1. Introduction
Cellular senescence is a critical hallmark of aging. Senescence is a stable proliferation arrest characterized by profound changes in cellular morphology and metabolism as well as by extensive chromatin reorganization in the nucleus. Cellular senescence was first discovered in cultured fibroblast cells, where prolonged passaging and replicative exhaustion led to growth arrest due to critically short telomeres [
1]. Senescence was eventually also observed in cells subjected to a variety of stressors, including acute DNA damage, oxidative stress, or oncogene induction. Senescent cells are characterized by cell-autonomous proliferative arrest with resistance to mitogenic signals, expression of senescence-associated beta-galactosidase (SA-β-Gal), formation of senescence-associated heterochromatic foci (SAHF), and release of inflammatory cytokines and chemokines, known as the senescence-associated secretory phenotype (SASP) [
2].
Normal cells undergo senescence in response to severe or irreparable DNA damage. Among the genome damage induced by ionizing radiation (IR), double-strand breaks (DSBs) represent the most biologically deleterious type of DNA lesion [
3]. To tackle these potentially lethally damaging lesions, cells have evolved orchestrated and conserved mechanisms known collectively as the DNA damage response (DDR) [
4]. The DDR coordinates cellular DSB repair activities immediately after the damage is detected, and multiple DSB repair proteins are recruited to DSB sites within seconds after IR exposure, forming radiation-induced foci detectable by immunofluorescence microscopy (IFM). Of particular importance in this context is the recruitment of 53BP1 to DSB-flanking chromatin [
5], supposed to stabilize the nucleosomal barrier to inhibit DNA end-resection [
6]. Most radiation-induced DSBs are repairable and these transient 53BP1-foci typically resolve within 24 h. However, severe or irreparable DNA damage, such as complex DSBs, cause persistent 53BP1-foci, detectable for days and weeks after their formation, suggesting that DSB repair mechanisms are incapable of resolving these lesions [
7,
8,
9,
10,
11]. The focal accumulation of DNA repair factors around DNA lesions is a characteristic feature of the cellular response to DNA damage.
One striking characteristic of senescent cells is the large-scale reorganization of their chromatin, exemplified by the formation of SAHFs [
12,
13]. SAHFs are microscopically discernible as condensed heterochromatic structures and have been widely used as a senescence marker [
14]. Previous studies indicate that chromatin restructuring at the onset of senescence implies the formation of so-called
‘DNA segments with chromatin alterations reinforcing senescence’ (DNA-SCARS) [
15]. Elevated concentrations of modified histones (e.g., γH2AX) and other proteins (e.g., 53BP1) are recruited to remodeled chromatin of DNA-SCARS. These DNA-SCARS are supposed to trigger constitutive DNA damage signaling, prolonged p53-dependent growth arrest, and eventually an irreversible senescence arrest [
16]. However, the precise relationship between radiation-induced DNA breaks, development of DNA-SCARS, and formation of SAHFs with higher-order chromatin rearrangements is currently unknown.
The fundamental repeating unit of chromatin is the nucleosome. The nucleosome core particle comprises 147 base pairs of DNA wrapped around the core histone octamer, containing two copies each of the canonical histone proteins H2A, H2B, H3, and H4. The linker histone H1 and non-histone proteins participate in the dynamic regulation of chromatin compaction within the nucleus. Post-translational histone modifications can directly modulate chromatin structure by altering the charge of histones, and are correlated with open/closed or active/repressed chromatin states [
17]. Additional diversity is provided by the incorporation of histone variants into chromatin [
18]. Histone variants can profoundly change chromatin properties by modulating nucleosome stability and function, which may affect their interaction with chromatin remodelers and modifiers [
19]. The deposition of canonical histones is coupled to DNA synthesis, whereby canonical histones assemble into nucleosomes behind replication forks and at sites of DNA repair. Incorporation of histone variants, by contrast, occurs throughout the cell cycle and is independent of DNA synthesis. In non-cycling senescent cells, canonical histone production declines (due to reduced synthesis and high turnover) and variant histones tend to accumulate [
20]. Histone modifications on canonical and variant histones may further affect the level of chromatin compaction, and may entail an imbalance of activating and repressive histone marks, thereby regulating gene expression [
19,
20,
21].
In previous studies, it could be shown that the histone variant H2A.J accumulates in human fibroblasts during replicative and oncogene-induced senescence and affects inflammatory gene expression of senescent cells [
22]. Recent work demonstrates the functional importance of H2A.J-specific residues and potential mechanisms for its function in promoting inflammatory gene expression in senescence. H2A.J accumulation contributes to weakening the association of histone H1 to chromatin and increasing its turnover. Decreased H1 in senescence is correlated with increased expression of some repeated DNA sequences, increased expression of STAT/IRF transcription factors, and transcriptional activation of interferon-stimulated genes [
23].
A better understanding of the molecular mechanisms that drive senescence and SASP expression may help to unravel the complicated role of senescence in age-related diseases, including cancer. Depending on the cell type and initiating event, the SASP can be tumor suppressive in normal cells and favorable for tumor treatment response by enforcing arrest and promoting immune clearance of damaged cells. Alternatively, some senescent cells can secrete factors that create an immunosuppressive environment that promotes tumor initiation and progression. The reduction of SASP might provide therapeutic opportunities that could complement cancer therapy to limit tumor progression and metastasis [
24].
