An Automated Functional Annotation Pipeline That Rapidly Prioritizes Clinically Relevant Genes for Autism Spectrum Disorder


 ,
, 

Abstract
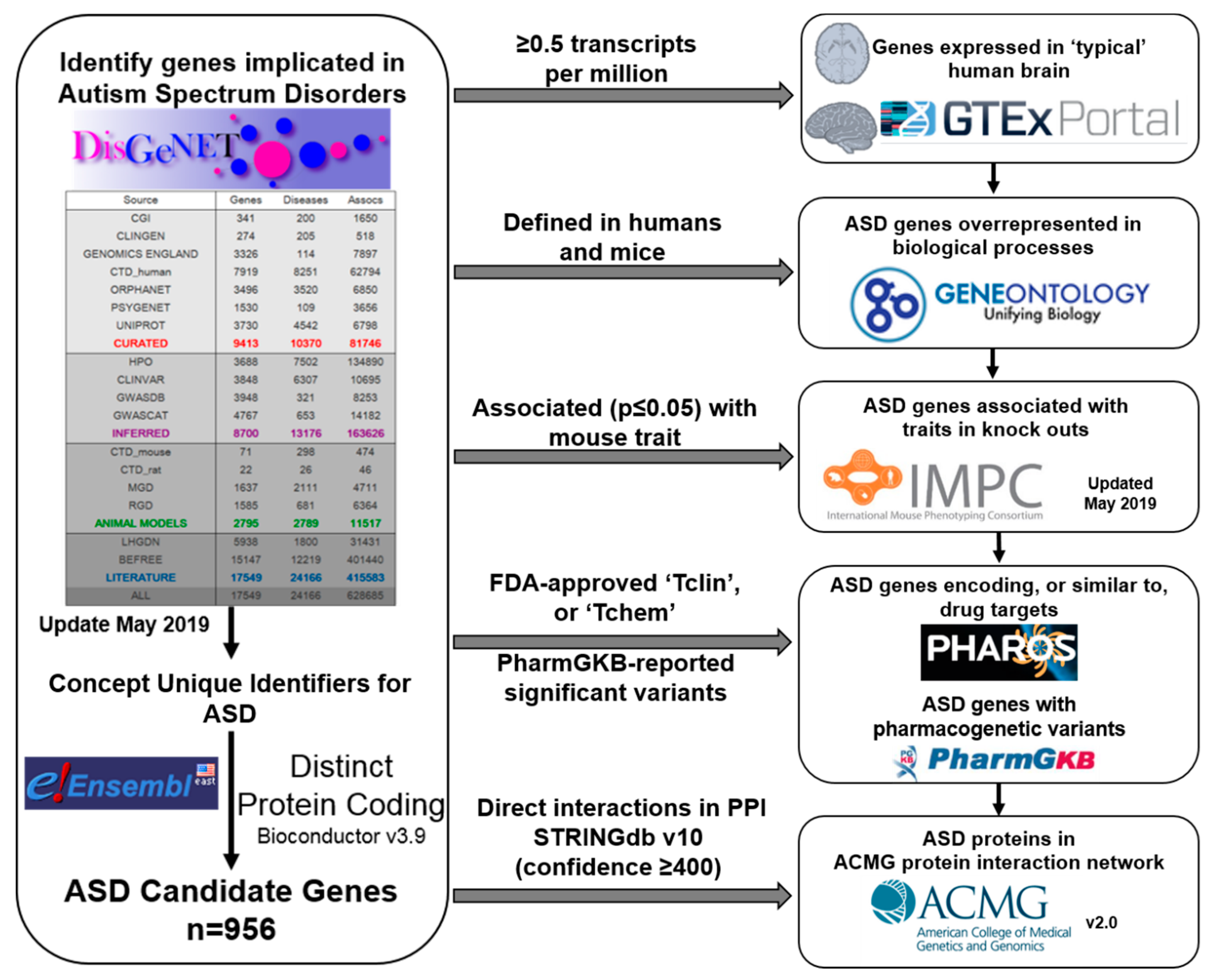
:1. Introduction
2. Results
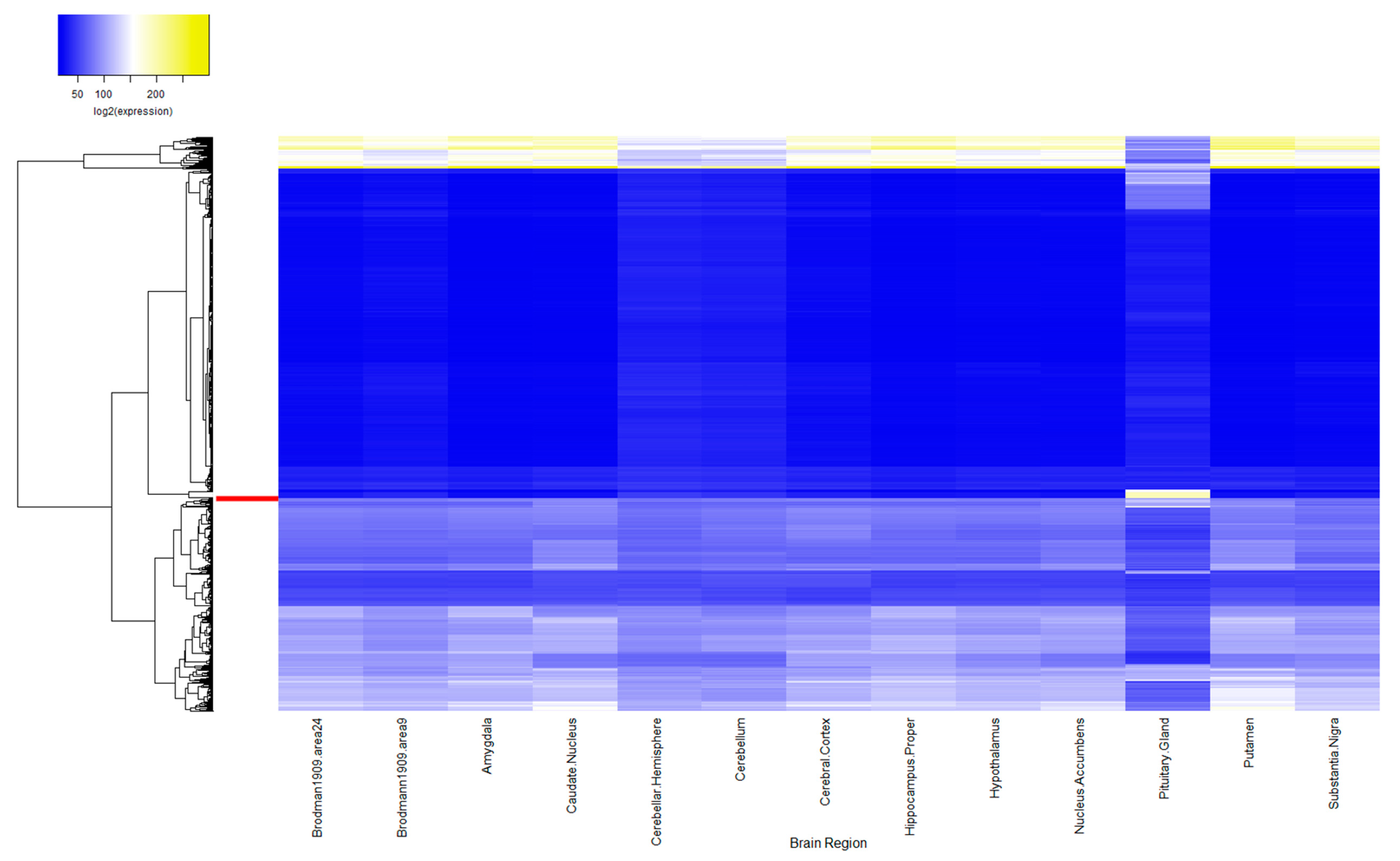
2.1. ASD Candidate Gene Expression in Human Brain
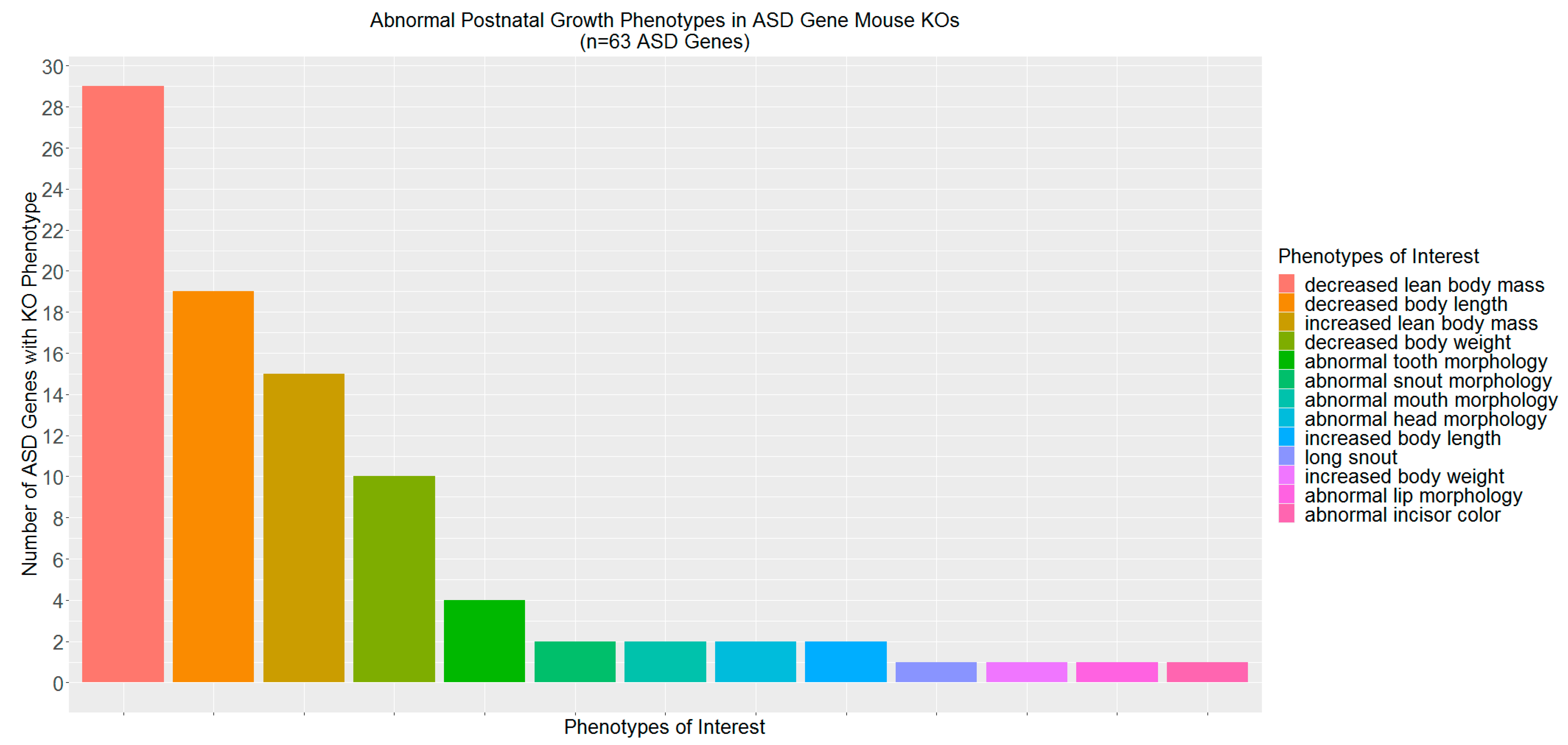
2.2. ASD Candidate Genes Associated with Mammalian Phenotypes
2.3. ASD Candidate Genes Influencing Drug Response
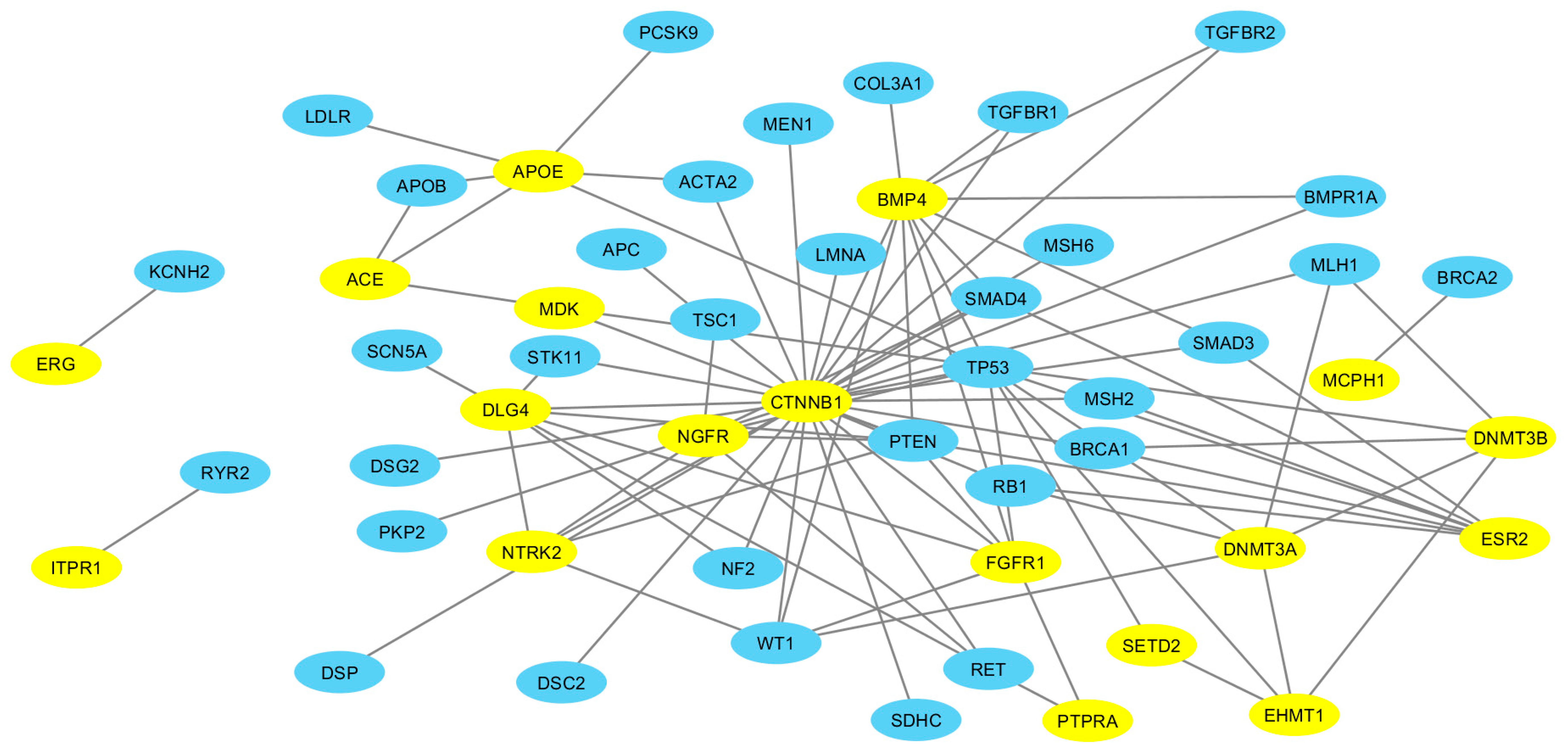
2.4. ASD-ACMG Protein Interactions
2.5. Evaluation of Pathogenic Variants in Prioritized ASD Candidate Genes
3. Discussion
3.1. ASD-Related Gene Expression in the Pituitary Is Increased Compared to Random Sets
3.2. ASD-Related Genes Associated with Abnormal Postnatal Growth in Mouse Knockouts
3.3. More ASD Genes Are Implicated in Drug Response
3.4. More ASD-Related Proteins Interact with ACMG Gene Encoded Proteins
3.5. Prioritized ASD Candidate Genes More Likely to Have Pathogenic Variants
3.6. Limitations and Future Directions
4. Materials and Methods
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ACMG | American College of Medical Genetics |
| API | Application program interface |
| ASD | Autism spectrum disorder |
| CUI | Concept Unique Identifiers |
| FDA | Food and Drug Administration |
| GO | Gene ontology |
| GTEx | Genotype tissue expression |
| GWAS | Genome-wide association study |
| IMPC | International Mouse Phenotyping Consortium |
| KOMP | Knockout Mouse Phenotyping project |
| MGI | Mouse Genome Informatics |
| MP | Mammalian phenotype |
| PharmGKB | Pharmacogenomics Knowledge Base |
References
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders, 5th ed.; American Psychiatric Association: Arlington, VA, USA, 2013. [Google Scholar]
- Hyman, S.L.; Levy, S.E.; Myers, S.M.; Council on children with disabilities, section on developmental and behavioral pediatrics. Identification, Evaluation, and Management of Children with Autism Spectrum Disorder. Pediatrics 2020, 145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bishop-Fitzpatrick, L.; Movaghar, A.; Greenberg, J.S.; Page, D.; DaWalt, L.S.; Brilliant, M.H.; Mailick, M.R. Using machine learning to identify patterns of lifetime health problems in decedents with autism spectrum disorder. Autism Res. Off. J. Int. Soc. Autism Res. 2018, 11, 1120–1128. [Google Scholar] [CrossRef] [PubMed]
- Robert, C.; Pasquier, L.; Cohen, D.; Fradin, M.; Canitano, R.; Damaj, L.; Odent, S.; Tordjman, S. Role of Genetics in the Etiology of Autistic Spectrum Disorder: Towards a Hierarchical Diagnostic Strategy. Int. J. Mol. Sci. 2017, 18, 618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hodges, H.; Fealko, C.; Soares, N. Autism spectrum disorder: Definition, epidemiology, causes, and clinical evaluation. Transl. Pediatr. 2020, 9, S55–S65. [Google Scholar] [CrossRef] [PubMed]
