Brain Endothelial P-Glycoprotein Level Is Reduced in Parkinson’s Disease via a Vitamin D Receptor-Dependent Pathway

 ,
, 
Abstract
:1. Introduction
2. Results
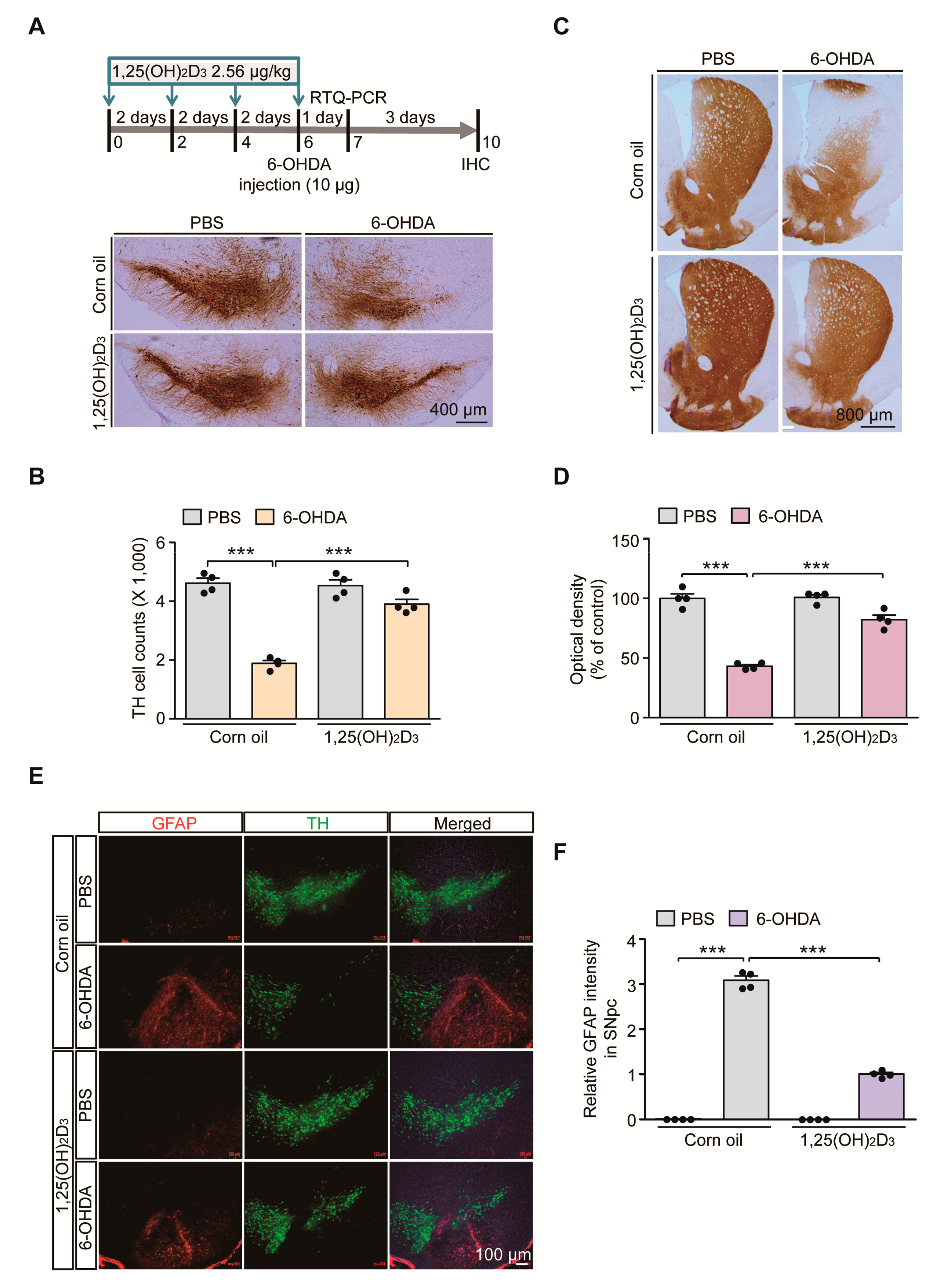
2.1. VDR Stimulation by 1,25(OH)2D3 Is Neuroprotective in 6-OHDA PD Mice
2.2. 1,25(OH)2D3 Treatment Restores 6-OHDA-Induced Impairment of VDR-Endothelial P-gp Signaling Pathway In Vivo
2.3. Clinical Relevance of Endothelial P-gp Downregulation with α-Synuclein Pathology in PD
2.4. PFF-Induced Repression of the VDR-P-gp Pathway in HUVECs Is Restored by 1,25(OH)2D3
3. Discussion
4. Materials and Methods
4.1. Chemicals and Antibodies
4.2. HUVEC Culture and Treatment
4.3. Real-Time Quantitative Polymerase Chain Reaction
| mGAPDH: |
| F- 5′ TGGCCTTCCGTGTTCCTAC 3′, R- 5′ GAGTTGCTGTTGAAGTCGCA 3′ |
| mVDR: |
| F- 5′ GAGGTGTCTGAAGCCTGGAG 3′, R- 5′ ACCTGCTTTCCTGGGTAGGT 3′ |
| mCYP24: |
| F- 5′ CTGCCCCATTGACAAAAGGC 3′, R- 5′ CTCACCGTCGGTCATCAGC 3′ |
| mMDR1a: |
| F- 5′ CAGCAGTCAGTGTGCTTACAA 3′, R- 5′ ATGGCTCTTTTATCGGCCTCA 3′ |
| hGAPDH: |
| F- 5′ AAACCCATCACCATCTTCCAG 3′, R- 5′ AGGGGCCATCCACAGTCTTCT 3′ |
| hVDR: |
| F- 5′ GTGGACATCGGCATGATGAAG 3′, R- 5′ GGTCGTAGGTCTTATGGTGGG 3′ |
| hCYP24: |
| F- 5′ CGACTACCGCAAAGAAGGCTA 3′, R- 5′ ACCATTTGTTCAGTTCGCTGT 3′ |
| hMDR1: |
| F- 5′ GGGAGCTTAACACCCGACTTA 3′, R- 5′ GCCAAAATCACAAGGGTTAGCTT 3′ |
4.4. Animal Experiments
4.5. Stereotaxic Injection of 6-OHDA or PFF/rAAV-αSyn
4.6. TH Stereological Cell Counting
4.7. Immunofluorescence
4.8. Doxorubicin Uptake and Clearance Assay
4.9. Acquisition and Handling of Human Postmortem Brains
4.10. Statistical Analyses
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| MDR1 | Multidrug resistance protein 1 |
| VDR | vitamin D receptor |
| PD | Parkinson’s disease |
| P-gp | P-glycoprotein |
| PFF | α-synuclein preformed fibril |
| 6-OHDA | 6-hydroxydopamine |
References
- Lang, A.E.; Lozano, A.M. Parkinson’s disease. First of two parts. N. Engl. J. Med. 1998, 339, 1044–1053. [Google Scholar] [CrossRef]
- Lang, A.E.; Lozano, A.M. Parkinson’s disease. Second of two parts. N. Engl. J. Med. 1998, 339, 1130–1143. [Google Scholar] [CrossRef]
- Luk, K.C.; Kehm, V.; Carroll, J.; Zhang, B.; O’Brien, P.; Trojanowski, J.Q.; Lee, V.M. Pathological alpha-synuclein transmission initiates Parkinson-like neurodegeneration in nontransgenic mice. Science 2012, 338, 949–953. [Google Scholar] [CrossRef] [Green Version]
- Spillantini, M.G.; Goedert, M. The alpha-synucleinopathies: Parkinson’s disease, dementia with Lewy bodies, and multiple system atrophy. Ann. N. Y. Acad. Sci. 2000, 920, 16–27. [Google Scholar] [CrossRef]
