Establishment of a Gene Signature to Predict Prognosis for Patients with Lung Adenocarcinoma

Abstract
:
1. Introduction
2. Results
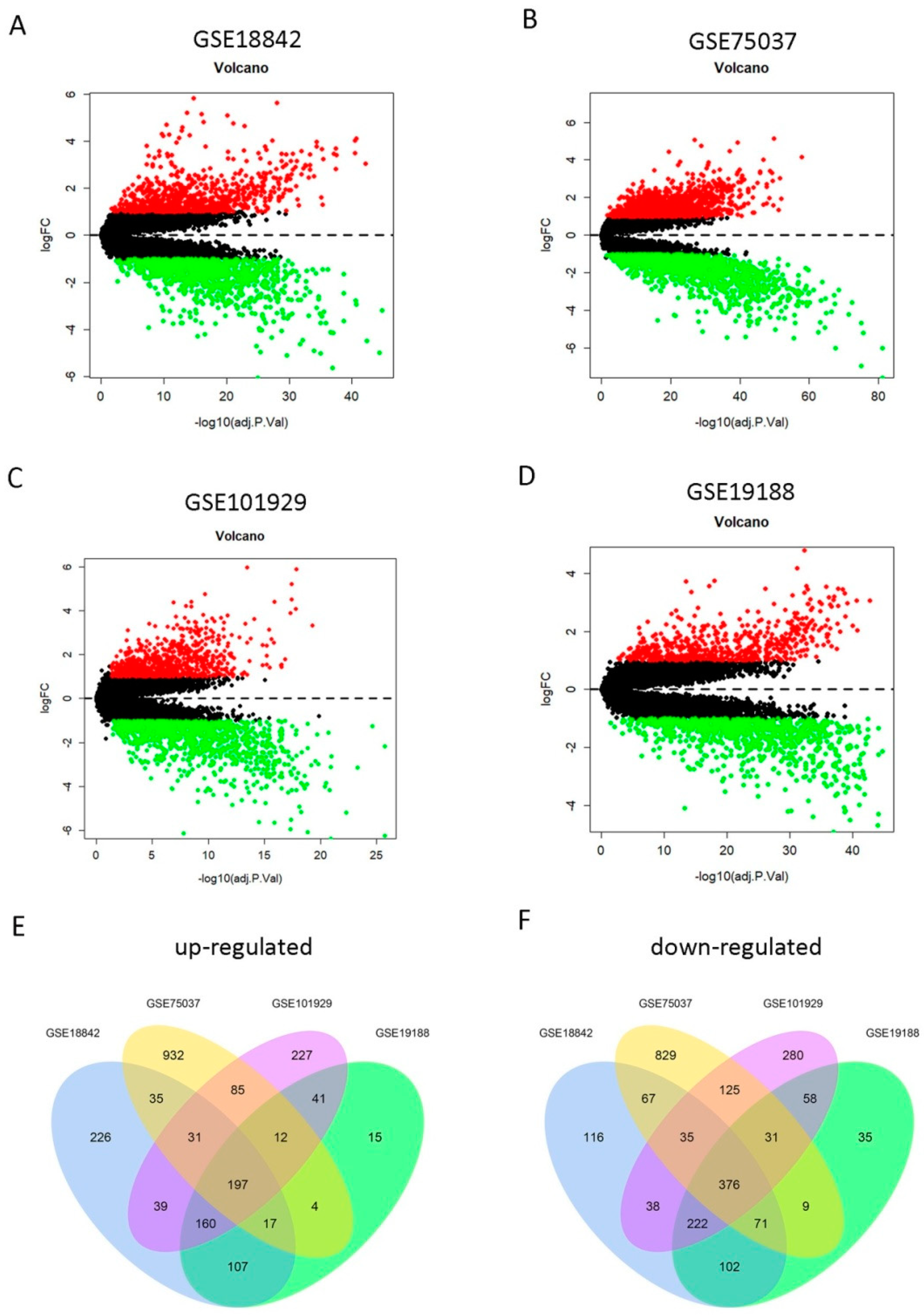
2.1. Identification of 573 DEGs Shared by Four GEO Profiles
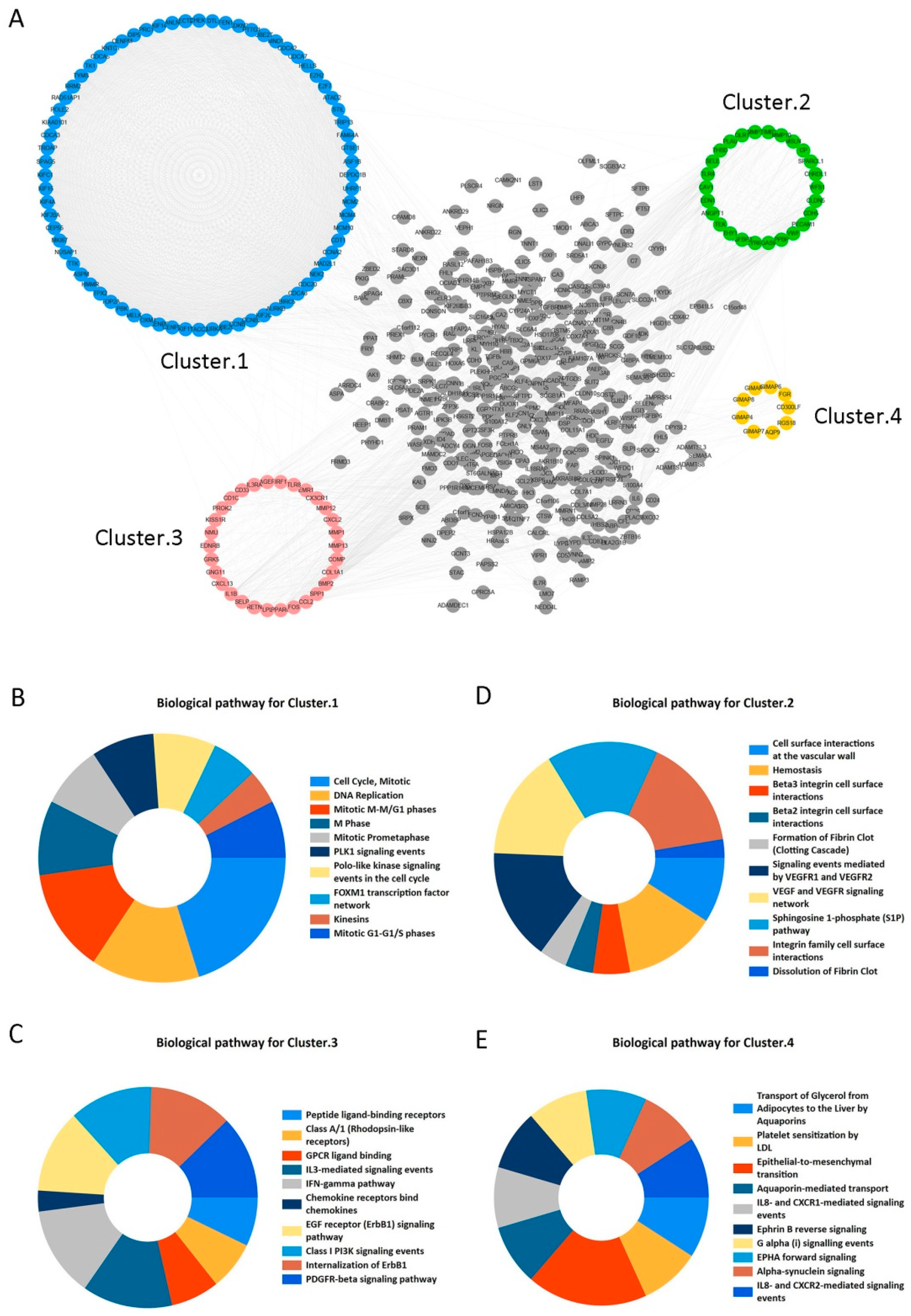
2.2. Candidate Core Genes Identification
2.3. The Risk Model Based on the Eight Genes is Verified as an Independent Prognosis Factor
2.4. Signature Genes Show High Expression in LUAD Samples
2.5. Signature Genes with High Expression in LUAD Cells
3. Discussion
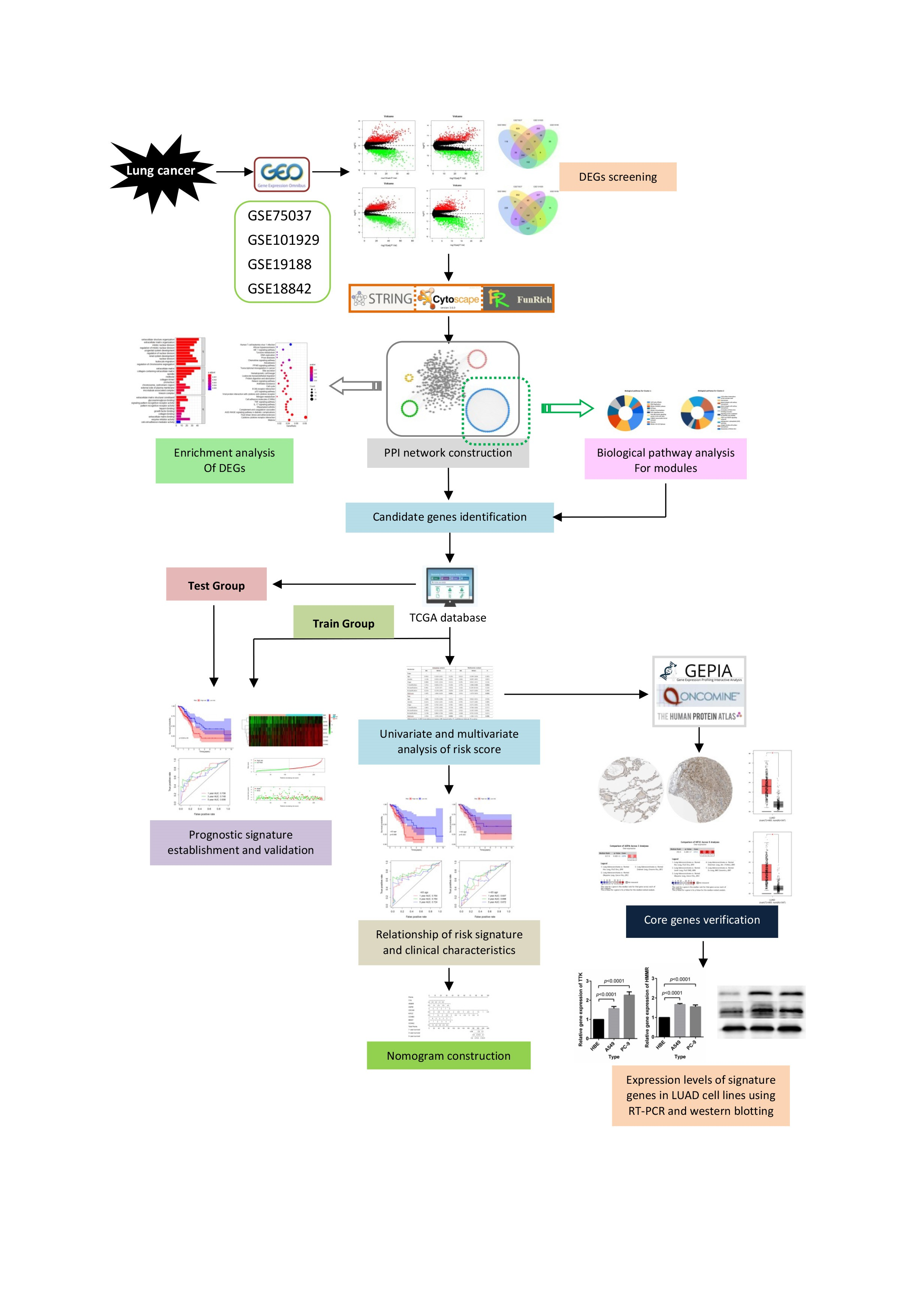
4. Materials and Methods
4.1. Data Collection
4.2. Data Processing and Identification of DEGs
4.3. PPI Network Construction and Clustering Module Analysis
4.4. Establishment and Validation of a Prognostic Signature
4.5. Validation of Corresponding Genes in Risk Score Signature
4.6. Cell Culture
4.7. Quantitative Real-Time Polymerase Chain Reaction
4.8. Western Blot Analysis
4.9. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| LUAD | Lung adenocarcinoma |
| DEGs | Differentially expressed genes |
| TNM | Tumor, node, and metastases |
| GEO | Gene Expression Omnibus |
| TCGA | The Cancer Genome Atlas |
| PPI | Protein-protein interaction |
| ROC | Receiver operating characteristic curve |
| STRING | Search Tool for the Retrieval of Interacting Genes database |
| MCODE | Molecularcomplex detection |
| OS | Overall survival |
| LASSO | Least absolute shrinkage and selection operator |
| GEPIA | Gene Expression Profiling Interactive Analysis |
| HPA | Human Protein Atlas |
| GO | Gene Ontology |
| KEGG | Kyoto Encyclopedia of Genes and Genomes |
References
- Ling, B.; Liao, X.; Huang, Y.; Liang, L.; Jiang, Y.; Pang, Y.; Qi, G. Identification of prognostic markers of lung cancer through bioinformatics analysis and in vitro experiments. Int. J. Oncol. 2020, 56, 193–205. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Zhang, Z.; Yu, Z. Identification of a novel glycolysis-related gene signature for predicting metastasis and survival in patients with lung adenocarcinoma. J. Transl. Med. 2019, 17, 423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, Q.; Shang, J.; Yang, Z.; Zhang, L.; Zhang, C.; Chen, J.; Wu, X. Identification of an immune signature predicting prognosis risk of patients in lung adenocarcinoma. J. Transl. Med. 2019, 17, 70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Denisenko, T.V.; Budkevich, I.N.; Zhivotovsky, B. Cell death-based treatment of lung adenocarcinoma. Cell Death Dis. 2018, 9, 117. [Google Scholar] [CrossRef] [PubMed]