- Elsabbagh, M.; Divan, G.; Koh, Y.-J.; Kim, Y.S.; Kauchali, S.; Marcín, C.; Montiel-Nava, C.; Patel, V.; Paula, C.S.; Wang, C.; et al. Global prevalence of autism and other pervasive developmental disorders. Autism Res. Off. J. Int. Soc. Autism Res. 2012, 5, 160–179. [Google Scholar] [CrossRef] [Green Version]
- Scott, L.J.; Dhillon, S. Spotlight on risperidone in irritability associated with autistic disorder in children and adolescents. CNS Drugs 2008, 22, 259–262. [Google Scholar] [CrossRef]
- Kolevzon, A.; Mathewson, K.A.; Hollander, E. Selective serotonin reuptake inhibitors in autism: A review of efficacy and tolerability. J. Clin. Psychiatry 2006, 67, 407–414. [Google Scholar] [CrossRef]
- McPheeters, M.L.; Warren, Z.; Sathe, N.; Bruzek, J.L.; Krishnaswami, S.; Jerome, R.N.; Veenstra-Vanderweele, J. A systematic review of medical treatments for children with autism spectrum disorders. Pediatrics 2011, 127, e1312–e1321. [Google Scholar] [CrossRef] [Green Version]
- Carter, T.C.; He, M.M. Challenges of Identifying Clinically Actionable Genetic Variants for Precision Medicine. J. Healthc. Eng. 2016, 2016. [Google Scholar] [CrossRef] [Green Version]
- Cooper, D.N.; Stenson, P.D.; Chuzhanova, N.A. The Human Gene Mutation Database (HGMD) and Its Exploitation in the Study of Mutational Mechanisms. Curr. Protoc. Bioinform. 2005, 12, 1–13. [Google Scholar] [CrossRef]
- Rouillard, A.D.; Gundersen, G.W.; Fernandez, N.F.; Wang, Z.; Monteiro, C.D.; McDermott, M.G.; Ma’ayan, A. The harmonizome: A collection of processed datasets gathered to serve and mine knowledge about genes and proteins. Database J. Biol. Databases Curation 2016, 2016. [Google Scholar] [CrossRef] [PubMed]
- GTEx Consortium. The Genotype-Tissue Expression (GTEx) project. Nat. Genet. 2013, 45, 580–585. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, D.-T.; Mathias, S.; Bologa, C.; Brunak, S.; Fernandez, N.; Gaulton, A.; Hersey, A.; Holmes, J.; Jensen, L.J.; Karlsson, A.; et al. Pharos: Collating protein information to shed light on the druggable genome. Nucleic Acids Res. 2017, 45, D995–D1002. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eppig, J.T.; Smith, C.L.; Blake, J.A.; Ringwald, M.; Kadin, J.A.; Richardson, J.E.; Bult, C.J. Mouse Genome Informatics (MGI): Resources for Mining Mouse Genetic, Genomic, and Biological Data in Support of Primary and Translational Research. In Systems Genetics; Schughart, K., Williams, R.W., Eds.; Methods in Molecular Biology; Springer: New York, NY, USA, 2017; Volume 1488, pp. 47–73. ISBN 978-1-4939-6425-3. [Google Scholar]
- Gene Ontology Consortium. Gene Ontology Consortium: Going forward. Nucleic Acids Res. 2015, 43, D1049–D1056. [Google Scholar] [CrossRef]