- Kim, C.; Ho, D.H.; Suk, J.E.; You, S.; Michael, S.; Kang, J.; Joong Lee, S.; Masliah, E.; Hwang, D.; Lee, H.J.; et al. Neuron-released oligomeric alpha-synuclein is an endogenous agonist of TLR2 for paracrine activation of microglia. Nat. Commun. 2013, 4, 1562. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.; Kang, S.J.; Jo, Y.M.; Park, S.; Yun, S.P.; Lee, Y.S.; Kim, H.T.; Lee, N.E.; Kim, Y.S.; Cho, S.H.; et al. Novel Nasal Epithelial Cell Markers of Parkinson’s Disease Identified Using Cells Treated with alpha-Synuclein Preformed Fibrils. J. Clin. Med. 2020, 9, 2128. [Google Scholar] [CrossRef]
- Mao, X.; Ou, M.T.; Karuppagounder, S.S.; Kam, T.I.; Yin, X.; Xiong, Y.; Ge, P.; Umanah, G.E.; Brahmachari, S.; Shin, J.H.; et al. Pathological alpha-synuclein transmission initiated by binding lymphocyte-activation gene 3. Science 2016, 353, 6307. [Google Scholar] [CrossRef] [Green Version]
- Yun, S.P.; Kam, T.I.; Panicker, N.; Kim, S.; Oh, Y.; Park, J.S.; Kwon, S.H.; Park, Y.J.; Karuppagounder, S.S.; Park, H.; et al. Block of A1 astrocyte conversion by microglia is neuroprotective in models of Parkinson’s disease. Nat. Med. 2018, 24, 931–938. [Google Scholar] [CrossRef]
- Guan, J.; Pavlovic, D.; Dalkie, N.; Waldvogel, H.J.; O’Carroll, S.J.; Green, C.R.; Nicholson, L.F. Vascular degeneration in Parkinson’s disease. Brain Pathol. 2013, 23, 154–164. [Google Scholar] [CrossRef]
- Yang, P.; Min, X.L.; Mohammadi, M.; Turner, C.; Faull, R. Endothelial Degeneration of Parkinson’s Disease is Related to Alpha-Synuclein Aggregation. J. Alzheimers Dis. Parkinsonism 2017, 7, 370. [Google Scholar] [CrossRef] [Green Version]
- Kuan, W.L.; Bennett, N.; He, X.; Skepper, J.N.; Martynyuk, N.; Wijeyekoon, R.; Moghe, P.V.; Williams-Gray, C.H.; Barker, R.A. Alpha-Synuclein pre-formed fibrils impair tight junction protein expression without affecting cerebral endothelial cell function. Exp. Neurol. 2016, 285, 72–81. [Google Scholar] [CrossRef] [Green Version]
- Durk, M.R.; Han, K.; Chow, E.C.; Ahrens, R.; Henderson, J.T.; Fraser, P.E.; Pang, K.S. 1alpha,25-Dihydroxyvitamin D3 reduces cerebral amyloid-beta accumulation and improves cognition in mouse models of Alzheimer’s disease. J. Neurosci. 2014, 34, 7091–7101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, S.F. Structure, function and regulation of P-glycoprotein and its clinical relevance in drug disposition. Xenobiotica 2008, 38, 802–832. [Google Scholar] [CrossRef] [PubMed]
- Bartels, A.L.; Willemsen, A.T.; Kortekaas, R.; de Jong, B.M.; de Vries, R.; de Klerk, O.; van Oostrom, J.C.; Portman, A.; Leenders, K.L. Decreased blood-brain barrier P-glycoprotein function in the progression of Parkinson’s disease, PSP and MSA. J. Neural Transm. 2008, 115, 1001–1009. [Google Scholar] [CrossRef] [Green Version]