Using IR to initiate DNA damage-induced senescence in human fibroblasts, we investigated the spatiotemporal dynamics of transient and persistent 53BP1-foci at DSBs sites, the formation of DNA-SCARS, and their impact on establishing and maintaining senescence-associated phenotypes. Here, we show that H2A.J is incorporated predominantly in radiation-induced DNA-SCARS during senescence progression and triggers decisively the development of the SASP.
3. Discussion
Cellular genomes are constantly exposed to DNA damage, such as IR, threatening not only genome stability but also the integrity of chromatin organization. The DDR coordinates DNA repair activities and chromatin dynamics following radiation-induced damage, arrests cell-cycle progression, and is the central mediator in triggering cellular senescence [
15,
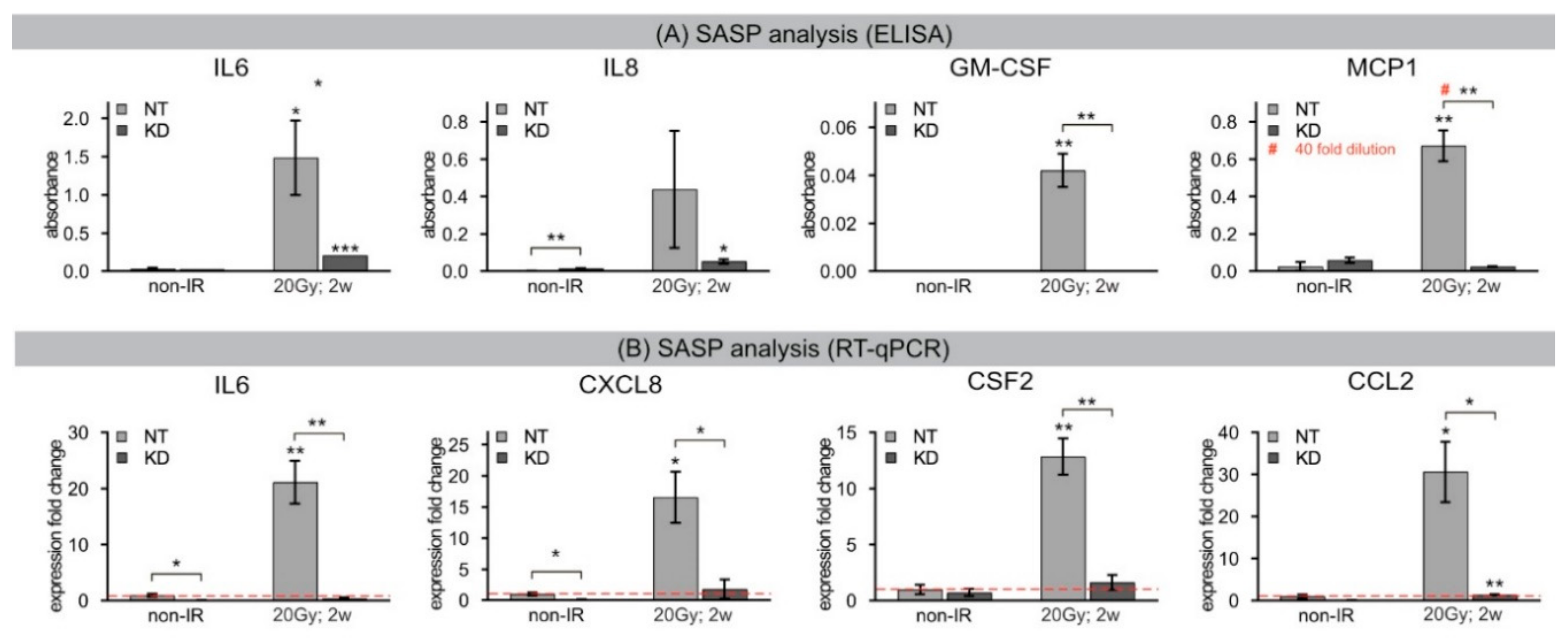
16]. To investigate the molecular mechanisms that eventually provoke continuous DDR leading to radiation-induced senescence, we analyzed chromatin dynamics of the histone variant H2A.J during the repair of radiation-induced DSBs in human fibroblasts. Senescence is associated with profound chromatin reorganization and in particular with the formation of SAHFs, where condensed chromatin is supposed to repress the expression of proliferation-promoting genes. Using high-resolution imaging techniques, we found that persisting 53BP1 foci after DNA-damaging IR transform into DNA-SCARS, which were consistently located in peripheral regions of SAHFs. Quantitative immuno-electron microscopic analysis revealed that H2A.J co-localizes preferentially with 53BP1 at damaged chromatin, and is steadily incorporated into these radiation-induced DNA-SCARS during senescence progression. H2A.J depletion in KD fibroblasts did not affect the stability of cell-cycle arrest nor the coarser re-structuring of senescence-associated chromatin with formation of SAHFs. However, H2A.J depletion appeared to influence the nanostructure of DNA-SCARS and to suppress the expression of SASP, and thereby inhibited the chronic production of inflammatory mediators. Accordingly, the incorporation of H2A.J at damaged chromatin may directly affect the transcriptional program of senescent cells. These findings suggest that H2A.J inhibition could be a reasonable strategy to ablate the SASP without affecting the senescence-associated growth arrest.