- Piñero, J.; Bravo, À.; Queralt-Rosinach, N.; Gutiérrez-Sacristán, A.; Deu-Pons, J.; Centeno, E.; García-García, J.; Sanz, F.; Furlong, L.I. DisGeNET: A comprehensive platform integrating information on human disease-associated genes and variants. Nucleic Acids Res. 2017, 45, D833–D839. [Google Scholar] [CrossRef] [PubMed]
- Landrum, M.J.; Lee, J.M.; Riley, G.R.; Jang, W.; Rubinstein, W.S.; Church, D.M.; Maglott, D.R. ClinVar: Public archive of relationships among sequence variation and human phenotype. Nucleic Acids Res. 2014, 42, D980–D985. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mooney, S.D.; Krishnan, V.G.; Evani, U.S. Bioinformatic tools for identifying disease gene and SNP candidates. Methods Mol. Biol. Clifton NJ 2010, 628, 307–319. [Google Scholar] [CrossRef] [Green Version]
- Ferreira, M.A.R.; Jansen, R.; Willemsen, G.; Penninx, B.; Bain, L.M.; Vicente, C.T.; Revez, J.A.; Matheson, M.C.; Hui, J.; Tung, J.Y.; et al. Gene-based analysis of regulatory variants identifies 4 putative novel asthma risk genes related to nucleotide synthesis and signaling. J. Allergy Clin. Immunol. 2017, 139, 1148–1157. [Google Scholar] [CrossRef] [PubMed]
- Pers, T.H.; Timshel, P.; Ripke, S.; Lent, S.; Sullivan, P.F.; O’Donovan, M.C.; Franke, L.; Hirschhorn, J.N.; Schizophrenia Working Group of the Psychiatric Genomics Consortium. Comprehensive analysis of schizophrenia-associated loci highlights ion channel pathways and biologically plausible candidate causal genes. Hum. Mol. Genet. 2016, 25, 1247–1254. [Google Scholar] [CrossRef]
- Petryszak, R.; Keays, M.; Tang, Y.A.; Fonseca, N.A.; Barrera, E.; Burdett, T.; Füllgrabe, A.; Fuentes, A.M.-P.; Jupp, S.; Koskinen, S.; et al. Expression Atlas update—An integrated database of gene and protein expression in humans, animals and plants. Nucleic Acids Res. 2016, 44, D746–D752. [Google Scholar] [CrossRef]
- Hardison, R.C. A guide to translation of research results from model organisms to human. Genome Biol. 2016, 17, 161. [Google Scholar] [CrossRef] [Green Version]
- Ecker, C.; Bookheimer, S.Y.; Murphy, D.G.M. Neuroimaging in autism spectrum disorder: Brain structure and function across the lifespan. Lancet Neurol. 2015, 14, 1121–1134. [Google Scholar] [CrossRef] [Green Version]
- Sanders, S.J.; He, X.; Willsey, A.J.; Ercan-Sencicek, A.G.; Samocha, K.E.; Cicek, A.E.; Murtha, M.T.; Bal, V.H.; Bishop, S.L.; Dong, S.; et al. Insights into Autism Spectrum Disorder Genomic Architecture and Biology from 71 Risk Loci. Neuron 2015, 87, 1215–1233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Voineagu, I.; Wang, X.; Johnston, P.; Lowe, J.K.; Tian, Y.; Horvath, S.; Mill, J.; Cantor, R.M.; Blencowe, B.J.; Geschwind, D.H. Transcriptomic analysis of autistic brain reveals convergent molecular pathology. Nature 2011, 474, 380–384. [Google Scholar] [CrossRef] [PubMed]