- Niemira, M.; Collin, F.; Szalkowska, A.; Bielska, A.; Chwialkowska, K.; Reszec, J.; Niklinski, J.; Kwasniewski, M.; Kretowski, A. Molecular Signature of Subtypes of Non-Small-Cell Lung Cancer by Large-Scale Transcriptional Profiling: Identification of Key Modules and Genes by Weighted Gene Co-Expression Network Analysis (WGCNA). Cancers 2019, 12, 37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barresi, V.; Cinnirella, G.; Valenti, G.; Spampinato, G.; Musso, N.; Castorina, S.; Condorelli, D.F. Gene expression profiles in genome instability-based classes of colorectal cancer. BMC Cancer 2018, 18, 1265. [Google Scholar] [CrossRef]
- Yoshimi, A.; Lin, K.T.; Wiseman, D.H.; Rahman, M.A.; Pastore, A.; Wang, B.; Lee, S.C.; Micol, J.B.; Zhang, X.J.; de Botton, S.; et al. Coordinated alterations in RNA splicing and epigenetic regulation drive leukaemogenesis. Nature 2019, 574, 273–277. [Google Scholar] [CrossRef]
- Sung, J.Y.; Shin, H.T.; Sohn, K.A.; Shin, S.Y.; Park, W.Y.; Joung, J.G. Assessment of intratumoral heterogeneity with mutations and gene expression profiles. PLoS ONE 2019, 14, e0219682. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.; Sheng, L.; Gong, Z.; Ru, S.; Bian, H. Investigation of the molecular mechanisms of hepatic injury upon naphthalene exposure in zebrafish (Danio rerio). Ecotoxicology 2018, 27, 650–660. [Google Scholar] [CrossRef]
- Zhou, C.; Ji, J.; Cai, Q.; Shi, M.; Chen, X.; Yu, Y.; Zhu, Z.; Zhang, J. MTA2 enhances colony formation and tumor growth of gastric cancer cells through IL-11. BMC Cancer 2015, 15, 343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, H.; Xie, C. A Six-Gene Signature Predicts Survival of Adenocarcinoma Type of Non-Small-Cell Lung Cancer Patients: A Comprehensive Study Based on Integrated Analysis and Weighted Gene Coexpression Network. BioMed Res. Int. 2019, 2019, 4250613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, J.; Wang, M.; Hu, D. Development of an autophagy-related gene prognostic signature in lung adenocarcinoma and lung squamous cell carcinoma. PeerJ 2020, 8, e8288. [Google Scholar] [CrossRef] [PubMed]
- Hou, J.; Aerts, J.; den-Hamer, B.; van-Ijcken, W.; den-Bakker, M.; Riegman, P.; van-der-Leest, C.; van-der-Spek, P.; Foekens, J.A.; Hoogsteden, H.C.; et al. Gene expression-based classification of non-small cell lung carcinomas and survival prediction. PLoS ONE 2010, 5, e10312. [Google Scholar] [CrossRef]
