New Cav1.2 Channelopathy with High-Functioning Autism, Affective Disorder, Severe Dental Enamel Defects, a Short QT Interval, and a Novel CACNA1C Loss-of-Function Mutation


 , ,
, , {kind=link}
{kind=link}
Abstract
:1. Introduction
2. Case Presentation
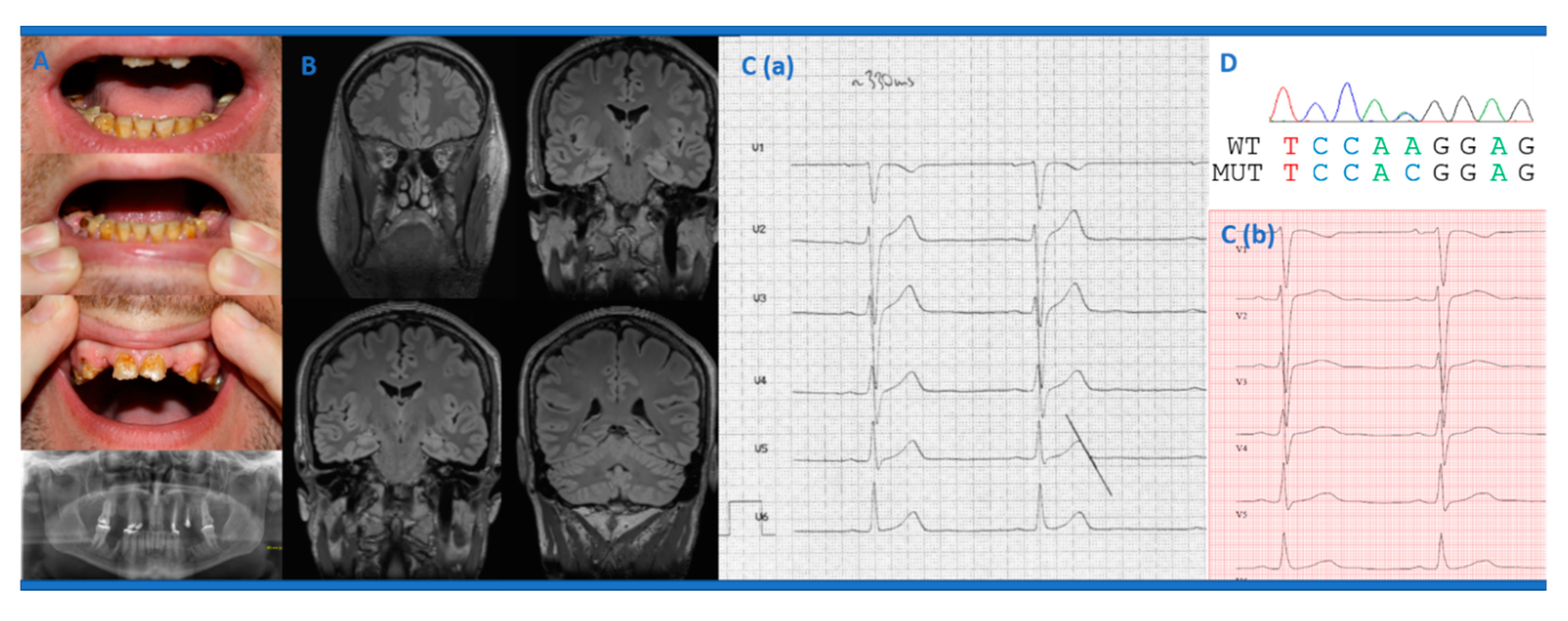
2.1. Clinical Case Description
2.2. Genetic Analyses
2.3. Electrophysiological Analyses
3. Discussion
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Tebartz van Elst, L.; Pick, M.; Biscaldi, M.; Fangmeier, T.; Riedel, A. High-functioning autism spectrum disorder as a basic disorder in adult psychiatry and psychotherapy: Psychopathological presentation, clinical relevance and therapeutic concepts. Eur. Arch. Psychiatry Clin. Neurosci. 2013, 263 (Suppl. S2), 189–196. [Google Scholar] [CrossRef] [PubMed]
- Splawski, I.; Timothy, K.W.; Sharpe, L.M.; Decher, N.; Kumar, P.; Bloise, R.; Napolitano, C.; Schwartz, P.J.; Joseph, R.M.; Condouris, K.; et al. CaV1.2 Calcium Channel Dysfunction Causes a Multisystem Disorder Including Arrhythmia and Autism. Cell 2004, 119, 19–31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Napolitano, C.; Splawski, I.; Timothy, K.W.; Bloise, R.; Priori, S.G. Timothy Syndrome. In GeneReviews®; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Stephens, K., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 2006. [Google Scholar]
- Walsh, M.A.; Turner, C.; Timothy, K.W.; Seller, N.; Hares, D.L.; James, A.F.; Hancox, J.C.; Uzun, O.; Boyce, D.; Stuart, A.G.; et al. A multicentre study of patients with Timothy syndrome. Europace 2018, 20, 377–385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wemhöner, K.; Friedrich, C.; Stallmeyer, B.; Coffey, A.J.; Grace, A.; Zumhagen, S.; Seebohm, G.; Ortiz-Bonnin, B.; Rinné, S.; Sachse, F.B.; et al. Gain-of-function mutations in the calcium channel CACNA1C (Cav1.2) cause non-syndromic long-QT but not Timothy syndrome. J. Mol. Cell. Cardiol. 2015, 80, 186–195. [Google Scholar] [CrossRef] [PubMed]
- Tebartz van Elst, L. Das Asperger-Syndrom im Erwachsenenalter und Andere Hochfunktionale Autismus-Spektrum-Störungen, 2nd ed.; Medizinisch Wissenschaftliche Verlagsgesellschaft: Berlin, Germany, 2015. [Google Scholar]
