Genetic Deletion of NOD1 Prevents Cardiac Ca2+ Mishandling Induced by Experimental Chronic Kidney Disease

 , ,
, ,  , and
, and
Abstract
:1. Introduction
2. Results
2.1. Macroscopic and Microscopic Cardiac Features and Biochemical Parameters of Renal Function in Wild-Type and Nod1−/− Mice at Baseline and after Experimental CKD
2.2. Deficiency of NOD1 Prevents both Systolic Ca2+ Release Impairment and the Decrease in SR Ca2+ Load Triggered by Experimental CKD
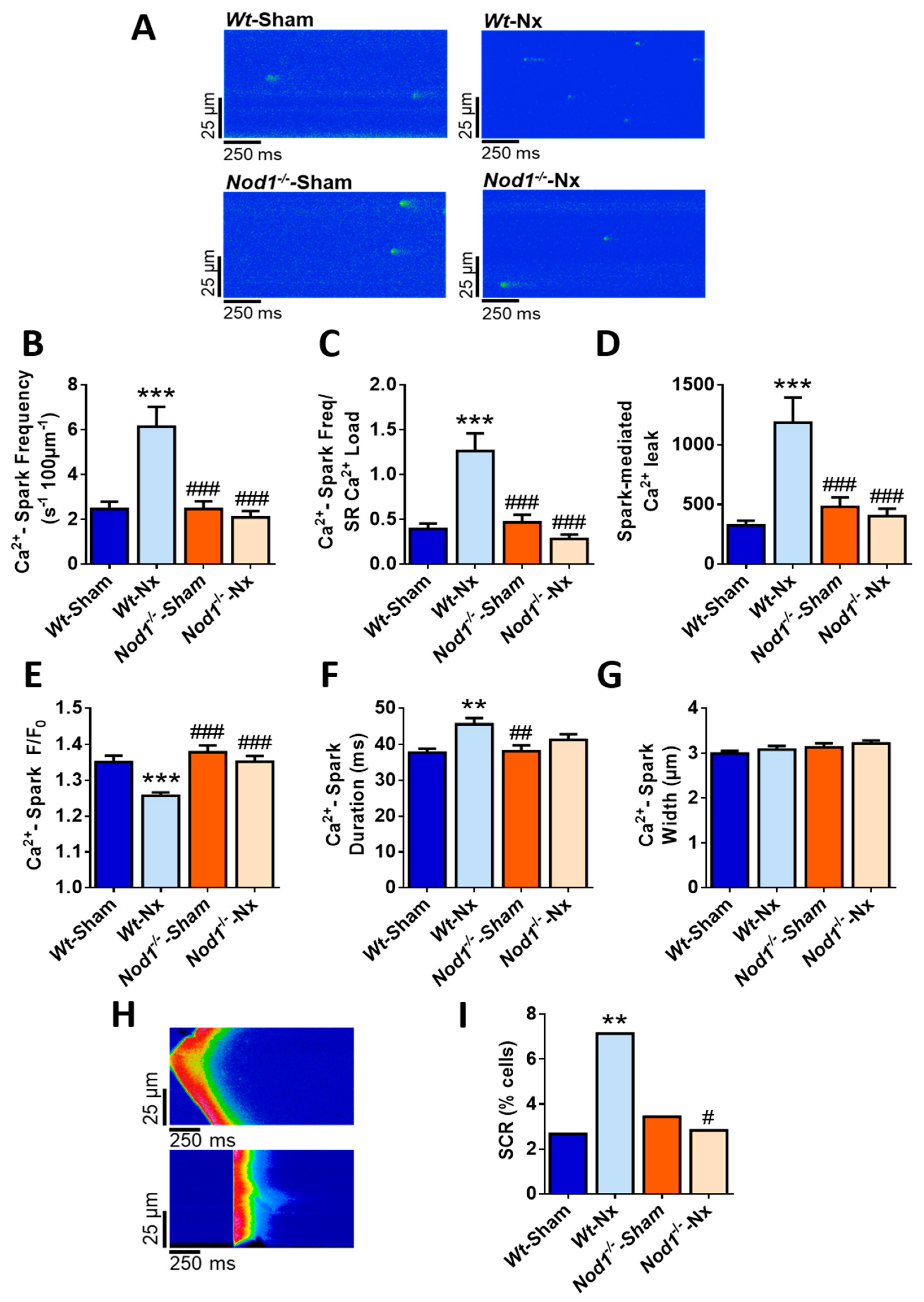
2.3. Deficiency of NOD1 Blunts the Increase in Diastolic Ca2+ Release Induced by Nx
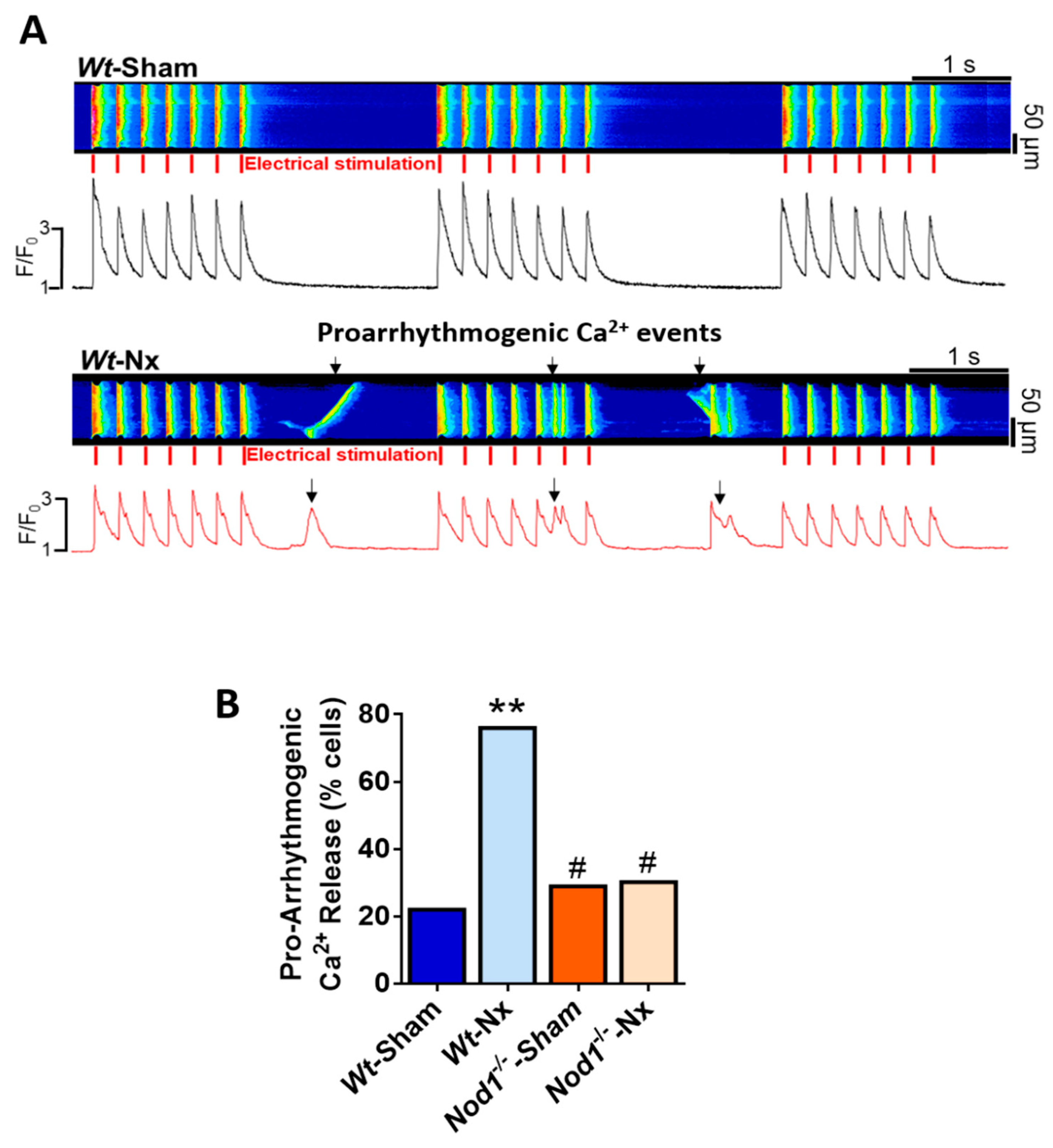
2.4. Deficiency of NOD1 Prevents the Increase in the Rate of Pro-Arrhythmogenic Ca2+ Events Induced by Nx
2.5. Macroscopic and Microscopic Cardiac Features and Biochemistry Parameters of Renal Function of Rip2−/− Mice at Baseline and after Experimental CKD
2.6. Deficiency of RIP2 Prevents Ca2+ Mishandling Induced by Experimental CKD
3. Discussion
4. Methods
4.1. Animal Care
4.2. Serology
4.3. Experimental CKD
4.4. Cardiomyocyte Isolation
4.5. Intracellular Calcium Imaging
4.6. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| BW | body weight |
| CARD | caspase activation and recruitment domain |
| CKD | chronic kidney disease |
| CVD | cardiovascular disease |
| EC | excitation-contraction |
| FGF23 | fibroblast growth factor-23 |
| HF | heart failure |
| HW | heart weight |
| KW | kidney weight |
| LTCCs | sarcolemma L-type Ca2+ channels |
| NCX | Na+/Ca2+ exchanger |
| NFκB | nuclear factor kappa B |
| NOD1 | nucleotide-binding oligomerization domain-containing protein 1 |
| NOD2 | nucleotide-binding oligomerization domain-containing protein 2 |
| Nx | five-sixth nephrectomy |
| RIP2 | receptor-interacting-serine/threonine-protein kinase 2 |
| RyR2 | ryanodine receptor type 2 |
| SCR | spontaneous Ca2+ release |
| SERCA2a | sarco/endoplasmic reticulum Ca2+ pump subtype |
| SR | sarcoplasmic reticulum |
| TL | tibia length |
| Wt | wild-type |
References
- Plantinga, L.C.; Boulware, L.E.; Coresh, J.; Stevens, L.A.; Miller, E.R.; Saran, R.; Messer, K.L.; Levey, A.S.; Powe, N.R. Patient awareness of chronic kidney disease: Trends and predictors. Arch. Intern. Med. 2008, 168, 2268–2275. [Google Scholar] [CrossRef] [PubMed]