- Selamat, S.A.; Chung, B.S.; Girard, L.; Zhang, W.; Zhang, Y.; Campan, M.; Siegmund, K.D.; Koss, M.N.; Hagen, J.A.; Lam, W.L.; et al. Genome-scale analysis of DNA methylation in lung adenocarcinoma and integration with mRNA expression. Genome Res. 2012, 22, 1197–1211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okayama, H.; Kohno, T.; Ishii, Y.; Shimada, Y.; Shiraishi, K.; Iwakawa, R.; Furuta, K.; Tsuta, K.; Shibata, T.; Yamamoto, S.; et al. Identification of genes upregulated in ALK-positive and EGFR/KRAS/ALK-negative lung adenocarcinomas. Cancer Res. 2012, 72, 100–111. [Google Scholar] [CrossRef] [Green Version]
- Landi, M.T.; Dracheva, T.; Rotunno, M.; Figueroa, J.D.; Liu, H.; Dasgupta, A.; Mann, F.E.; Fukuoka, J.; Hames, M.; Bergen, A.W.; et al. Gene expression signature of cigarette smoking and its role in lung adenocarcinoma development and survival. PLoS ONE 2008, 3, e1651. [Google Scholar] [CrossRef]
- Stearman, R.S.; Dwyer-Nield, L.; Zerbe, L.; Blaine, S.A.; Chan, Z.; Bunn, P.A.J.; Johnson, G.L.; Hirsch, F.R.; Merrick, D.T.; Franklin, W.A.; et al. Analysis of orthologous gene expression between human pulmonary adenocarcinoma and a carcinogen-induced murine model. Am. J. Pathol. 2005, 167, 1763–1775. [Google Scholar] [CrossRef] [Green Version]
- Su, L.J.; Chang, C.W.; Wu, Y.C.; Chen, K.C.; Lin, C.J.; Liang, S.C.; Lin, C.H.; Whang-Peng, J.; Hsu, S.L.; Chen, C.H.; et al. Selection of DDX5 as a novel internal control for Q-RT-PCR from microarray data using a block bootstrap re-sampling scheme. BMC Genom. 2007, 8, 140. [Google Scholar] [CrossRef] [Green Version]
- Wu, T.; Wang, M.C.; Jing, L.; Liu, Z.Y.; Guo, H.; Liu, Y.; Bai, Y.Y.; Cheng, Y.Z.; Nan, K.J.; Liang, X. Autophagy facilitates lung adenocarcinoma resistance to cisplatin treatment by activation of AMPK/mTOR signaling pathway. Drug Des. Dev. Ther. 2015, 9, 6421–6431. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.; Zhang, J.; Xu, F.P.; Wang, Y.G.; Xie, Z.; Su, J.; Dong, S.; Nie, Q.; Shao, Y.; Zhou, Q.; et al. Genomic Landscape and Immune Microenvironment Features of Preinvasive and Early Invasive Lung Adenocarcinoma. J. Thorac. Oncol. 2019, 14, 1912–1923. [Google Scholar] [CrossRef]
- Guo, D.; Wang, M.; Shen, Z.; Zhu, J. A new immune signature for survival prediction and immune checkpoint molecules in lung adenocarcinoma. J. Transl. Med. 2020, 18, 123. [Google Scholar] [CrossRef] [Green Version]