- Tebartz van Elst, L.; Maier, S.E.; Fangmeier, T.; Endres, D.; Mueller, G.T.; Nickel, K.; Ebert, D.; Lange, T.; Hennig, J.; Biscaldi, M.; et al. Disturbed cingulate glutamate metabolism in adults with high-functioning autism spectrum disorder: Evidence in support of the excitatory/inhibitory imbalance hypothesis. Mol. Psychiatry 2014, 19, 1314–1325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Endres, D.; Tebartz van Elst, L.; Meyer, S.A.; Feige, B.; Nickel, K.; Bubl, A.; Riedel, A.; Ebert, D.; Lange, T.; Glauche, V.; et al. Glutathione metabolism in the prefrontal brain of adults with high-functioning autism spectrum disorder: An MRS study. Mol. Autism 2017, 8, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lord, C.; Risi, S.; Lambrecht, E.H., Jr.; Leventhal, B.; DiLavore, P.C.; Pickles, A.; Rutter, M. The Autism Diagnostic Observation Schedule—Generic: A Standard Measure of Social and Communication Deficits Associated with the Spectrum of Autism. J. Autism Dev. Disord. 2000, 30, 205–223. [Google Scholar] [CrossRef] [PubMed]
- Dziobek, I.; Fleck, S.; Kalbe, E.; Rogers, K.; Hassenstab, J.; Brand, M.; Kessler, J.; Woike, J.K.; Wolf, O.T.; Convit, A. Introducing MASC: A Movie for the Assessment of Social Cognition. J. Autism Dev. Disord. 2006, 36, 623–636. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–423. [Google Scholar] [CrossRef] [PubMed]
- Papineau, S.D.; Wilson, S. Dentition abnormalities in a Timothy syndrome patient with a novel genetic mutation: A case report. Pediatr. Dent. 2014, 36, 245–249. [Google Scholar] [PubMed]
- Campuzano, O.; Sarquella-Brugada, G.; Cesar, S.; Arbelo, E.; Brugada, J.; Brugada, R. Recent Advances in Short QT Syndrome. Front. Cardiovasc. Med. 2018, 5, 149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campuzano, O.; Fernandez-Falgueras, A.; Maulen, X.L.; Sarquella-Brugada, G.; Cesar, S.; Coll, M.; Mates, J.; Arbelo, E.; Jordà, P.; Pérez-Serra, A.; et al. Short QT Syndrome: A Comprehensive Genetic Interpretation and Clinical Translation of Rare Variants. J. Clin. Med. 2019, 8, 1035. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Barajas-Martinez, H.; Zhu, D.; Wang, X.; Chen, C.; Zhuang, R.; Shi, J.; Wu, X.; Tao, Y.; Jin, W.; et al. Novel trigenic CACNA1C/DES/MYPN mutations in a family of hypertrophic cardiomyopathy with early repolarization and short QT syndrome. J. Transl. Med. 2017, 15, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Liao, P.; Soong, T.W. CaV1.2 channelopathies: From arrhythmias to autism, bipolar disorder, and immunodeficiency. Pflug. Arch. 2010, 460, 353–359. [Google Scholar] [CrossRef] [PubMed]