- Gansevoort, R.T.; Correa-Rotter, R.; Hemmelgarn, B.R.; Jafar, T.H.; Heerspink, H.J.L.; Mann, J.F.; Matsushita, K.; Wen, C.P. Chronic kidney disease and cardiovascular risk: Epidemiology, mechanisms, and prevention. Lancet 2013, 382, 339–352. [Google Scholar] [CrossRef]
- Schefold, J.C.; Filippatos, G.; Hasenfuss, G.; Anker, S.D.; von Haehling, S. Heart failure and kidney dysfunction: Epidemiology, mechanisms and management. Nat. Rev. Nephrol. 2016, 12, 610–623. [Google Scholar] [CrossRef] [PubMed]
- House, A.A.; Wanner, C.; Sarnak, M.J.; Piña, I.L.; McIntyre, C.W.; Komenda, P.; Kasiske, B.L.; Deswal, A.; deFilippi, C.R.; Cleland, J.G.F.; et al. Heart failure in chronic kidney disease: Conclusions from a Kidney Disease: Improving Global Outcomes (KDIGO) Controversies Conference. Kidney Int. 2019, 95, 1304–1317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarnak, M.J.; Levey, A.S.; Schoolwerth, A.C.; Coresh, J.; Culleton, B.; Hamm, L.L.; McCullough, P.A.; Kasiske, B.L.; Kelepouris, E.; Klag, M.J.; et al. Kidney disease as a risk factor for development of cardiovascular disease: A statement from the American Heart Association Councils on Kidney in Cardiovascular Disease, High Blood Pressure Research, Clinical Cardiology, and Epidemiology and Prevention. Circulation 2003, 108, 2154–2169. [Google Scholar] [CrossRef]
- Rantanen, J.M.; Riahi, S.; Schmidt, E.B.; Johansen, M.B.; Søgaard, P.; Christensen, J.H. Arrhythmias in Patients on Maintenance Dialysis: A Cross-sectional Study. Am. J. Kidney Dis. 2020, 75, 214–224. [Google Scholar] [CrossRef]
- Di Lullo, L.; Rivera, R.; Barbera, V.; Bellasi, A.; Cozzolino, M.; Russo, D.; De Pascalis, A.; Banerjee, D.; Floccari, F.; Ronco, C. Sudden cardiac death and chronic kidney disease: From pathophysiology to treatment strategies. Int. J. Cardiol. 2016, 217, 16–27. [Google Scholar] [CrossRef]
- Navarro-García, J.A.; Fernández-Velasco, M.; Delgado, C.; Delgado, J.F.; Kuro-O, M.; Ruilope, L.M.; Ruiz-Hurtado, G. PTH, vitamin D, and the FGF-23-klotho axis and heart: Going beyond the confines of nephrology. Eur. J. Clin. Investig. 2018, 48, e12902. [Google Scholar] [CrossRef] [Green Version]
- Ruiz-Hurtado, G.; Ruilope, L.M. Hypertension and obesity: Correlates with renin-angiotensin-aldosterone system and uric acid. J. Clin. Hypertens. 2014, 16, 559–560. [Google Scholar] [CrossRef] [Green Version]