- Johnson, M.R.; Shkura, K.; Langley, S.R.; Delahaye-Duriez, A.; Srivastava, P.; Hill, W.D.; Rackham, O.J.L.; Davies, G.; Harris, S.E.; Moreno-Moral, A.; et al. Systems genetics identifies a convergent gene network for cognition and neurodevelopmental disease. Nat. Neurosci. 2016, 19, 223–232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mi, H.; Muruganujan, A.; Casagrande, J.T.; Thomas, P.D. Large-scale gene function analysis with the PANTHER classification system. Nat. Protoc. 2013, 8, 1551–1566. [Google Scholar] [CrossRef] [PubMed]
- Rutherford, K.D.; Mazandu, G.K.; Mulder, N.J. A systems-level analysis of drug-target-disease associations for drug repositioning. Brief. Funct. Genom. 2018, 17, 34–41. [Google Scholar] [CrossRef]
- Mohs, R.C.; Greig, N.H. Drug discovery and development: Role of basic biological research. Alzheimers Dement. 2017, 3, 651–657. [Google Scholar] [CrossRef]
- Shulman, J.M.; Shulman, L.M.; Weiner, W.J.; Feany, M.B. From fruit fly to bedside: Translating lessons from Drosophila models of neurodegenerative disease. Curr. Opin. Neurol. 2003, 16, 443–449. [Google Scholar] [CrossRef]
- Wang, J.; Al-Ouran, R.; Hu, Y.; Kim, S.-Y.; Wan, Y.-W.; Wangler, M.F.; Yamamoto, S.; Chao, H.-T.; Comjean, A.; Mohr, S.E.; et al. MARRVEL: Integration of Human and Model Organism Genetic Resources to Facilitate Functional Annotation of the Human Genome. Am. J. Hum. Genet. 2017, 100, 843–853. [Google Scholar] [CrossRef] [Green Version]
- Ji, X.; Kember, R.L.; Brown, C.D.; Bućan, M. Increased burden of deleterious variants in essential genes in autism spectrum disorder. Proc. Natl. Acad. Sci. USA 2016, 113, 15054–15059. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sacca, R.; Engle, S.J.; Qin, W.; Stock, J.L.; McNeish, J.D. Genetically Engineered Mouse Models in Drug Discovery Research. In Mouse Models for Drug Discovery; Proetzel, G., Wiles, M.V., Eds.; Methods in Molecular Biology; Humana Press: Totowa, NJ, USA, 2010; Volume 602, pp. 37–54. ISBN 978-1-60761-057-1. [Google Scholar]
- Koscielny, G.; Yaikhom, G.; Iyer, V.; Meehan, T.F.; Morgan, H.; Atienza-Herrero, J.; Blake, A.; Chen, C.-K.; Easty, R.; Di Fenza, A.; et al. The International Mouse Phenotyping Consortium Web Portal, a unified point of access for knockout mice and related phenotyping data. Nucleic Acids Res. 2014, 42, D802–D809. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Collins, F.S.; Varmus, H. A new initiative on precision medicine. N. Engl. J. Med. 2015, 372, 793–795. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Evans, W.E.; Relling, M.V. Pharmacogenomics: Translating functional genomics into rational therapeutics. Science 1999, 286, 487–491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Butler, M.G. Pharmacogenetics and Psychiatric Care: A Review and Commentary. J. Ment. Health Clin. Psychol. 2018, 2, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Hopkins, A.L.; Groom, C.R. The druggable genome. Nat. Rev. Drug Discov. 2002, 1, 727–730. [Google Scholar] [CrossRef]