- Kabitzke, P.; Brunner, D.; He, D.; Fazio, P.; Cox, K.; Sutphen, J.; Thiede, L.; Sabath, E.; Hanania, T.; Alexandrov, V.; et al. Comprehensive analysis of two Shank3 and the Cacna1c mouse models of autism spectrum disorder. Genes Brain Behav. 2017, 17, 4–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moon, A.L.; Haan, N.; Wilkinson, L.S.; Thomas, K.L.; Hall, J. CACNA1C: Association With Psychiatric Disorders, Behavior, and Neurogenesis. Schizophr. Bull. 2018, 44, 958–965. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Zhao, L.; You, Y.; Lu, T.; Jia, M.; Yu, H.; Ruan, Y.; Yue, W.; Liu, J.; Lu, L.; et al. Schizophrenia Related Variants in CACNA1C also Confer Risk of Autism. PLoS ONE 2015, 10, e0133247. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Endres, D.; Decher, N.; Röhr, I.; Vowinkel, K.; Domschke, K.; Komlosi, K.; Tzschach, A.; Gläser, B.; Schiele, M.A.; Runge, K.; et al. New Cav1.2 Channelopathy with High-Functioning Autism, Affective Disorder, Severe Dental Enamel Defects, a Short QT Interval, and a Novel CACNA1C Loss-of-Function Mutation. Int. J. Mol. Sci. 2020, 21, 8611. https://doi.org/10.3390/ijms21228611
Endres D, Decher N, Röhr I, Vowinkel K, Domschke K, Komlosi K, Tzschach A, Gläser B, Schiele MA, Runge K, et al. New Cav1.2 Channelopathy with High-Functioning Autism, Affective Disorder, Severe Dental Enamel Defects, a Short QT Interval, and a Novel CACNA1C Loss-of-Function Mutation. International Journal of Molecular Sciences. 2020; 21(22):8611. https://doi.org/10.3390/ijms21228611
Chicago/Turabian StyleEndres, Dominique, Niels Decher, Isabell Röhr, Kirsty Vowinkel, Katharina Domschke, Katalin Komlosi, Andreas Tzschach, Birgitta Gläser, Miriam A. Schiele, Kimon Runge, and et al. 2020. "New Cav1.2 Channelopathy with High-Functioning Autism, Affective Disorder, Severe Dental Enamel Defects, a Short QT Interval, and a Novel CACNA1C Loss-of-Function Mutation" International Journal of Molecular Sciences 21, no. 22: 8611. https://doi.org/10.3390/ijms21228611
APA StyleEndres, D., Decher, N., Röhr, I., Vowinkel, K., Domschke, K., Komlosi, K., Tzschach, A., Gläser, B., Schiele, M. A., Runge, K., Süß, P., Schuchardt, F., Nickel, K., Stallmeyer, B., Rinné, S., Schulze-Bahr, E., & Tebartz van Elst, L. (2020). New Cav1.2 Channelopathy with High-Functioning Autism, Affective Disorder, Severe Dental Enamel Defects, a Short QT Interval, and a Novel CACNA1C Loss-of-Function Mutation. International Journal of Molecular Sciences, 21(22), 8611. https://doi.org/10.3390/ijms21228611