- Bers, D.M. Cardiac excitation-contraction coupling. Nature 2002, 415, 198–205. [Google Scholar] [CrossRef]
- Val-Blasco, A.; Piedras, M.J.G.M.; Ruiz-Hurtado, G.; Suarez, N.; Prieto, P.; Gonzalez-Ramos, S.; Gómez-Hurtado, N.; Delgado, C.; Pereira, L.; Benito, G.; et al. Role of NOD1 in Heart Failure Progression via Regulation of Ca 2+ Handling. J. Am. Coll. Cardiol. 2017, 69, 423–433. [Google Scholar] [CrossRef] [PubMed]
- Yao, C.; Veleva, T.; Scott, L.; Cao, S.; Li, L.; Chen, G.; Jeyabal, P.; Pan, X.; Alsina, K.M.; Abu-Taha, I.; et al. Enhanced Cardiomyocyte NLRP3 Inflammasome Signaling Promotes Atrial Fibrillation. Circulation 2018, 138, 2227–2242. [Google Scholar] [CrossRef] [PubMed]
- Mann, D.L. Innate immunity and the failing heart: The cytokine hypothesis revisited. Circ. Res. 2015, 116, 1254–1268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shigeoka, A.A.; Kambo, A.; Mathison, J.C.; King, A.J.; Hall, W.F.; da Silva Correia, J.; Ulevitch, R.J.; McKay, D.B. Nod1 and nod2 are expressed in human and murine renal tubular epithelial cells and participate in renal ischemia reperfusion injury. J. Immunol. 2010, 184, 2297–2304. [Google Scholar] [CrossRef] [PubMed]
- Laman, J.D.; Schoneveld, A.H.; Moll, F.L.; Van Meurs, M.; Pasterkamp, G. Significance of peptidoglycan, a proinflammatory bacterial antigen in atherosclerotic arteries and its association with vulnerable plaques. Am. J. Cardiol. 2002, 90, 119–123. [Google Scholar] [CrossRef]
- Inohara, N.; Koseki, T.; del Peso, L.; Hu, Y.; Yee, C. Nod1, an Apaf-1-like activator of caspase-9 and nuclear factor-κB. J. Biol. Chem. 1999, 274, 14560–14567. [Google Scholar] [CrossRef] [Green Version]
- Moreno, L.; Gatheral, T. Therapeutic targeting of NOD1 receptors. Br. J. Pharmacol. 2013, 170, 475–485. [Google Scholar] [CrossRef] [Green Version]
- Park, J.; Kim, Y.; Shaw, M.; Kanneganti, T.; Fujimoto, Y. Nod1/RICK and TLR signaling regulate chemokine and antimicrobial innate immune responses in mesothelial cells. J. Immunol. 2007, 179, 514–521. [Google Scholar] [CrossRef] [Green Version]
- Ogura, Y.; Inohara, N.; Benito, A.; Chen, F.F.; Yamaoka, S.; Nunez, G. Nod2, a Nod1/Apaf-1 family member that is restricted to monocytes and activates NF-kappaB. J. Biol. Chem. 2001, 276, 4812–4818. [Google Scholar] [CrossRef] [Green Version]