- Yang, H.; Qin, C.; Li, Y.H.; Tao, L.; Zhou, J.; Yu, C.Y.; Xu, F.; Chen, Z.; Zhu, F.; Chen, Y.Z. Therapeutic target database update 2016: Enriched resource for bench to clinical drug target and targeted pathway information. Nucleic Acids Res. 2016, 44, D1069–D1074. [Google Scholar] [CrossRef]
- Thorn, C.F.; Klein, T.E.; Altman, R.B. Pharmacogenomics and bioinformatics: PharmGKB. Pharmacogenomics 2010, 11, 501–505. [Google Scholar] [CrossRef] [Green Version]
- Kalia, S.S.; Adelman, K.; Bale, S.J.; Chung, W.K.; Eng, C.; Evans, J.P.; Herman, G.E.; Hufnagel, S.B.; Klein, T.E.; Korf, B.R.; et al. Recommendations for reporting of secondary findings in clinical exome and genome sequencing, 2016 update (ACMG SF v2.0): A policy statement of the American College of Medical Genetics and Genomics. Genet. Med. Off. J. Am. Coll. Med. Genet. 2017, 19, 249–255. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.Y.; Youn, E.; Mooney, S.D. Connecting protein interaction data, mutations, and disease using bioinformatics. Methods Mol. Biol. Clifton NJ 2009, 541, 449–461. [Google Scholar] [CrossRef] [Green Version]
- Raghavan, S.; Vassy, J.L. Do physicians think genomic medicine will be useful for patient care? Pers. Med. 2014, 11, 424–433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCabe, M.J.; Dattani, M.T. Genetic aspects of hypothalamic and pituitary gland development. Handb. Clin. Neurol. 2014, 124, 3–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iovino, M.; Messana, T.; De Pergola, G.; Iovino, E.; Dicuonzo, F.; Guastamacchia, E.; Giagulli, V.A.; Triggiani, V. The Role of Neurohypophyseal Hormones Vasopressin and Oxytocin in Neuropsychiatric Disorders. Endocr. Metab. Immune Disord. Drug Targets 2018, 18, 341–347. [Google Scholar] [CrossRef] [PubMed]
- Harony, H.; Wagner, S. The contribution of oxytocin and vasopressin to mammalian social behavior: Potential role in autism spectrum disorder. Neurosignals 2010, 18, 82–97. [Google Scholar] [CrossRef]
- Weston, C.S.E. Four Social Brain Regions, Their Dysfunctions, and Sequelae, Extensively Explain Autism Spectrum Disorder Symptomatology. Brain Sci. 2019, 9, 130. [Google Scholar] [CrossRef] [Green Version]
- Raznahan, A. Sizing up the search for autism spectrum disorder (ASD) risk markers during prenatal and early postnatal life. J. Am. Acad. Child Adolesc. Psychiatry 2014, 53, 1045–1047. [Google Scholar] [CrossRef] [Green Version]