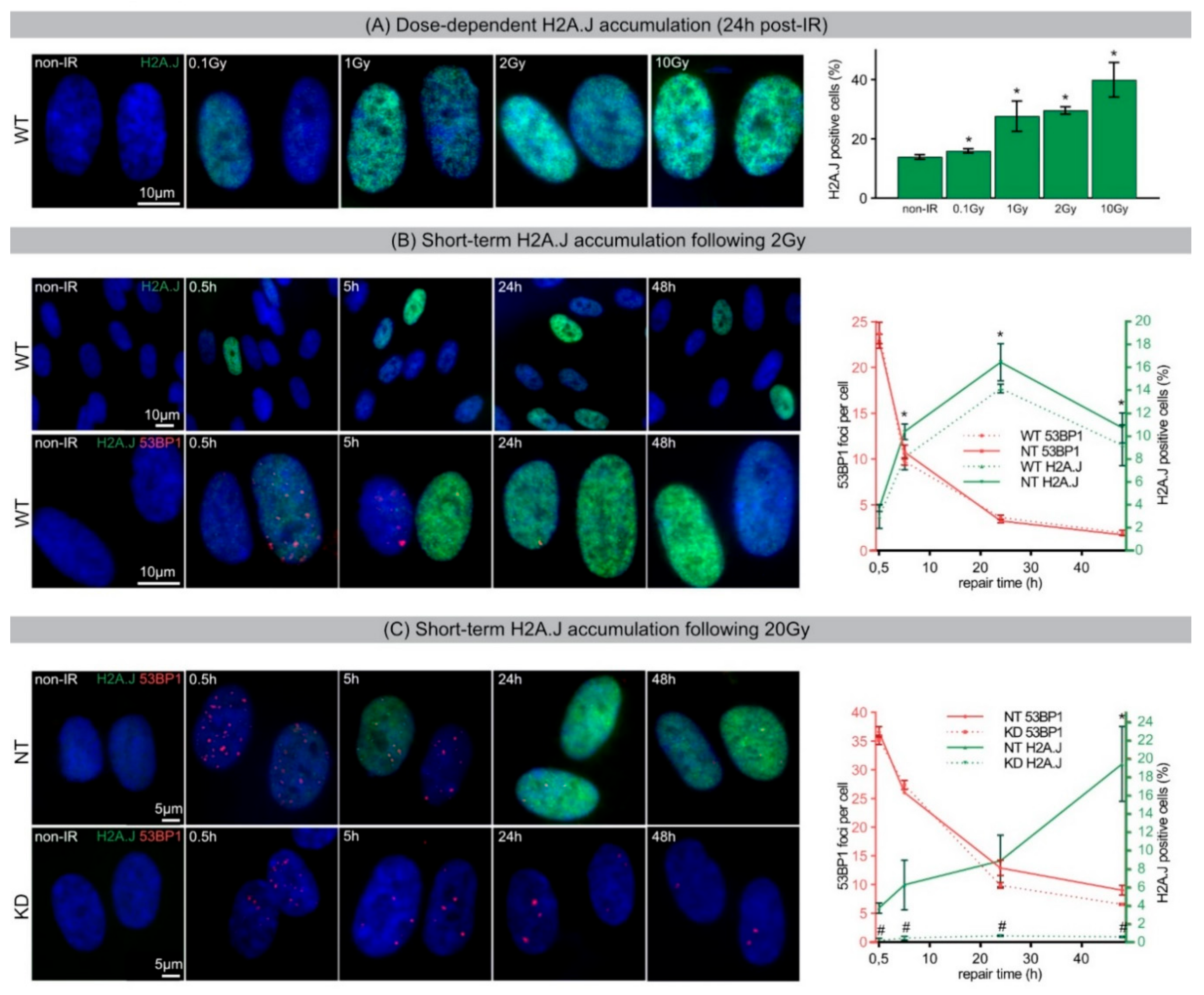
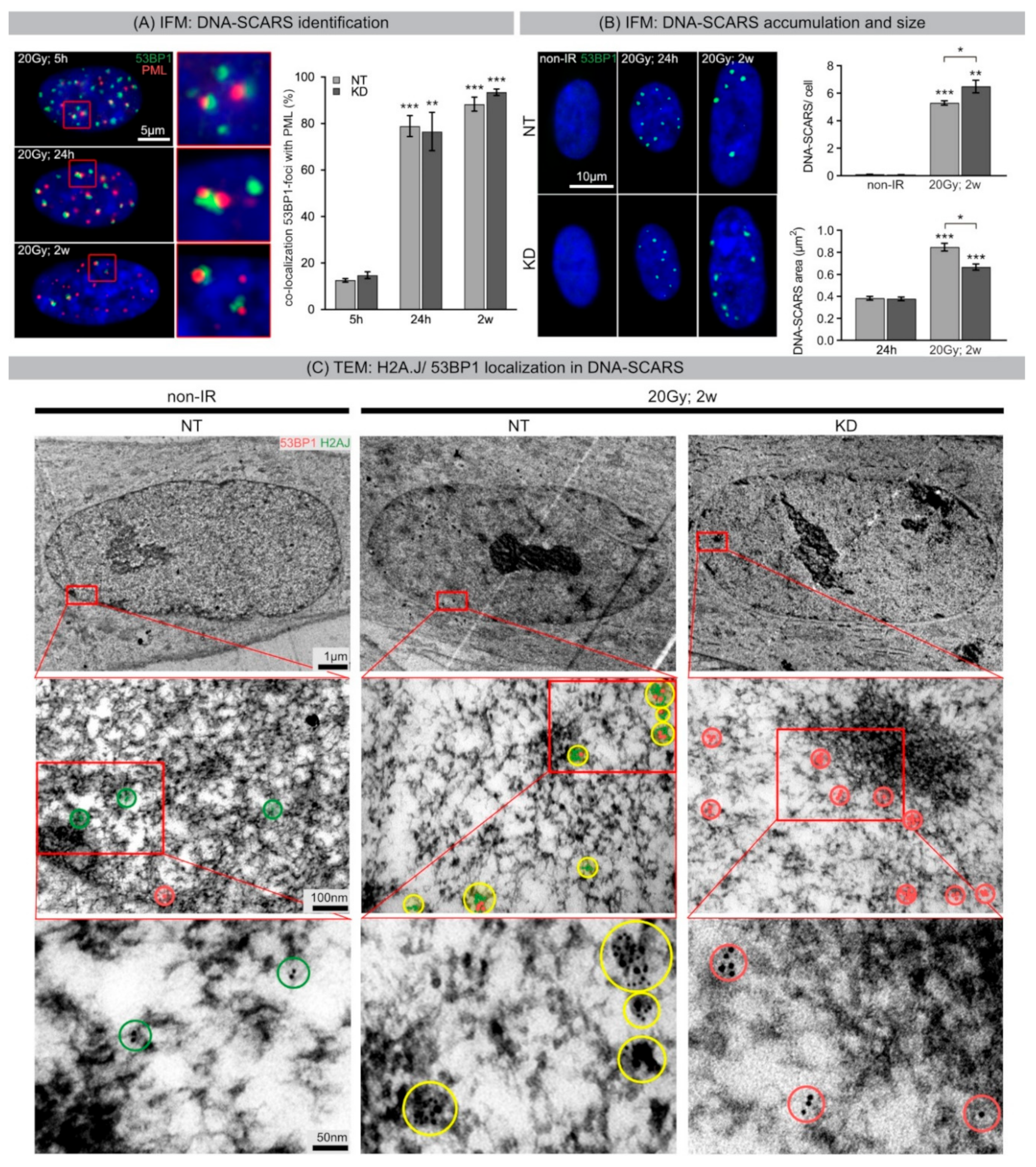
During acute DDR, most radiation-induced 53BP1 foci resolved within 24 h post-IR, but ~10% of 53BP1 foci persist for days and weeks, particularly after high-dose exposure. The focal accumulation of DNA repair factors, including 53BP1 and PML nuclear bodies, is a key cytological signature of DDR [
25,
26]. Although persistent DNA damage foci have been studied by super-resolution localization microcopy [
27,
28,
29], our knowledge of persistent DNA damage triggering SASP is still incomplete. In previous light and electron microscopic studies, we characterized the ultrastructure of radiation-induced foci in the chromatin context within the nuclei of normal human fibroblasts. By time course experiments with immuno-gold labeling of 53BP1, γH2AX, and other key repair factors, such as Ku70-Ku80, we could show that DNA repair foci are highly dynamic, with early and late repair foci exhibiting distinct compositions of repair factors and chromatin arrangements [
8,
30]. During acute DDR, 53BP1 accumulates on chromatin regions in the vicinity of radiation-induced DSBs. These early 53BP1-foci typically contain core components of DSB repair pathways, such as the Ku70-Ku80 heterodimer of non-homologous end-joining (NHEJ) [
8]. However, persistent 53BP1 foci usually lack these key repair factors, and thus are no longer sites of active DNA damage repair. Instead, they may reflect stable chromatin alterations remaining from radiation-induced DNA lesions [
8,
15,
16,
25,
26]. Notably, at the early stage of DDR, our TEM studies revealed no global chromatin restructuring, and particularly no specific chromatin domains with circumscribed H2A.J incorporation. These findings suggest that the characteristic remodeling of the nuclear architecture in senescent cells may rely on progressive chromatin maturation events after completion of DNA repair.