- Ogura, Y.; Lala, S.; Xin, W.; Smith, E.; Dowds, T.A.; Chen, F.F.; Zimmermann, E.; Tretiakova, M.; Cho, J.H.; Hart, J.; et al. Expression of NOD2 in Paneth cells: A possible link to Crohn’s ileitis. Gut 2003, 52, 1591–1597. [Google Scholar] [CrossRef]
- Davey, M.P.; Martin, T.M.; Planck, S.R.; Lee, J.; Zamora, D.; Rosenbaum, J.T. Human endothelial cells express NOD2/CARD15 and increase IL-6 secretion in response to muramyl dipeptide. Microvasc. Res. 2006, 71. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.Q.; Zhang, X.Y.; Edfeldt, K.; Nijhuis, M.O.; Idborg, H.; Bäck, M.; Roy, J.; Hedin, U.; Jakobsson, P.J.; Laman, J.D.; et al. NOD2-mediated innate immune signaling regulates the eicosanoids in atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 2193–2201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prieto, P.; Vallejo-Cremades, M.T.; Benito, G.; González-Peramato, P.; Francés, D.; Agra, N.; Terrón, V.; Gónzalez-Ramos, S.; Delgado, C.; Ruiz-Gayo, M.; et al. NOD1 receptor is up-regulated in diabetic human and murine myocardium. Clin. Sci. 2014, 127, 665–677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loyer, X.; Gómez, A.M.; Milliez, P.; Fernandez-Velasco, M.; Vangheluwe, P.; Vinet, L.; Charue, D.; Vaudin, E.; Zhang, W.; Sainte-Marie, Y.; et al. Cardiomyocyte overexpression of neuronal nitric oxide synthase delays transition toward heart failure in response to pressure overload by preserving calcium cycling. Circulation 2008, 117, 3187–3198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferrand, A.; Al Nabhani, Z.; Tapias, N.S.; Mas, E.; Hugot, J.P.; Barreau, F. NOD2 Expression in Intestinal Epithelial Cells Protects Toward the Development of Inflammation and Associated Carcinogenesis. CMGH 2019, 7, 357–369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al Nabhani, Z.; Dietrich, G.; Hugot, J.P.; Barreau, F. Nod2: The intestinal gate keeper. PLoS Pathog. 2017, 13, e1006177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mirkov, M.U.; Verstockt, B.; Cleynen, I. Genetics of inflammatory bowel disease: Beyond NOD2. Lancet Gastroenterol. Hepatol. 2017, 2, 224–234. [Google Scholar] [CrossRef]
- Berlin, P.; Reiner, J.; Witte, M.; Wobar, J.; Lindemann, S.; Barrantes, I.; Kreikemeyer, B.; Bastian, M.; Schäffler, H.; Bannert, K.; et al. Nod2 deficiency functionally impairs adaptation to short bowel syndrome via alterations of the epithelial barrier function. Am. J. Physiol. Gastrointest. Liver Physiol. 2019, 317, G727–G738. [Google Scholar] [CrossRef]