- Li, R.; Yang, Y.E.; Yin, Y.H.; Zhang, M.Y.; Li, H.; Qu, Y.Q. Methylation and transcriptome analysis reveal lung adenocarcinoma-specific diagnostic biomarkers. J. Transl. Med. 2019, 17, 324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Yuan, Y.; Li, Y.; Zhang, P.; Chen, P.; Sun, S. An inverse interaction between HOXA11 and HOXA11-AS is associated with cisplatin resistance in lung adenocarcinoma. Epigenetics 2019, 14, 949–960. [Google Scholar] [CrossRef]
- Stutvoet, T.S.; Kol, A.; de Vries, E.G.; de Bruyn, M.; Fehrmann, R.S.; Terwisscha van Scheltinga, A.G.; de Jong, S. MAPK pathway activity plays a key role in PD-L1 expression of lung adenocarcinoma cells. J. Pathol. 2019, 249, 52–64. [Google Scholar] [CrossRef] [Green Version]
- Cao, P.; Liu, B.; Du, F.; Li, D.; Wang, Y.; Yan, X.; Li, X.; Li, Y. Scutellarin suppresses proliferation and promotes apoptosis in A549 lung adenocarcinoma cells via AKT/mTOR/4EBP1 and STAT3 pathways. Thorac. Cancer 2019, 10, 492–500. [Google Scholar] [CrossRef]
- Zhao, C.; Zhang, J.; Ma, L.; Wu, H.; Zhang, H.; Su, J.; Geng, B.; Yao, Q.; Zheng, J. GOLPH3 Promotes Angiogenesis of Lung Adenocarcinoma by Regulating the Wnt/beta-Catenin Signaling Pathway. OncoTargets Ther. 2020, 13, 6265–6277. [Google Scholar] [CrossRef]
- Cui, Y.; Fang, W.; Li, C.; Tang, K.; Zhang, J.; Lei, Y.; He, W.; Peng, S.; Kuang, M.; Zhang, H.; et al. Development and Validation of a Novel Signature to Predict Overall Survival in “Driver Gene-negative” Lung Adenocarcinoma (LUAD): Results of a Multicenter Study. Clin. Cancer Res. 2019, 25, 1546–1556. [Google Scholar] [CrossRef] [Green Version]
- Li, B.; Cui, Y.; Diehn, M.; Li, R. Development and Validation of an Individualized Immune Prognostic Signature in Early-Stage Nonsquamous Non-Small Cell Lung Cancer. JAMA Oncol. 2017, 3, 1529–1537. [Google Scholar] [CrossRef]
- Li, S.; Xuan, Y.; Gao, B.; Sun, X.; Miao, S.; Lu, T.; Wang, Y.; Jiao, W. Identification of an eight-gene prognostic signature for lung adenocarcinoma. Cancer Manag. Res. 2018, 10, 3383–3392. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Jiang, B.; Zhu, N.; Tao, M.; Jun, Y.; Chen, X.; Wang, Q.; Luo, C. Mitotic checkpoint kinase Mps1/TTK predicts prognosis of colon cancer patients and regulates tumor proliferation and differentiation via PKCalpha/ERK1/2 and PI3K/Akt pathway. Med. Oncol. 2019, 37, 5. [Google Scholar] [CrossRef] [PubMed]
- King, J.L.; Zhang, B.; Li, Y.; Li, K.P.; Ni, J.J.; Saavedra, H.I.; Dong, J.T. TTK promotes mesenchymal signaling via multiple mechanisms in triple negative breast cancer. Oncogenesis 2018, 7, 69. [Google Scholar] [CrossRef] [PubMed]