Recent reports emphasize the great importance of chromatin remodeling during DSB repair in response to DNA-damaging IR [
3,
4,
31,
32]. Etoposide, another widely used anticancer agent, inhibits topoisomerase activity, impairs DNA strand rejoining functions, and thereby induces single- and double-strand breaks [
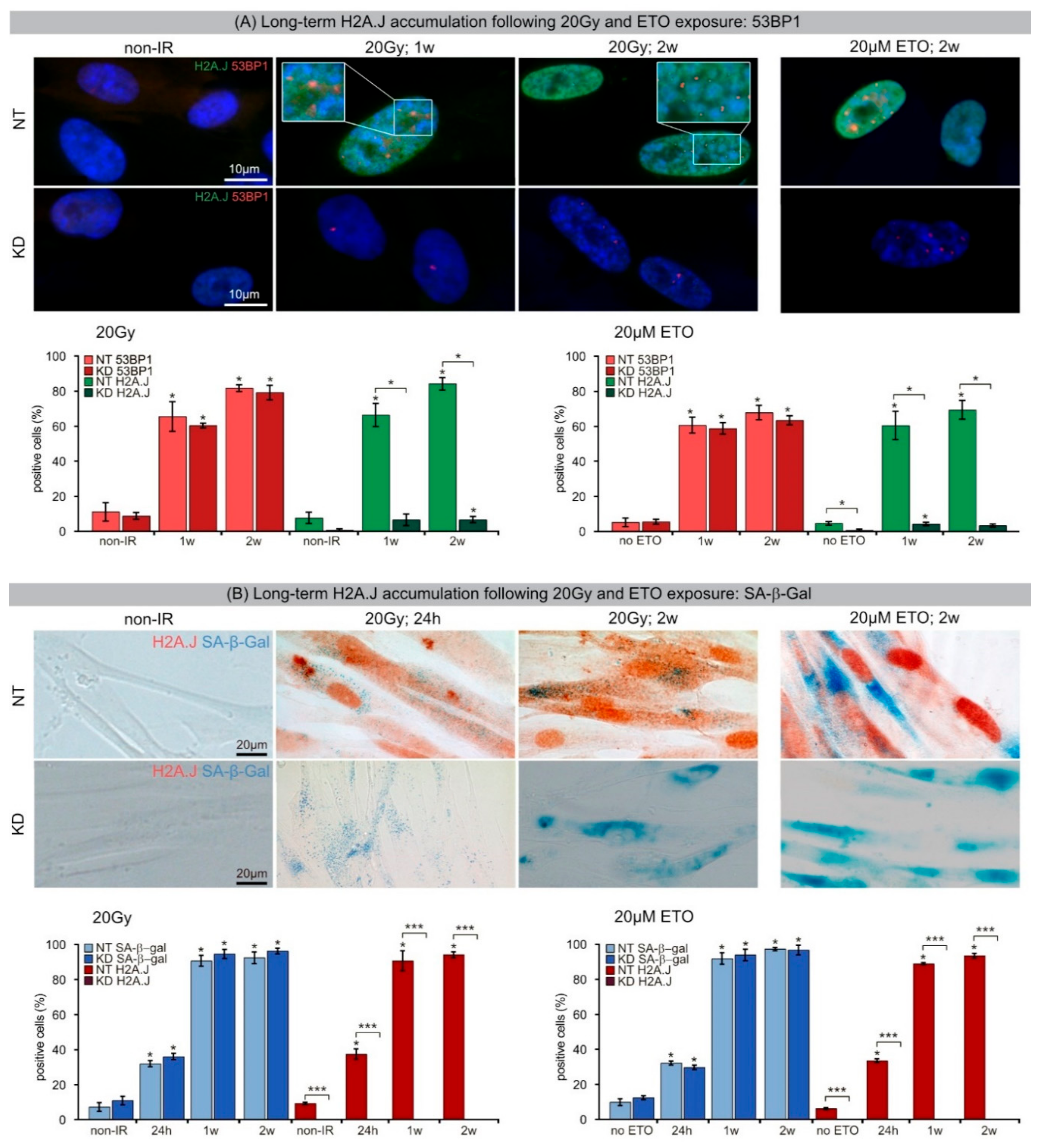
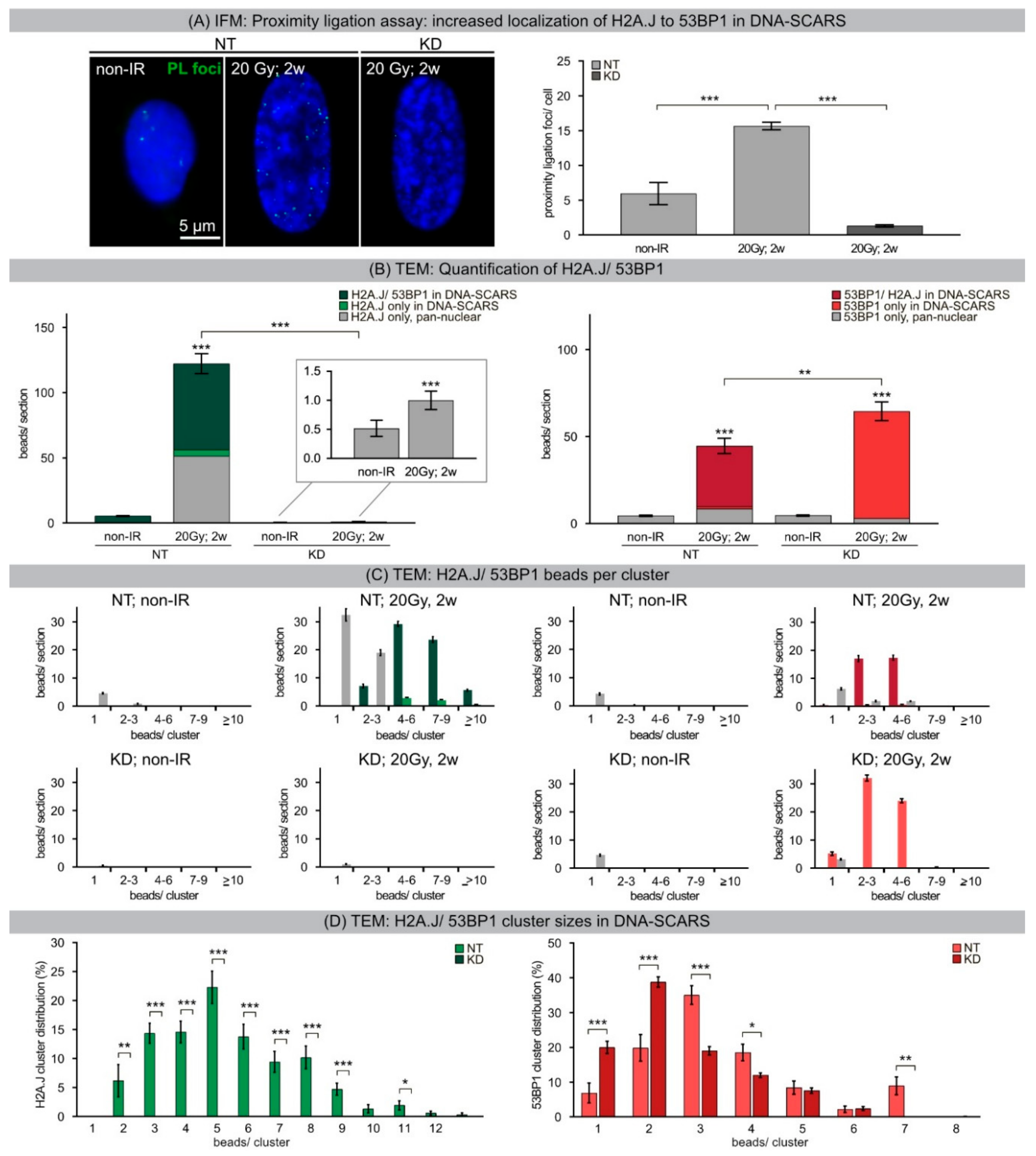
33]. Our results show that chromatin restoration after different DNA-damaging agents similarly involves the deposition of the H2A.J variant, which may not only replace damaged canonical histones but leave marks of damage experience. Following IR exposure with moderate doses (2 Gy), the reversible H2A.J incorporation correlated temporally with the transient growth arrest. Exposure to high doses of IR (≥10 Gy) or to high concentrations of etoposide resulted in progressive irreversible H2A.J incorporation and correlated with the senescence-associated permanent cell-cycle arrest. Significantly, H2A.J depletion had no obvious effects on DNA damage repair, nor on the induction of the senescent state with permanent cell-cycle arrest and increased SA-β-Gal expression. However, we did observe a modification of 53BP1 foci in senescent cells depleted for H2A.J; the number of 53BP1 clusters per nuclear section increased, but the 53BP1 density within these clusters was decreased. H2A.J depletion thus perturbs the normal ultrastructure of DNA-SCARS.
Histone dynamics coupled to DNA damage repair may contribute to the maintenance of epigenome integrity during the response to DNA damage. However, destabilization of nucleosomes during the DDR allows histone exchange and may result in the replacement of canonical histones with variants that carry out specialized functions. Histone variant H2A.J differs from canonical H2A protein by the presence of an SQK motif near the C-terminus and an A11V substitution in the N-terminal tail that do not impart any considerable structural alterations to nucleosomes [