- Di Cristofano, A.; Pesce, B.; Cordon-Cardo, C.; Pandolfi, P.P. Pten is essential for embryonic development and tumour suppression. Nat. Genet. 1998, 19, 348–355. [Google Scholar] [CrossRef]
- Meehan, T.F.; Conte, N.; West, D.B.; Jacobsen, J.O.; Mason, J.; Warren, J.; Chen, C.-K.; Tudose, I.; Relac, M.; Matthews, P.; et al. Disease model discovery from 3,328 gene knockouts by The International Mouse Phenotyping Consortium. Nat. Genet. 2017, 49, 1231–1238. [Google Scholar] [CrossRef] [Green Version]
- Brommage, R.; Powell, D.R.; Vogel, P. Predicting human disease mutations and identifying drug targets from mouse gene knockout phenotyping campaigns. Dis. Model. Mech. 2019, 12. [Google Scholar] [CrossRef] [Green Version]
- Pushpakom, S.; Iorio, F.; Eyers, P.A.; Escott, K.J.; Hopper, S.; Wells, A.; Doig, A.; Guilliams, T.; Latimer, J.; McNamee, C.; et al. Drug repurposing: Progress, challenges and recommendations. Nat. Rev. Drug Discov. 2019, 18, 41–58. [Google Scholar] [CrossRef]
- Van Driest, S.L.; Shi, Y.; Bowton, E.A.; Schildcrout, J.S.; Peterson, J.F.; Pulley, J.; Denny, J.C.; Roden, D.M. Clinically actionable genotypes among 10,000 patients with preemptive pharmacogenomic testing. Clin. Pharmacol. Ther. 2014, 95, 423–431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Green, R.C.; Berg, J.S.; Grody, W.W.; Kalia, S.S.; Korf, B.R.; Martin, C.L.; McGuire, A.L.; Nussbaum, R.L.; O’Daniel, J.M.; Ormond, K.E.; et al. ACMG recommendations for reporting of incidental findings in clinical exome and genome sequencing. Genet. Med. Off. J. Am. Coll. Med. Genet. 2013, 15, 565–574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gabrielli, A.P.; Manzardo, A.M.; Butler, M.G. GeneAnalytics Pathways and Profiling of Shared Autism and Cancer Genes. Int. J. Mol. Sci. 2019, 20, 1166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- GTEx Consortium. The GTEx Consortium atlas of genetic regulatory effects across human tissues. Science 2020, 369, 1318–1330. [Google Scholar] [CrossRef]
- Aken, B.L.; Ayling, S.; Barrell, D.; Clarke, L.; Curwen, V.; Fairley, S.; Fernandez Banet, J.; Billis, K.; García Girón, C.; Hourlier, T.; et al. The Ensembl gene annotation system. Database J. Biol. Databases Curation 2016, 2016. [Google Scholar] [CrossRef]
- Grossmann, S.; Bauer, S.; Robinson, P.N.; Vingron, M. Improved detection of overrepresentation of Gene-Ontology annotations with parent child analysis. Bioinformatics 2007, 23, 3024–3031. [Google Scholar] [CrossRef]
- Alexa, A.; Rahnenführer, J.; Lengauer, T. Improved scoring of functional groups from gene expression data by decorrelating GO graph structure. Bioinformatics 2006, 22, 1600–1607. [Google Scholar] [CrossRef] [Green Version]