- Kobayashi, K.S.; Chamaillard, M.; Ogura, Y.; Henegariu, O.; Inohara, N.; Nuñez, G.; Flavell, R.A. Nod2-dependent regulation of innate and adaptive immunity in the intestinal tract. Science 2005, 307, 731–734. [Google Scholar] [CrossRef]
- Caruso, R.; Warner, N.; Inohara, N.; Núñez, G. NOD1 and NOD2: Signaling, Host Defense, and Inflammatory Disease. Immunity 2014, 41, 898–908. [Google Scholar] [CrossRef] [Green Version]
- Marchitti, S.A.; Brocker, C.; Stagos, D.; Vasiliou, V. Non-P450 aldehyde oxidizing enzymes: The aldehyde dehydrogenase superfamily. Expert Opin. Drug Metab. Toxicol. 2008, 4, 697–720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Motomura, Y.; Kanno, S.; Asano, K.; Tanaka, M.; Hasegawa, Y.; Katagiri, H.; Saito, T.; Hara, H.; Nishio, H.; Hara, T.; et al. Identification of Pathogenic Cardiac CD11c+ Macrophages in Nod1-Mediated Acute Coronary Arteritis. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 1423–1433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delgado, C.; Ruiz-Hurtado, G.; Gómez-Hurtado, N.; González-Ramos, S.; Rueda, A.; Benito, G.; Prieto, P.; Zaragoza, C.; Delicado, E.G.; Pérez-Sen, R.; et al. NOD1, a new player in cardiac function and calcium handling. Cardiovasc. Res. 2015, 106, 375–386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Val-Blasco, A.; Navarro-García, J.A.; Tamayo, M.; Piedras, M.J.; Prieto, P.; Delgado, C.; Ruiz-Hurtado, G.; Rozas-Romero, L.; Gil-Fernández, M.; Zaragoza, C.; et al. Deficiency of NOD1 improves the β-adrenergic modulation of Ca2+ handling in a mouse model of heart failure. Front. Physiol. 2018, 9. [Google Scholar] [CrossRef] [Green Version]
- Val-Blasco, A.; Prieto, P.; Gonzalez-Ramos, S.; Benito, G.; Vallejo-Cremades, M.T.; Pacheco, I.; González-Peramato, P.; Agra, N.; Terrón, V.; Delgado, C.; et al. NOD1 activation in cardiac fibroblasts induces myocardial fibrosis in a murine model of type 2 diabetes. Biochem. J. 2017, 474, 399–410. [Google Scholar] [CrossRef]
- Uehara, A.; Fujimoto, Y.; Fukase, K.; Takada, H. Various human epithelial cells express functional Toll-like receptors, NOD1 and NOD2 to produce anti-microbial peptides, but not proinflammatory cytokines. Mol. Immunol. 2007, 44, 3100–3111. [Google Scholar] [CrossRef]