- Ishigami, S.; Ueno, S.; Nishizono, Y.; Matsumoto, M.; Kurahara, H.; Arigami, T.; Uchikado, Y.; Setoyama, T.; Arima, H.; Yoshiaki, K.; et al. Prognostic impact of CD168 expression in gastric cancer. BMC Cancer 2011, 11, 106. [Google Scholar] [CrossRef] [Green Version]
- Ye, S.; Liu, Y.; Fuller, A.M.; Katti, R.; Ciotti, G.E.; Chor, S.; Alam, M.Z.; Devalaraja, S.; Lorent, K.; Weber, K.; et al. TGFbeta and Hippo Pathways Cooperate to Enhance Sarcomagenesis and Metastasis through the Hyaluronan-Mediated Motility Receptor (HMMR). Mol. Cancer Res. 2020, 18, 560–573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pai, V.C.; Hsu, C.C.; Chan, T.S.; Liao, W.Y.; Chuu, C.P.; Chen, W.Y.; Li, C.R.; Lin, C.Y.; Huang, S.P.; Chen, L.T.; et al. ASPM promotes prostate cancer stemness and progression by augmenting Wnt-Dvl-3-beta-catenin signaling. Oncogene 2019, 38, 1340–1353. [Google Scholar] [CrossRef]
- Liu, X.; Liu, X.; Li, J.; Ren, F. Identification and Integrated Analysis of Key Biomarkers for Diagnosis and Prognosis of Non-Small Cell Lung Cancer. Med. Sci. Monit. 2019, 25, 9280–9289. [Google Scholar] [CrossRef]
- Yu, D.; Shi, L.; Bu, Y.; Li, W. Cell Division Cycle Associated 8 Is a Key Regulator of Tamoxifen Resistance in Breast Cancer. J. Breast Cancer 2019, 22, 237–247. [Google Scholar] [CrossRef]
- Bu, Y.; Shi, L.; Yu, D.; Liang, Z.; Li, W. CDCA8 is a key mediator of estrogen-stimulated cell proliferation in breast cancer cells. Gene 2019, 703, 1–6. [Google Scholar] [CrossRef]
- Ci, C.; Tang, B.; Lyu, D.; Liu, W.; Qiang, D.; Ji, X.; Qiu, X.; Chen, L.; Ding, W. Overexpression of CDCA8 promotes the malignant progression of cutaneous melanoma and leads to poor prognosis. Int. J. Mol. Med. 2019, 43, 404–412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gnjatic, S.; Cao, Y.; Reichelt, U.; Yekebas, E.F.; Nolker, C.; Marx, A.H.; Erbersdobler, A.; Nishikawa, H.; Hildebrandt, Y.; Bartels, K.; et al. NY-CO-58/KIF2C is overexpressed in a variety of solid tumors and induces frequent T cell responses in patients with colorectal cancer. Int. J. Cancer 2010, 127, 381–393. [Google Scholar] [CrossRef]
- Song, X.; Zhang, T.; Wang, X.; Liao, X.; Han, C.; Yang, C.; Su, K.; Cao, W.; Gong, Y.; Chen, Z.; et al. Distinct Diagnostic and Prognostic Values of Kinesin Family Member Genes Expression in Patients with Breast Cancer. Med. Sci. Monit. 2018, 24, 9442–9464. [Google Scholar] [CrossRef]