22]. However, the incorporation of H2A.J instead of canonical H2A may imply the disruption of original epigenetic information at damaged chromatin sites. Our findings are in line with previous studies suggesting that high-order heterochromatin formation and epigenetic remodeling of the genome can be discrete events [
34]. Previous ChIP-seq studies could not show any preferential incorporation of H2A.J in the promoter regions of SASP genes [
22]. Our imaging data establish a close connection between persisting 53BP1 foci later transforming into DNA-SCARS, likely reflecting remodeled chromatin at radiation-induced DNA lesions. These DNA-SCARS were consistently localized to the periphery of SAHFs, representing the main characteristic of senescence-related chromatin disorganization. SAHF formation results generally in concentric chromatin architectures, segregating H3K9me3-enriched compact cores from H3K27me3-enriched less densely packed ring structures [
34]. The segregation of distinct epigenetic components into specific chromatin compartments is believed to promote marked changes in the gene expression program, such as downregulation of cell cycle genes and upregulation of SASP genes. High-resolution TEM imaging of senescent fibroblasts shows that H2A.J was incorporated preferentially in damaged chromatin of DNA-SCARS at the periphery of SAHFs. The profound chromatin reorganization during senescence progression is an essential epigenetic mechanism for controlling the different aspects of the senescence program. However, our data indicate that the incorporation of H2A.J in radiation-induced DNA-SCARS is not necessary for the establishment of SAHFs but indispensable to promote the SASP with the production of inflammatory mediators. RNA interference-mediated knock-down of H2A.J suppressed SASP expression but did not affect senescence-related chromatin reorganization nor the stability of senescence-associated cell-cycle arrest.