- Kortekaas, R.; Leenders, K.L.; van Oostrom, J.C.; Vaalburg, W.; Bart, J.; Willemsen, A.T.; Hendrikse, N.H. Blood-brain barrier dysfunction in parkinsonian midbrain in vivo. Ann. Neurol. 2005, 57, 176–179. [Google Scholar] [CrossRef] [PubMed]
- Rimmelzwaan, L.M.; van Schoor, N.M.; Lips, P.; Berendse, H.W.; Eekhoff, E.M. Systematic Review of the Relationship between Vitamin D and Parkinson’s Disease. J. Parkinsons Dis. 2016, 6, 29–37. [Google Scholar] [CrossRef] [Green Version]
- Lima, L.A.R.; Lopes, M.J.P.; Costa, R.O.; Lima, F.A.V.; Neves, K.R.T.; Calou, I.B.F.; Andrade, G.M.; Viana, G.S.B. Vitamin D protects dopaminergic neurons against neuroinflammation and oxidative stress in hemiparkinsonian rats. J. Neuroinflamm. 2018, 15, 249. [Google Scholar] [CrossRef]
- Garcion, E.; Wion-Barbot, N.; Montero-Menei, C.N.; Berger, F.; Wion, D. New clues about vitamin D functions in the nervous system. Trends Endocrinol. Metab. 2002, 13, 100–105. [Google Scholar] [CrossRef]
- Naveilhan, P.; Neveu, I.; Wion, D.; Brachet, P. 1,25-Dihydroxyvitamin D3, an inducer of glial cell line-derived neurotrophic factor. Neuroreport 1996, 7, 2171–2175. [Google Scholar] [CrossRef]
- Durk, M.R.; Chan, G.N.; Campos, C.R.; Peart, J.C.; Chow, E.C.; Lee, E.; Cannon, R.E.; Bendayan, R.; Miller, D.S.; Pang, K.S. 1alpha,25-Dihydroxyvitamin D3-liganded vitamin D receptor increases expression and transport activity of P-glycoprotein in isolated rat brain capillaries and human and rat brain microvessel endothelial cells. J. Neurochem. 2012, 123, 944–953. [Google Scholar] [CrossRef] [Green Version]
- Maeng, H.J.; Chapy, H.; Zaman, S.; Pang, K.S. Effects of 1alpha,25-dihydroxyvitamin D3 on transport and metabolism of adefovir dipivoxil and its metabolites in Caco-2 cells. Eur. J. Pharm. Sci. 2012, 46, 149–166. [Google Scholar] [CrossRef] [PubMed]
- Chow, E.C.; Maeng, H.J.; Liu, S.; Khan, A.A.; Groothuis, G.M.; Pang, K.S. 1alpha,25-Dihydroxyvitamin D(3) triggered vitamin D receptor and farnesoid X receptor-like effects in rat intestine and liver in vivo. Biopharm. Drug Dispos. 2009, 30, 457–475. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.C.; Kim, I.B.; Noh, C.K.; Quach, H.P.; Yoon, I.S.; Chow, E.C.Y.; Kim, M.; Jin, H.E.; Cho, K.H.; Chung, S.J.; et al. Effects of 1alpha,25-dihydroxyvitamin D3, the natural vitamin D receptor ligand, on the pharmacokinetics of cefdinir and cefadroxil, organic anion transporter substrates, in rat. J. Pharm. Sci. 2014, 103, 3793–3805. [Google Scholar] [CrossRef] [PubMed]
- Durk, M.R.; Fan, J.; Sun, H.; Yang, Y.; Pang, H.; Pang, K.S.; de Lannoy, I.A. Vitamin D receptor activation induces P-glycoprotein and increases brain efflux of quinidine: An intracerebral microdialysis study in conscious rats. Pharm. Res. 2015, 32, 1128–1140. [Google Scholar] [CrossRef] [PubMed]