- Gwon, M.R.; Cho, J.H.; Kim, J.R. Mitotic centromere-associated kinase (MCAK/Kif2C) regulates cellular senescence in human primary cells through a p53-dependent pathway. FEBS Lett. 2012, 586, 4148–4156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arsic, N.; Bendris, N.; Peter, M.; Begon-Pescia, C.; Rebouissou, C.; Gadea, G.; Bouquier, N.; Bibeau, F.; Lemmers, B.; Blanchard, J.M. A novel function for Cyclin A2: Control of cell invasion via RhoA signaling. J. Cell. Biol. 2012, 196, 147–162. [Google Scholar] [CrossRef] [Green Version]
- Bendris, N.; Arsic, N.; Lemmers, B.; Blanchard, J.M. Cyclin A2, Rho GTPases and EMT. Small GTPases 2012, 3, 225–228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Tian, X.; Wang, P.; Huang, M.; Xu, R.; Nie, T. MicroRNA-582-3p negatively regulates cell proliferation and cell cycle progression in acute myeloid leukemia by targeting cyclin B2. Cell. Mol. Biol. Lett. 2019, 24, 66. [Google Scholar] [CrossRef]
- Gao, C.L.; Wang, G.W.; Yang, G.Q.; Yang, H.; Zhuang, L. Karyopherin subunit-alpha 2 expression accelerates cell cycle progression by upregulating CCNB2 and CDK1 in hepatocellular carcinoma. Oncol. Lett. 2018, 15, 2815–2820. [Google Scholar]
- Gao, X.; Chen, Y.; Chen, M.; Wang, S.; Wen, X.; Zhang, S. Identification of key candidate genes and biological pathways in bladder cancer. PeerJ 2018, 6, e6036. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristic | TCGA Data (n, %) | GSE18842 (n, %) | GSE75037 (n, %) | GSE101929 (n, %) | GSE19188 (n, %) | |
|---|---|---|---|---|---|---|
| Platform | Illumina HiSeq2000 RNA sequencing platform | Affymetrix Human Genome U133 Plus 2.0 Array | Illumina HumanWG-6 v3.0 expression beadchip | Affymetrix Human Genome U133 Plus 2.0 Array | Affymetrix Human Genome U133 Plus 2.0 Array | |
| Samples | 551 (100.0%) | 91 (100.0%) | 166 (100.0%) | 66 (100.0%) | 156 (100.0%) | |
| Normal | 54 (9.8%) | 45 (49.5%) | 83 (50.0%) | 34 (51.5%) | 65 (41.7%) | |
| Tumor | 497 (90.2%) | 46 (50.5%) | 83 (50.0%) | 32 (48.5%) | 91 (58.3%) | |
| Survival Status | 486 (88.2%) | NA | NA | 66 (100.0%) | 82 (52.6%) | |
| Death | 162 (29.4%) | NA | NA | 40 (60.6%) | 50 (32.1%) | |
| Survival | 324 (58.8%) | NA | NA | 26 (39.4%) | 32 (20.5%) | |
| Age | 467 (84.8%) | NA | 166 (100.0%) | 66 (100.0%) | NA | |
| <=65 | 227 (41.2%) | NA | 58 (34.9%) | 43 (65.2%) | NA | |
| >65 | 240 (43.6%) | NA | 108 (65.1%) | 23 (34.8%) | NA | |
| Gender | 486 (88.2%) | NA | 166 (100.0%) | 66 (100.0%) | 134 (85.9%) | |
| Female | 264 (47.9%) | NA | 118 (71.1%) | 38 (57.6%) | 34 (21.8%) | |
| Male | 222 (40.3%) | NA | 48 (28.9%) | 28 (42.4%) | 100 (64.1%) | |
| Stage | 478 (86.8%) | NA | 33 (19.9%) | NA | NA | |
| I | 262 (47.5%) | NA | 20 (12.0%) | NA | NA | |
