trends Cell Biol. 2017, 27, 322–339. [Google Scholar] [CrossRef] [PubMed]
- Boice, A.; Bouchier-Hayes, L. Targeting apoptotic caspases in cancer. Biochim. Biophys. Acta Mol. Cell Res. 2020, 1867, 118688. [Google Scholar] [CrossRef]
- Huang, Z.; Pinto, J.T.; Deng, H.; Richie, J.P., Jr. Inhibition of caspase-3 activity and activation by protein glutathionylation. Biochem. Pharmacol. 2008, 75, 2234–2244. [Google Scholar] [CrossRef] [Green Version]
- Pan, S.; Berk, B.C. Glutathiolation regulates tumor necrosis factor-alpha-induced caspase-3 cleavage and apoptosis: Key role for glutaredoxin in the death pathway. Circ. Res. 2007, 100, 213–219. [Google Scholar] [CrossRef] [Green Version]
- Musaogullari, A.; Mandato, A.; Chai, Y.-C. Role of Glutathione Depletion and Reactive Oxygen Species Generation of Caspase-3 Activation: A Study with the Kinase Inhibitor Staurosporine. Front. Physiol. 2020, 11, 998. [Google Scholar] [CrossRef]
- Byrne, C.D.; Targher, G. NAFLD: A multisystem disease. J. Hepatol. 2015, 62, S47–S64. [Google Scholar] [CrossRef] [Green Version]
- Friedman, S.L.; Neuschwander-Tetri, B.A.; Rinella, M.; Sanyal, A.J. Mechanisms of NAFLD development and therapeutic strategies. Nat. Med. 2018, 24, 908–922. [Google Scholar] [CrossRef]
- Videla, L.A.; Rodrigo, R.; Araya, J.; Poniachik, J. Insulin resistance and oxidative stress interdependency in non-alcoholic fatty liver disease. Trends Mol. Med. 2006, 12, 555–558. [Google Scholar] [CrossRef] [PubMed]
- Dou, X.; Li, S.; Hu, L.; Ding, L.; Ma, Y.; Ma, W.; Chai, H.; Song, Z. Glutathione disulfide sensitizes hepatocytes to TNFα-mediated cytotoxicity via IKK-β S-glutathionylation: A potential mechanism underlying non-alcoholic fatty liver disease. Exp. Mol. Med. 2018, 50, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rufini, A.; Tucci, P.; Celardo, I.; Melino, G. Senescence and aging: The critical roles of p53. Oncogene 2013, 32, 5129–5143. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Zhang, L. PUMA, a potent killer with or without p53. Oncogene 2008, 27, S71–S83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deng, X.Q.; Chen, L.L.; Li, N.X. The expression of SIRT1 in nonalcoholic fatty liver disease induced by high-fat diet in rats. Liver Int. 2007, 27, 708–715. [Google Scholar] [CrossRef] [PubMed]
- Yin, H.; Hu, M.; Liang, X.; Ajmo, J.M.; Li, X.; Bataller, R.; You, M. Deletion of SIRT1 from hepatocytes in mice disrupts lipin-1 signaling and aggravates alcoholic fatty liver. Gastroenterology 2014, 146, 801–811. [Google Scholar] [CrossRef] [Green Version]
- Seo, Y.Y.; Cho, Y.K.; Bae, J.C.; Seo, M.H.; Park, S.E.; Rhee, E.J.; Park, C.Y.; Oh, K.W.; Park, S.W.; Lee, W.Y. Tumor Necrosis Factor-α as a Predictor for the Development of Nonalcoholic Fatty Liver Disease: A 4-Year Follow-Up Study. Endocrinol. Metab. 2013, 28, 41–45. [Google Scholar] [CrossRef] [Green Version]
- Kakino, S.; Ohki, T.; Nakayama, H.; Yuan, X.; Otabe, S.; Hashinaga, T.; Wada, N.; Kurita, Y.; Tanaka, K.; Hara, K.; et al. Pivotal Role of TNF-α in the Development and Progression of Nonalcoholic Fatty Liver Disease in a Murine Model. Horm. Metab. Res. 2018, 50, 80–87. [Google Scholar] [CrossRef]
- Hayden, M.S.; Ghosh, S. Regulation of NF-κB by TNF family cytokines. Semin. Immunol. 2014, 26, 253–266. [Google Scholar] [CrossRef] [Green Version]
- Solt, L.A.; May, M.J. The IkappaB kinase complex: Master regulator of NF-kappaB signaling. Immunol. Res. 2008, 42, 3–18. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| Protein | Reported Effect of S-Glutathionylation on Function | References |
|---|---|---|
| c-Jun | Inhibition | [52] |
| Protein tyrosine phosphatase 1B (PTP1B) | Inhibition | [58] |
| Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) | Inhibition | [78] |
| Estrogen receptor α | Inhibition | [92] |
| AMP-activated protein kinase (AMPK) | Activation | [94] |
| Sirtuin-1 | Inhibition | [112] |
| Antigen 85C * | Inhibition | [113] |
| Transcriptional Co-activator with PDZ-binding Motif (TAZ) | Activation | [114] |
| Catalase | Inhibition | [115] |
| Ryanodine receptor 2 (RyR2) | Activation | [116] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Musaogullari, A.; Chai, Y.-C. Redox Regulation by Protein S-Glutathionylation: From Molecular Mechanisms to Implications in Health and Disease. Int. J. Mol. Sci. 2020, 21, 8113. https://doi.org/10.3390/ijms21218113
Musaogullari A, Chai Y-C. Redox Regulation by Protein S-Glutathionylation: From Molecular Mechanisms to Implications in Health and Disease. International Journal of Molecular Sciences. 2020; 21(21):8113. https://doi.org/10.3390/ijms21218113
Chicago/Turabian StyleMusaogullari, Aysenur, and Yuh-Cherng Chai. 2020. "Redox Regulation by Protein S-Glutathionylation: From Molecular Mechanisms to Implications in Health and Disease" International Journal of Molecular Sciences 21, no. 21: 8113. https://doi.org/10.3390/ijms21218113
APA StyleMusaogullari, A., & Chai, Y. -C. (2020). Redox Regulation by Protein S-Glutathionylation: From Molecular Mechanisms to Implications in Health and Disease. International Journal of Molecular Sciences, 21(21), 8113. https://doi.org/10.3390/ijms21218113