- Saeki, M.; Kurose, K.; Tohkin, M.; Hasegawa, R. Identification of the functional vitamin D response elements in the human MDR1 gene. Biochem. Pharmacol. 2008, 76, 531–542. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.W.; Cho, H.J.; Lee, H.J.; Jin, H.E.; Maeng, H.J. Polyethylene glycol-decorated doxorubicin/carboxymethyl chitosan/gold nanocomplex for reducing drug efflux in cancer cells and extending circulation in blood stream. Int. J. Biol. Macromol. 2019, 125, 61–71. [Google Scholar] [CrossRef]
- Ozvegy, C.; Litman, T.; Szakacs, G.; Nagy, Z.; Bates, S.; Varadi, A.; Sarkadi, B. Functional characterization of the human multidrug transporter, ABCG2, expressed in insect cells. Biochem. Biophys. Res. Commun. 2001, 285, 111–117. [Google Scholar] [CrossRef]
- Stride, B.D.; Grant, C.E.; Loe, D.W.; Hipfner, D.R.; Cole, S.P.; Deeley, R.G. Pharmacological characterization of the murine and human orthologs of multidrug-resistance protein in transfected human embryonic kidney cells. Mol. Pharmacol. 1997, 52, 344–353. [Google Scholar] [CrossRef] [Green Version]
- Coppola-Segovia, V.; Cavarsan, C.; Maia, F.G.; Ferraz, A.C.; Nakao, L.S.; Lima, M.M.; Zanata, S.M. ER Stress Induced by Tunicamycin Triggers alpha-Synuclein Oligomerization, Dopaminergic Neurons Death and Locomotor Impairment: A New Model of Parkinson’s Disease. Mol. Neurobiol. 2016, 54, 5798–5806. [Google Scholar] [CrossRef]
- Olzscha, H.; Schermann, S.M.; Woerner, A.C.; Pinkert, S.; Hecht, M.H.; Tartaglia, G.G.; Vendruscolo, M.; Hayer-Hartl, M.; Hartl, F.U.; Vabulas, R.M. Amyloid-like aggregates sequester numerous metastable proteins with essential cellular functions. Cell 2011, 144, 67–78. [Google Scholar] [CrossRef] [Green Version]
- Di Maio, R.; Barrett, P.J.; Hoffman, E.K.; Barrett, C.W.; Zharikov, A.; Borah, A.; Hu, X.; McCoy, J.; Chu, C.T.; Burton, E.A.; et al. Alpha-Synuclein binds to TOM20 and inhibits mitochondrial protein import in Parkinson’s disease. Sci. Transl. Med. 2016, 8, 342ra78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jehan, F.; DeLuca, H.F. Cloning and characterization of the mouse vitamin D receptor promoter. Proc. Natl. Acad. Sci. USA 1997, 94, 10138–10143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, F.H.; Li, S.S.; Li, X.X.; Wang, S.; Li, M.G.; Guan, L.; Luan, T.G.; Liu, Z.G.; Liu, Z.J.; Yang, P.C. Vitamin D3 induces vitamin D receptor and HDAC11 binding to relieve the promoter of the tight junction proteins. Oncotarget 2017, 8, 58781–58789. [Google Scholar] [CrossRef] [PubMed]
- Thakur, P.; Breger, L.S.; Lundblad, M.; Wan, O.W.; Mattsson, B.; Luk, K.C.; Lee, V.M.Y.; Trojanowski, J.Q.; Bjorklund, A. Modeling Parkinson’s disease pathology by combination of fibril seeds and alpha-synuclein overexpression in the rat brain. Proc. Natl. Acad. Sci. USA 2017, 114, E8284–E8293. [Google Scholar] [CrossRef] [Green Version]