| II | 112 (20.3%) | NA | 8 (4.8%) | NA | NA | |
| III | 79 (14.3%) | NA | 5 (3.0%) | NA | NA | |
| IV | 25 (4.5%) | NA | NA | NA | NA | |
| T classification | 483 (87.7%) | NA | NA | NA | NA | |
| T1 | 163 (29.6%) | NA | NA | NA | NA | |
| T2 | 260 (47.2%) | NA | NA | NA | NA | |
| T3 | 41 (7.4%) | NA | NA | NA | NA | |
| T4 | 19 (3.4%) | NA | NA | NA | NA | |
| N classification | 474 (86.0%) | NA | NA | NA | NA | |
| N0 | 312 (56.6%) | NA | NA | NA | NA | |
| N1 | 90 (16.3%) | NA | NA | NA | NA | |
| N2 | 70 (12.7%) | NA | NA | NA | NA | |
| N3 | 2 (0.4%) | NA | NA | NA | NA | |
| M classification | 357 (64.8%) | NA | NA | NA | NA | |
| M0 | 333 (60.4%) | NA | NA | NA | NA | |
| M1 | 24 (4.4%) | NA | NA | NA | NA |
| NO. | Gene | Degree | NO. | Gene | Degree | NO. | Gene | Degree |
|---|---|---|---|---|---|---|---|---|
| 1 | UBE2C | 71 | 11 | ASPM | 71 | 21 | KIAA0101 | 70 |
| 2 | NUSAP1 | 71 | 12 | CENPF | 71 | 22 | SPAG5 | 70 |
| 3 | TPX2 | 71 | 13 | CDCA8 | 71 | 23 | KIF15 | 70 |
| 4 | PBK | 71 | 14 | KIF2C | 71 | 24 | CEP55 | 70 |
| 5 | MELK | 71 | 15 | AURKB | 71 | 25 | CENPE | 70 |
| 6 | TTK | 71 | 16 | CCNB2 | 71 | 26 | CDC20 | 70 |
| 7 | KIF11 | 71 | 17 | KIF20A | 71 | 27 | BIRC5 | 70 |
| 8 | TOP2A | 71 | 18 | MKI67 | 71 | 28 | MCM10 | 70 |
| 9 | HMMR | 71 | 19 | CCNA2 | 71 | 29 | MAD2L1 | 70 |
| 10 | RRM2 | 71 | 20 | CCNB1 | 71 | 30 | AURKA | 70 |
| NO. | Gene | Univariate Analysis * | Multivariate Analysis ** | ||||
|---|---|---|---|---|---|---|---|
| HR | 95%CI | p | HR | 95%CI | Coef. | ||
| 1 | UBE2C | 1.145 | 1.033–1.270 | 0.010 | --- | --- | --- |
| 2 | TPX2 | 1.226 | 1.089–1.381 | 0.001 | --- | --- | --- |
| 3 | PBK | 1.264 | 1.100–1.453 | 0.001 | --- | --- | --- |
| 4 | MELK | 1.233 | 1.069–1.422 | 0.004 | --- | --- | --- |
| 5 | TTK | 1.247 | 1.053–1.477 | 0.010 | 0.630 | 0.341–1.165 | −0.462 |
| 6 | KIF11 | 1.358 | 1.148–1.608 | <0.001 | --- | --- | --- |
| 7 | TOP2A | 1.178 | 1.043–1.331 | 0.008 | --- | --- | --- |
| 8 | HMMR | 1.472 | 1.243–1.742 | <0.001 | 1.883 | 1.153–3.074 | 0.633 |
| 9 | RRM2 | 1.298 | 1.128–1.493 | <0.001 | --- | --- | --- |
| 10 | ASPM | 1.409 | 1.169–1.698 | <0.001 | 0.577 | 0.287–1.159 | −0.550 |
| 11 | CENPF | 1.293 | 1.112–1.503 | 0.001 | --- | --- | --- |
| 12 | CDCA8 | 1.206 | 1.039–1.401 | 0.014 | 0.270 | 0.100–0.730 | −1.309 |
| 13 | KIF2C | 1.234 | 1.074–1.417 | 0.003 | 3.281 | 1.232–8.738 | 1.188 |
| 14 | AURKB | 1.188 | 1.039–1.358 | 0.012 | --- | --- | --- |
| 15 | CCNB2 | 1.258 | 1.085–1.458 | 0.002 | 0.622 | 0.329–1.178 | −0.474 |