Further investigations are necessary to capture the dynamic process of senescence progression in response to radiation-induced DNA damage. Remodeling of chromatin during the progression of radiation-induced senescence might be associated with varying production of different SASP factors. However, our current understanding of the interdependence of chromatin structure and nuclear function in senescent cells is extremely limited. In previous studies, Contrepois et al. examined the effect of ectopic H2A.J expression in proliferating and senescent cells in which the endogenous H2AFJ mRNA was depleted. This ectopic H2A.J overexpression did not overtly affect cell proliferation, nor did it inhibit entry into senescence in response to etoposide treatment. Significantly, this ectopic H2A.J overexpression increased the expression of inflammatory genes in both senescent and proliferating cells, independent from their different chromatin structure [
22].
Collectively, our results suggest that H2A.J inhibition could be a reasonable strategy to ablate the SASP, without affecting senescence-associated growth arrest. Using epigenetic mechanisms to target H2A.J incorporation could serve to uncouple the long-term deleterious effects of SASP from the beneficial effects connected to senescence-associated growth arrest [
2]. Targeting proteins controlling the senescent state has many therapeutic implications. Senescent cells accumulate with age and are detrimental for tissue structure and function, and the SASP is likely the most important contributor to the negative effects of senescent cells on tissue homeostasis [
35]. Our data enhances our understanding of the epigenomic regulation of the SASP and sets the stage for the development of new therapies aimed at suppressing the inflammatory component of senescence.
4. Experimental Procedures
4.1. Cell Culture
WI-38 human embryonic fibroblasts (WI-38 wild-type, WT) were obtained from the ATCC. Lentiviral shRNA constructs were used to knock-down H2A.J in immortalized fibroblasts (WI-38hTERT/ptet-on-sh3-H2AFJ knock-down, KD) and non-targeted controls (WI-38hTERT/ptet-on-sh-no-target, NT). NT and KD fibroblasts were provided by C. Mann and prepared as described previously [
22]. KD shRNA targets positions 265–283 of the human H2AFJ coding sequence: 50-CGCAACGACGAGGAGTTAA-3. Fibroblasts were cultured at 5% O
2 and 5% CO
2 in MEM (Invitrogen, Karlsruhe, Germany) with 10% fetal bovine serum, 1 mM sodium pyruvate, 2 mM L-glutamine, 0.1 mM non-essential amino acids, and 1% penicillin/streptomycin. Then, 1 µg/mL doxycycline was added to medium for 1 week prior to experimental begin to activate sh inserts. Cells were then grown on coverslips and used once 90% confluent.
4.2. Radiation Exposure
Cells were exposed to ionizing radiation (IR) using the linear accelerator Artiste™ (Siemens, Munich, Germany) (6-MV photons; dose-rate 2 Gy/min). Cells were analyzed following different doses (2 Gy; 20 Gy) and at different time-points (0.5 h, 5 h 24 h, 48 h, 1 week, and 2 weeks post-IR) and compared to non-irradiated controls.
4.3. Etoposide Exposure
Cells were incubated with 20 µM Etoposide for the duration of experiments (1 or 2 weeks) and compared to non-exposed confluent controls.
4.4. IFM Analysis
Cells were fixed with 4% paraformaldehyde and permeabilized with 0.5% Triton X-100, washed with 0.1% Tween®-20, and incubated overnight with primary antibody (anti-H2A.J supplied by C. Mann; anti-53BP1, Merck, Darmstadt, Germany; anti-H3K27me3, anti-H3K9me3, and anti-p21, Abcam, Berlin, Germany; anti-ki67, Thermo Fisher, Waltham, MA, USA; anti-lamine B1, Proteintech, Manchester, UK; anti-PML, Santa Cruz, CA, USA) followed by Alexa-Fluor®488 or Alexa-Fluor®568 secondary antibody (Invitrogen, Karlsruhe, Germany). Subsequently, cells were mounted in VECTAshield™ mounting medium with 4′, 6-diamidino-2-phenylindole (DAPI, Vector Laboratories, Burlingame, CA, USA). Fluorescence images were captured with a Nikon-Eclipse Ni fluorescence microscope equipped with a Nikon DS-Qi2 camera (Nikon, Düsseldorf, Germany).
For evaluating H2A.J, 53BP1, SAHF, Ki67, and p21 positivity, at least 200 cells were captured for each sample (positive cells in %). For counting 53BP1 foci per cell, at least 50 foci and/or 50 cells were analyzed per sample. For identification of DNA-SCARS, 53BP1/PML co-localization was analyzed in 25 nuclei and numbers of 53BP1/PML co-localizing foci relative to total 53BP1 foci were expressed in percentages. Area of nuclei, area of 53BP1-foci, and lamine B1 fluorescence intensity (FITC signal normalized to DAPI) were measured based on their DAPI/FITC signal in 200 cells using Nikon NIS-Elements Basic Research acquisition software. Region of Interests (ROIs) were defined by appropriate fluorescence thresholds to measure the structure size accurately in an unbiased and transparent manner.