- Hu, Y.; Flockhart, I.; Vinayagam, A.; Bergwitz, C.; Berger, B.; Perrimon, N.; Mohr, S.E. An integrative approach to ortholog prediction for disease-focused and other functional studies. BMC Bioinform. 2011, 12, 357. [Google Scholar] [CrossRef] [Green Version]
- Perez-Riverol, Y.; Ternent, T.; Koch, M.; Barsnes, H.; Vrousgou, O.; Jupp, S.; Vizcaíno, J.A. OLS Client and OLS Dialog: Open Source Tools to Annotate Public Omics Datasets. Proteomics 2017, 17. [Google Scholar] [CrossRef] [Green Version]
- Dickinson, M.E.; Flenniken, A.M.; Ji, X.; Teboul, L.; Wong, M.D.; White, J.K.; Meehan, T.F.; Weninger, W.J.; Westerberg, H.; Adissu, H.; et al. High-throughput discovery of novel developmental phenotypes. Nature 2016, 537, 508–514. [Google Scholar] [CrossRef]
- Ooms, J. The jsonlite Package: A Practical and Consistent Mapping Between JSON Data and R Objects. arXiv 2014, arXiv:1403.2805. [Google Scholar]
- Szklarczyk, D.; Franceschini, A.; Wyder, S.; Forslund, K.; Heller, D.; Huerta-Cepas, J.; Simonovic, M.; Roth, A.; Santos, A.; Tsafou, K.P.; et al. STRING v10: Protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 2015, 43, D447–D452. [Google Scholar] [CrossRef] [PubMed]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Smedley, D.; Haider, S.; Ballester, B.; Holland, R.; London, D.; Thorisson, G.; Kasprzyk, A. BioMart—Biological queries made easy. BMC Genom. 2009, 10, 22. [Google Scholar] [CrossRef] [Green Version]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Attribute | ASD (n) | Random (mean ± sd) | Χ2 (95%CI) | p-Value | p-Valuecorrected |
|---|---|---|---|---|---|
| Brain Expressed | 861 | 808.94 ± 11.01 | 21.36 (0.88, 0.92) | 3.80 × 10−6 | 5.07 × 10−6 |
| Associated Mouse Trait | 88 | 68.89 ± 7.83 | 5.42 (0.07, 0.11) | 1.99 × 10−2 | 1.99 × 10−2 |
| Encodes Tclin | 113 | 31.19 ± 5.45 | 219.18 (0.10, 0.14) | 1.37 × 10−49 | 6.98 × 10−49 |
| Encodes Tchem | 148 | 80.76 ± 8.43 | 60.25 (0.13, 0.18) | 8.35 × 10−15 | 1.34 × 10−14 |
| Encodes Tbio | 615 | 574.56 ± 14.99 | 6.96 (0.61, 0.67) | 8.34 × 10−3 | 9.53 × 10−3 |
| Encodes Tdark | 51 | 253.28 ± 13.02 | 218.69 (0.04, 0.07) | 1.74 × 10−49 | 6.98 × 10−49 |
| Pharmacogenomic | 124 | 52.15 ± 50.23 | 103.25 (0.11, 0.15) | 2.96 × 10−24 | 5.92 × 10−24 |
| ACMG Network | 475 | 313.53 ± 13.25 | 122.98 (0.46, 0.53) | 1.41 × 10−28 | 3.76 × 10−28 |
| All Attributes | 18 | 8.02 ± 2.56 | 11.30 (0.01, 0.03) | 7.75 × 10−4 | - |
| Associated Mouse Trait | ASD (n) | Random (mean ± sd) | Χ2 (95%CI) | p-Value | p-Valuecorrected |
|---|---|---|---|---|---|
| Growth phenotype | 63 | 43.59 ± 6.32 | 8.60 (0.05, 0.08) | 3.37 × 10−3 | 1.01 × 10−2 |
| Nervous system phenotype | 18 | 12.16 ± 3.36 | 2.37 (0.01, 0.03) | 1.23 × 10−1 | 1.85 × 10−1 |
| Embryo phenotype | 17 | 18.46 ± 4.18 | 0.05 (0.01, 0.03) | 8.21 × 10−1 | 8.21 × 10−1 |