- Stroo, I.; Emal, D.; Butter, L.M.; Teske, G.J.; Claessen, N.; Dessing, M.C.; Girardin, S.E.; Florquin, S.; Leemans, J.C. No difference in renal injury and fibrosis between wild-type and NOD1/NOD2 double knockout mice with chronic kidney disease induced by ureteral obstruction. BMC Nephrol. 2018, 19, 78. [Google Scholar] [CrossRef] [Green Version]
- Park, J.-H.; Kim, Y.-G.; McDonald, C.; Kanneganti, T.-D.; Hasegawa, M.; Body-Malapel, M.; Inohara, N.; Núñez, G. RICK/RIP2 Mediates Innate Immune Responses Induced through Nod1 and Nod2 but Not TLRs. J. Immunol. 2007, 178, 2380–2386. [Google Scholar] [CrossRef]
- Rafiq, K.; Noma, T.; Fujisawa, Y.; Ishihara, Y.; Arai, Y.; Nabi, A.H.M.N.; Suzuki, F.; Nagai, Y.; Nakano, D.; Hitomi, H.; et al. Renal sympathetic denervation suppresses de novo podocyte injury and albuminuria in rats with aortic regurgitation. Circulation 2012, 125, 1402–1413. [Google Scholar] [CrossRef]
- Wolf, M. Forging forward with 10 burning questions on FGF23 in kidney disease. J. Am. Soc. Nephrol. 2010, 21, 1427–1435. [Google Scholar] [CrossRef] [Green Version]
- Brown, S.J.; Ruppe, M.D.; Tabatabai, L.S. The Parathyroid Gland and Heart Disease. Methodist DeBakey Cardiovasc. J. 2017, 13, 49–54. [Google Scholar] [CrossRef] [Green Version]
- Ridker, P.M.; MacFadyen, J.G.; Glynn, R.J.; Koenig, W.; Libby, P.; Everett, B.M.; Lefkowitz, M.; Thuren, T.; Cornel, J.H. Inhibition of Interleukin-1β by Canakinumab and Cardiovascular Outcomes in Patients With Chronic Kidney Disease. J. Am. Coll. Cardiol. 2018, 71, 2405–2414. [Google Scholar] [CrossRef] [PubMed]
- Ridker, P.M. Clinician’s Guide to Reducing Inflammation to Reduce Atherothrombotic Risk: JACC Review Topic of the Week. J. Am. Coll. Cardiol. 2018, 72, 3320–3331. [Google Scholar] [CrossRef] [PubMed]
- Tourneur, E.; Ben Mkaddem, S.; Chassin, C.; Bens, M.; Goujon, J.-M.; Charles, N.; Pellefigues, C.; Aloulou, M.; Hertig, A.; Monteiro, R.C.; et al. Cyclosporine A Impairs Nucleotide Binding Oligomerization Domain (Nod1)-Mediated Innate Antibacterial Renal Defenses in Mice and Human Transplant Recipients. PLoS Pathog. 