- Sui, Y.T.; Bullock, K.M.; Erickson, M.A.; Zhang, J.; Banks, W.A. Alpha synuclein is transported into and out of the brain by the blood-brain barrier. Peptides 2014, 62, 197–202. [Google Scholar] [CrossRef] [Green Version]
- Garretti, F.; Agalliu, D.; Lindestam Arlehamn, C.S.; Sette, A.; Sulzer, D. Autoimmunity in Parkinson’s Disease: The Role of alpha-Synuclein-Specific T Cells. Front Immunol. 2019, 10, 303. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.; Lu, R.; Zhang, Y.G.; Sun, J. Vitamin D Receptor Deletion Leads to the Destruction of Tight and Adherens Junctions in Lungs. Tissue Barriers 2018, 6, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ham, S.; Kim, H.; Yoon, J.H.; Song, B.R.; Choi, J.Y.; Lee, Y.S.; Paek, S.M.; Maeng, H.J.; Lee, Y. Therapeutic Evaluation of Synthetic Peucedanocoumarin III in an Animal Model of Parkinson’s Disease. Int. J. Mol. Sci. 2019, 20, 5481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Volpicelli-Daley, L.A.; Luk, K.C.; Lee, V.M. Addition of exogenous alpha-synuclein preformed fibrils to primary neuronal cultures to seed recruitment of endogenous alpha-synuclein to Lewy body and Lewy neurite-like aggregates. Nat. Protoc. 2014, 9, 2135–2146. [Google Scholar] [CrossRef] [PubMed] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Control | PD | p Value | |
|---|---|---|---|
| N | 6 | 6 | |
| Male: female | 3:3 | 3:3 | 0.56 |
| Age (years) | 80.83 ± 3.32 | 79.17 ± 2.06 | 0.68 |
| Postmortem interval (days) | 2.79 ± 0.49 | 3.47 ± 0.26 | 0.25 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, H.; Shin, J.-Y.; Lee, Y.-S.; Yun, S.P.; Maeng, H.-J.; Lee, Y. Brain Endothelial P-Glycoprotein Level Is Reduced in Parkinson’s Disease via a Vitamin D Receptor-Dependent Pathway. Int. J. Mol. Sci. 2020, 21, 8538. https://doi.org/10.3390/ijms21228538
Kim H, Shin J-Y, Lee Y-S, Yun SP, Maeng H-J, Lee Y. Brain Endothelial P-Glycoprotein Level Is Reduced in Parkinson’s Disease via a Vitamin D Receptor-Dependent Pathway. International Journal of Molecular Sciences. 2020; 21(22):8538. https://doi.org/10.3390/ijms21228538
Chicago/Turabian StyleKim, Hyojung, Jeong-Yong Shin, Yun-Song Lee, Seung Pil Yun, Han-Joo Maeng, and Yunjong Lee. 2020. "Brain Endothelial P-Glycoprotein Level Is Reduced in Parkinson’s Disease via a Vitamin D Receptor-Dependent Pathway" International Journal of Molecular Sciences 21, no. 22: 8538. https://doi.org/10.3390/ijms21228538
APA StyleKim, H., Shin, J. -Y., Lee, Y. -S., Yun, S. P., Maeng, H. -J., & Lee, Y. (2020). Brain Endothelial P-Glycoprotein Level Is Reduced in Parkinson’s Disease via a Vitamin D Receptor-Dependent Pathway. International Journal of Molecular Sciences, 21(22), 8538. https://doi.org/10.3390/ijms21228538