| ASD Gene | Brain Region (TPM) | Associated Mouse Trait (s) | Mapped GO BP | Drug Development | PharmGKB |
|---|---|---|---|---|---|
| APOE | Substantia Nigra (1141) | postnatal growth | growth | NA | PA55 |
| DLG4 | Cerebellar Hemisphere (232) | postnatal growth | growth | Tchem | NA |
| CTNNB1 | Cerebellar Hemisphere (188) | postnatal growth | growth | Tchem | PA27013 |
| FGFR1 | Cerebellum (92) | embryo development | organism development | Tclin | NA |
| NTRK2 | Brodman1909 area24 (91) | postnatal growth | growth | Tchem | PA31818 |
| nervous system morphology | CNS development | ||||
| ITPR1 | Cerebellum (73) | embryo development | organism development | Tchem | NA |
| SETD2 | Cerebellar Hemisphere (41) | postnatal growth | growth | Tchem | NA |
| nervous system morphology | CNS development | ||||
| DNMT3A | Cerebellum (21) | postnatal growth | growth | Tclin | PA27445 |
| embryo development | organism development | ||||
| EHMT1 | Cerebellum (14) | postnatal growth | growth | Tchem | NA |
| BMP4 | Cerebellar Hemisphere (8) | nervous system morphology | CNS development | Tchem | NA |
| ACE | Pituitary (7) | embryo development | organism development | Tclin | PA139 |
| DNMT3B | Cerebellar Hemisphere (6) | embryo development | organism development | Tchem | NA |
| MCPH1 | Cerebellar Hemisphere (6) | postnatal growth | growth | NA | PA30701 |
| ESR2 | Pituitary (0.8) | postnatal growth | growth | Tclin | PA27886 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Veatch, O.J.; Butler, M.G.; Elsea, S.H.; Malow, B.A.; Sutcliffe, J.S.; Moore, J.H. An Automated Functional Annotation Pipeline That Rapidly Prioritizes Clinically Relevant Genes for Autism Spectrum Disorder. Int. J. Mol. Sci. 2020, 21, 9029. https://doi.org/10.3390/ijms21239029
Veatch OJ, Butler MG, Elsea SH, Malow BA, Sutcliffe JS, Moore JH. An Automated Functional Annotation Pipeline That Rapidly Prioritizes Clinically Relevant Genes for Autism Spectrum Disorder. International Journal of Molecular Sciences. 2020; 21(23):9029. https://doi.org/10.3390/ijms21239029
Chicago/Turabian StyleVeatch, Olivia J., Merlin G. Butler, Sarah H. Elsea, Beth A. Malow, James S. Sutcliffe, and Jason H. Moore. 2020. "An Automated Functional Annotation Pipeline That Rapidly Prioritizes Clinically Relevant Genes for Autism Spectrum Disorder" International Journal of Molecular Sciences 21, no. 23: 9029. https://doi.org/10.3390/ijms21239029
APA StyleVeatch, O. J., Butler, M. G., Elsea, S. H., Malow, B. A., Sutcliffe, J. S., & Moore, J. H. (2020). An Automated Functional Annotation Pipeline That Rapidly Prioritizes Clinically Relevant Genes for Autism Spectrum Disorder. International Journal of Molecular Sciences, 21(23), 9029. https://doi.org/10.3390/ijms21239029