2013, 9, e1003152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stroo, I.; Butter, L.M.; Claessen, N.; Teske, G.J.; Rubino, S.J.; Girardin, S.E.; Florquin, S.; Leemans, J.C. Phenotyping of Nod1/2 double deficient mice and characterization of Nod1/2 in systemic inflammation and associated renal disease. Biol. Open 2012, 1, 1239–1247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gava, A.L.; Freitas, F.P.; Balarini, C.M.; Vasquez, E.C.; Meyrelles, S.S. Effects of 5/6 nephrectomy on renal function and blood pressure in mice. Int. J. Physiol. Pathophysiol. Pharmacol. 2012, 4, 167–173. [Google Scholar] [PubMed]
- Kren, S.; Hostetter, T.H. The course of the remnant kidney model in mice. Kidney Int. 1999, 56, 333–337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Navarro-García, J.A.; Rueda, A.; Romero-García, T.; Aceves-Ripoll, J.; Rodríguez-Sánchez, E.; González-Lafuente, L.; Zaragoza, C.; Fernández-Velasco, M.; Kuro-o, M.; Ruilope, L.M.; et al. Enhanced Klotho availability protects against cardiac dysfunction induced by uraemic cardiomyopathy by regulating Ca2+ handling. Br. J. Pharmacol. 2020. [Google Scholar] [CrossRef]
- Ruiz-Hurtado, G.; Li, L.; Fernández-Velasco, M.; Rueda, A.; Lefebvre, F.; Wang, Y.; Mateo, P.; Cassan, C.; Gellen, B.; Benitah, J.P.; et al. Reconciling depressed Ca2+ sparks occurrence with enhanced RyR2 activity in failing mice cardiomyocytes. J. Gen. Physiol. 2015, 146, 295–306. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, K.; Inohara, N.; Hernandez, L.D.; Galán, J.E.; Núñez, G.; Janeway, C.A.; Medzhitov, R.; Flavell, R.A. RICK/Rip2/CARDIAK mediates signalling for receptors of the innate and adaptive immune systems. Nature 2002, 416, 194–199. [Google Scholar] [CrossRef] [Green Version]
- Shioya, T. A Simple Technique for Isolating Healthy Heart Cells from Mouse Models. J. Physiol. Sci. 2007, 57, 327–335. [Google Scholar] [CrossRef] [PubMed] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Wt-sham | Wt-Nx | Nod1−/−-sham | Nod1−/−-Nx | |
|---|---|---|---|---|
| HW (mg) | 186.89 ± 6.08 (10) | 169.63 ± 4.65 (8) | 198.27 ± 6.66 # (10) | 185.03 ± 12.05 (9) |
| BW (g) | 26.24 ± 0.25 (10) | 23.11 ± 0.83 (8) | 26.99 ± 0.76 ## (10) | 25.26 ± 0.76 (9) |
| HW/BW (mg/g) | 7.11 ± 0.19 (10) | 7.43 ± 0.42 (8) | 7.38 ± 0.28 (10) | 7.31 ± 0.37 (9) |
| KW (mg) | 183.80 ± 4.78 (10) | 155.83 ± 8.62 * (8) | 190.96 ± 7.02 ## (10) | 165.90 ± 7.39 (9) |