4.5. BrdU Labeling
To test the stability of senescence-associated growth arrest, non-irradiated (non-IR) and senescent cells (20 Gy; 2 weeks post-IR) were pulsed with 10 µmol/L 5-bromo-2′-deoxyuridine (BrdU) in culture medium for 24 h. After medium removal and several PBS washing steps, cells were fixed and permeabilized as described for standard IFM. DNA denaturation was completed through 1-h incubation in 2 M HCl succeeded by PBS washes and 2-h incubation with primary anti-BrdU antibody (Bio-Rad Laboratories, Munich, Germany) in 0.1% Tween, 1% BSA in PBS. After washes of Tween/BSA/PBS, samples were incubated with fluorescence-coupled anti-rat secondary antibody in Tween/BSA/PBS for 2 h. After PBS washes, coverslips were mounted with hard-set mounting medium containing DAPI.
4.6. Proximity Ligation Assay
A Duolink® PLA system (DUO92002, -004, -014, -040, -049, Sigma-Aldrich, St. Louis, MO, USA) was used to detect co-localizations of H2A.J and 53BP1 molecules with spacing distances ≤40 nm, according to supplier protocol. Subsequently, samples were prepared and fixed as for standard IFM and PLA foci for 50 cells were quantified.
4.7. IHC Analysis (SA-β-Gal)
Following 5-min fixation with 2% paraformaldehyde and 0.2% glutaraldehyde, cells were incubated with X-Gal staining solution (AppliChem GmbH, Darmstadt, Germany) at 37 °C overnight. After a 30-s methanol incubation, dried samples were permeabilized with 0.2% Triton X-100 and washed with 1% BSA. Samples were blocked with 4% BSA for 1 h, followed by an overnight incubation with H2A.J primary antibody (anti-H2A.J supplied by C. Mann). Incubation with Dako immunoglobulin/bioatinylated secondary antibody (Agilent, Waldbronn, Germany) was followed by Vectastain ABC Peroxidase standard (Vector, Burlingame, CA, USA) and SIGMAFAST™ 3.3′ Diaminobenzidine (Merck, Darmstadt, Germany) incubations, respectively. Samples were finally mounted in Dako Faramount Mounting Medium (Agilent, Waldbronn, Germany).
4.8. TEM Analysis
Cells were fixed overnight using 2% paraformaldehyde and 0.05% glutaraldehyde in PBS. Samples were dehydrated in increasing ethanol concentrations and permeated overnight with LR White resin (EMS, Hatfield, PA, USA) followed by overnight embedding at 50 °C with fresh LR White resin containing LR White Accelerator (EMS, Hatfield, PA, USA). Microtome Ultracut UCT (Leica, Wetzlar, Germany) and diamond knife (Diatome, Biel, Switzerland) were implemented to acquire ultrathin sections (70 nm) picked up on pioloform-coated nickel grids and prepared for immunogold-labeling. Non-specific staining was blocked using Aurion blocking solution (Aurion, Wageningen, The Netherlands), sections were rinsed and incubated overnight with primary antibodies (anti-H2A.J supplied by C. Mann; anti-53BP1, Merck, Darmstadt, Germany; anti-H3K27me3 and anti-H3K9me3, Abcam, Berlin, Germany) at 4 °C, followed by incubation with 6- or 10-nm gold particle-conjugated secondary antibodies (Aurion, Wageningen, The Netherlands) for 1.5 h. Finally, sections were fixed with 2% glutaraldehyde and contrasted with uranyl acetate. A Tecnai Biotwin™ transmission electron microscope (FEI, Eindhoven, The Netherlands) was employed for visual analysis. For quantification, single beads and bead clusters were counted in 25 nuclear sections, additionally noting co-localizations as well as chromatin localization (hetero-/euchromatin; inside/outside DNA-SCARS).
4.9. ELISA Analysis
Conditioned media was isolated from three independent cultures of NT and KD fibroblasts. Media was analyzed from fibroblasts after inducing senescence by IR with 20 Gy or incubation with 20 μM etoposide for 2 weeks. Cell numbers in culture dishes were determined at the same time that media was collected and frozen at −80 °C until analysis. A Multi-Analyte ELISArray™ Kit (Qiagen, Hilden, Germany) was used to screen SASP factors (IL6, IL8, GM-CSF, MCP1) with a standard ELISA plate reader, according to supplier protocol. Conditions were optimized to ensure readings fell within the linear range of the assay for each cytokine.
4.10. RT-qPCR Analysis
RNA extraction was completed using TRIzol™ (Thermo Fisher Scientific, Darmstadt, Germany) and phenol/chloroform phase separation. A QuantiTect
® Reverse Transcription Kit (Qiagen, Hilden, Germany) was used for cDNA synthesis from RNA and a QuantiTect
® SYBR
® Green kit (Qiagen, Hilden, Germany) for subsequent quantitative PCR in Roche LightCycler II (Roche, Mannheim, Germany). Custom primers (
Table 1) were manufactured by Thermo Fisher Scientific (Darmstadt, Germany). Values were normalized to
GAPDH.
4.11. Statistical Analysis
All data were presented as mean ±SEM, where normally distributed data were analyzed by Student’s T-Test and non-parametric data were analyzed by Mann–Whitney-U-Test to evaluate differences between time-points and cell lines. All statistical analyses were performed by statistical software SPSS (SPSS Statistics25, IBM, Armonk, New York, NY, USA). Statistical significance was presented as * p < 0.05, ** p < 0.01, *** p < 0.001.









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}