| 16 | KIF20A | 1.350 | 1.136–1.605 | 0.001 | --- | --- | --- |
| 17 | MKI67 | 1.309 | 1.129–1.518 | <0.001 | 1.768 | 1.103–2.835 | 0.570 |
| 18 | CCNA2 | 1.328 | 1.150–1.533 | <0.001 | 1.622 | 0.889–2.959 | 0.484 |
| 19 | CCNB1 | 1.321 | 1.136–1.535 | <0.001 | --- | --- | --- |
| 20 | NUSAP1 | 1.293 | 1.107–1.511 | 0.001 | --- | --- | --- |
| Parameter | Univariate Analysis | Multivariate Analysis | ||||
|---|---|---|---|---|---|---|
| HR | 95%CI | p | HR | 95%CI | p | |
| Training Group | ||||||
| Age | 0.492 | 0.169–1.431 | 0.193 | 0.612 | 0.204–1.838 | 0.381 |
| Gender | 1.128 | 0.656–1.940 | 0.663 | 1.062 | 0.605–1.865 | 0.833 |
| Stage | 0.808 | 0.421–1.554 | 0.523 | 0.295 | 0.061–1.412 | 0.126 |
| T classification | 1.753 | 0.826–3.721 | 0.144 | 2.755 | 1.086–6.988 | 0.033 |
| M classification | 0.961 | 0.233–3.97 | 0.956 | 3.162 | 0.328–30.501 | 0.320 |
| N classification | 0.914 | 0.578–1.444 | 0.699 | 1.534 | 0.637–3.690 | 0.340 |
| RiskScore | 3.285 | 1.681–6.420 | 0.001 | 2.931 | 1.474–5.829 | 0.002 |
| Testing group | ||||||
| Age | 1.008 | 0.978–1.038 | 0.612 | 0.991 | 0.962–1.021 | 0.556 |
| Gender | 0.623 | 0.352–1.105 | 0.106 | 0.595 | 0.327–1.083 | 0.089 |
| Stage | 1.039 | 0.792–1.363 | 0.782 | 0.861 | 0.371–2.001 | 0.728 |
| T classification | 1.063 | 0.755–1.496 | 0.726 | 1.083 | 0.704–1.664 | 0.717 |
| M classification | 0.943 | 0.372–2.393 | 0.902 | 0.916 | 0.142–5.909 | 0.926 |
| N classification | 1.178 | 0.802–1.730 | 0.404 | 1.628 | 0.751–3.529 | 0.217 |
| RiskScore | 1.594 | 1.256–2.022 | <0.001 | 1.662 | 1.284–2.152 | <0.001 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, Z.; Qi, F.; Li, F. Establishment of a Gene Signature to Predict Prognosis for Patients with Lung Adenocarcinoma. Int. J. Mol. Sci. 2020, 21, 8479. https://doi.org/10.3390/ijms21228479
Li Z, Qi F, Li F. Establishment of a Gene Signature to Predict Prognosis for Patients with Lung Adenocarcinoma. International Journal of Molecular Sciences. 2020; 21(22):8479. https://doi.org/10.3390/ijms21228479
Chicago/Turabian StyleLi, Zhaodong, Fangyuan Qi, and Fan Li. 2020. "Establishment of a Gene Signature to Predict Prognosis for Patients with Lung Adenocarcinoma" International Journal of Molecular Sciences 21, no. 22: 8479. https://doi.org/10.3390/ijms21228479
APA StyleLi, Z., Qi, F., & Li, F. (2020). Establishment of a Gene Signature to Predict Prognosis for Patients with Lung Adenocarcinoma. International Journal of Molecular Sciences, 21(22), 8479. https://doi.org/10.3390/ijms21228479