| KW/BW (mg/g) | 7.01 ± 0.18 (10) | 6.76 ± 0.33 (8) | 7.09 ± 0.24 (10) | 6.58 ± 0.26 (9) |
| Cell area (μm2) | 3482.21 ± 109.25 (72 cells/10) | 3215.90 ± 119.73 (54 cells/8) | 3396.71 ± 94.36 (67 cells/10) | 3271.32 ± 116.62 (59 cells/9) |
| Wt-sham | Wt-Nx | Nod1−/−-sham | Nod1−/−-Nx | |
|---|---|---|---|---|
| Urea (mg/dL) | 37.24 ± 3.55 (5) | 83.30 ± 5.45 *** (8) | 39.80 ± 4.78 ### (6) | 83.68 ± 8.04 ***,&&& (8) |
| BUN (mg/dL) | 17.40 ± 1.66 (5) | 38.93 ± 2.55 *** (8) | 18.60 ± 2.23 ### (6) | 39.10 ± 3.76 ***,&&& (8) |
| Pi (mg/dL) | 6.19 ± 0.65 (7) | 6.91 ± 0.55 (8) | 6.21 ± 0.90 (6) | 6.92 ± 1.01 (8) |
| FGF-23 (pg/mL) | 140.3 ± 22.72 (7) | 294.00 ± 47.47 * (8) | 137.30 ± 24.66 (5) | 256.90 ± 38.51 (8) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gil-Fernández, M.; Navarro-García, J.A.; Val-Blasco, A.; González-Lafuente, L.; Martínez, J.C.; Rueda, A.; Tamayo, M.; Morgado, J.L.; Zaragoza, C.; Ruilope, L.M.; et al. Genetic Deletion of NOD1 Prevents Cardiac Ca2+ Mishandling Induced by Experimental Chronic Kidney Disease. Int. J. Mol. Sci. 2020, 21, 8868. https://doi.org/10.3390/ijms21228868
Gil-Fernández M, Navarro-García JA, Val-Blasco A, González-Lafuente L, Martínez JC, Rueda A, Tamayo M, Morgado JL, Zaragoza C, Ruilope LM, et al. Genetic Deletion of NOD1 Prevents Cardiac Ca2+ Mishandling Induced by Experimental Chronic Kidney Disease. International Journal of Molecular Sciences. 2020; 21(22):8868. https://doi.org/10.3390/ijms21228868
Chicago/Turabian StyleGil-Fernández, Marta, José Alberto Navarro-García, Almudena Val-Blasco, Laura González-Lafuente, José Carlos Martínez, Angélica Rueda, Maria Tamayo, José Luis Morgado, Carlos Zaragoza, Luis Miguel Ruilope, and et al. 2020. "Genetic Deletion of NOD1 Prevents Cardiac Ca2+ Mishandling Induced by Experimental Chronic Kidney Disease" International Journal of Molecular Sciences 21, no. 22: 8868. https://doi.org/10.3390/ijms21228868
APA StyleGil-Fernández, M., Navarro-García, J. A., Val-Blasco, A., González-Lafuente, L., Martínez, J. C., Rueda, A., Tamayo, M., Morgado, J. L., Zaragoza, C., Ruilope, L. M., Delgado, C., Ruiz-Hurtado, G., & Fernández-Velasco, M. (2020). Genetic Deletion of NOD1 Prevents Cardiac Ca2+ Mishandling Induced by Experimental Chronic Kidney Disease. International Journal of Molecular Sciences, 21(22), 8868. https://doi.org/10.3390/ijms21228868