Enteric Glia at the Crossroads between Intestinal Immune System and Epithelial Barrier: Implications for Parkinson Disease

 , ,
, ,  ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Methods
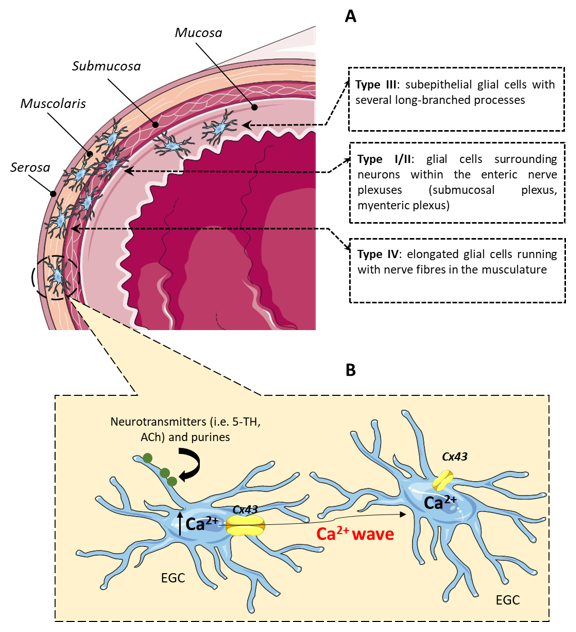
3. The Enteric Glial Cells
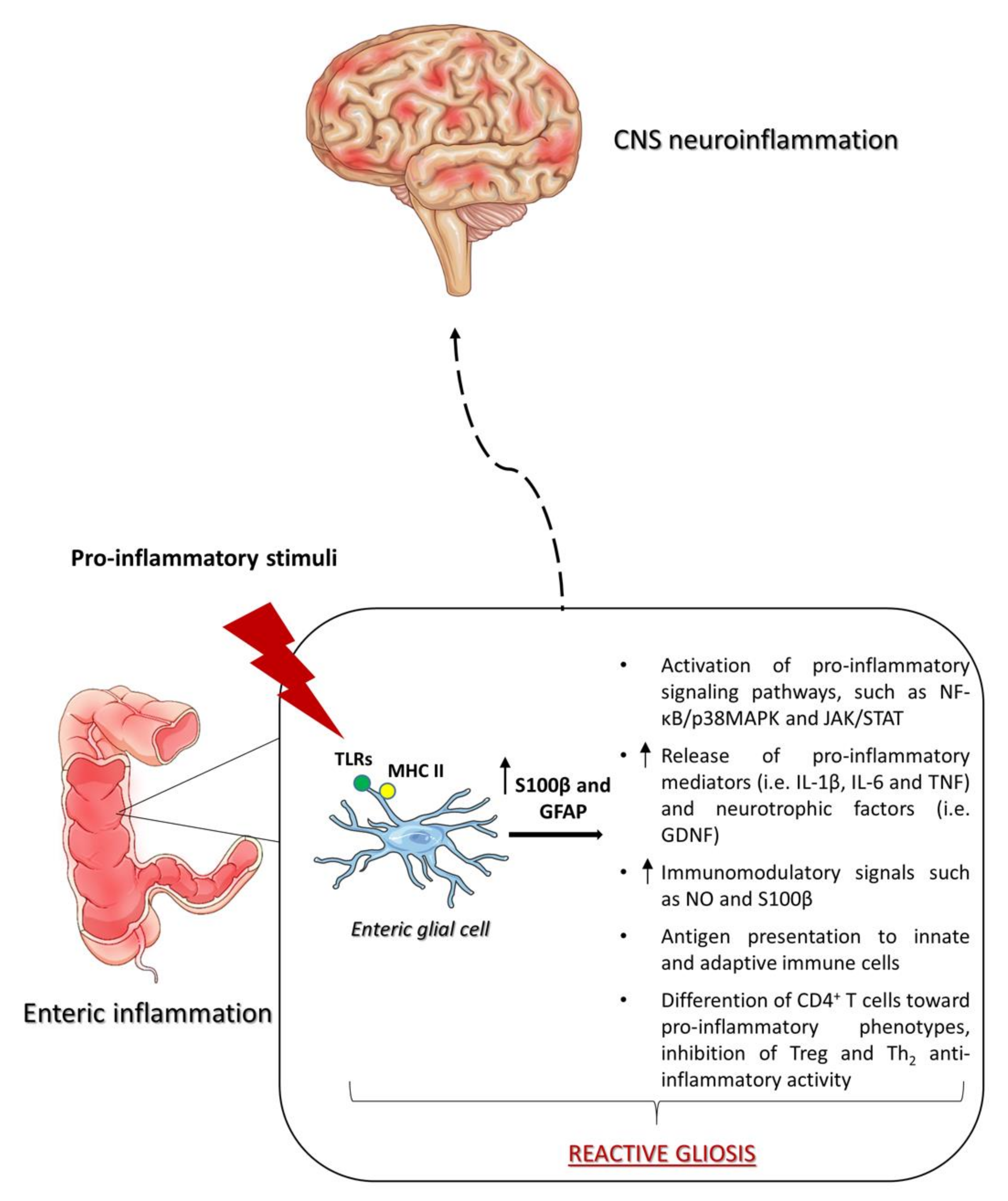
4. Role of Enteric Glia in Intestinal and CNS Inflammation
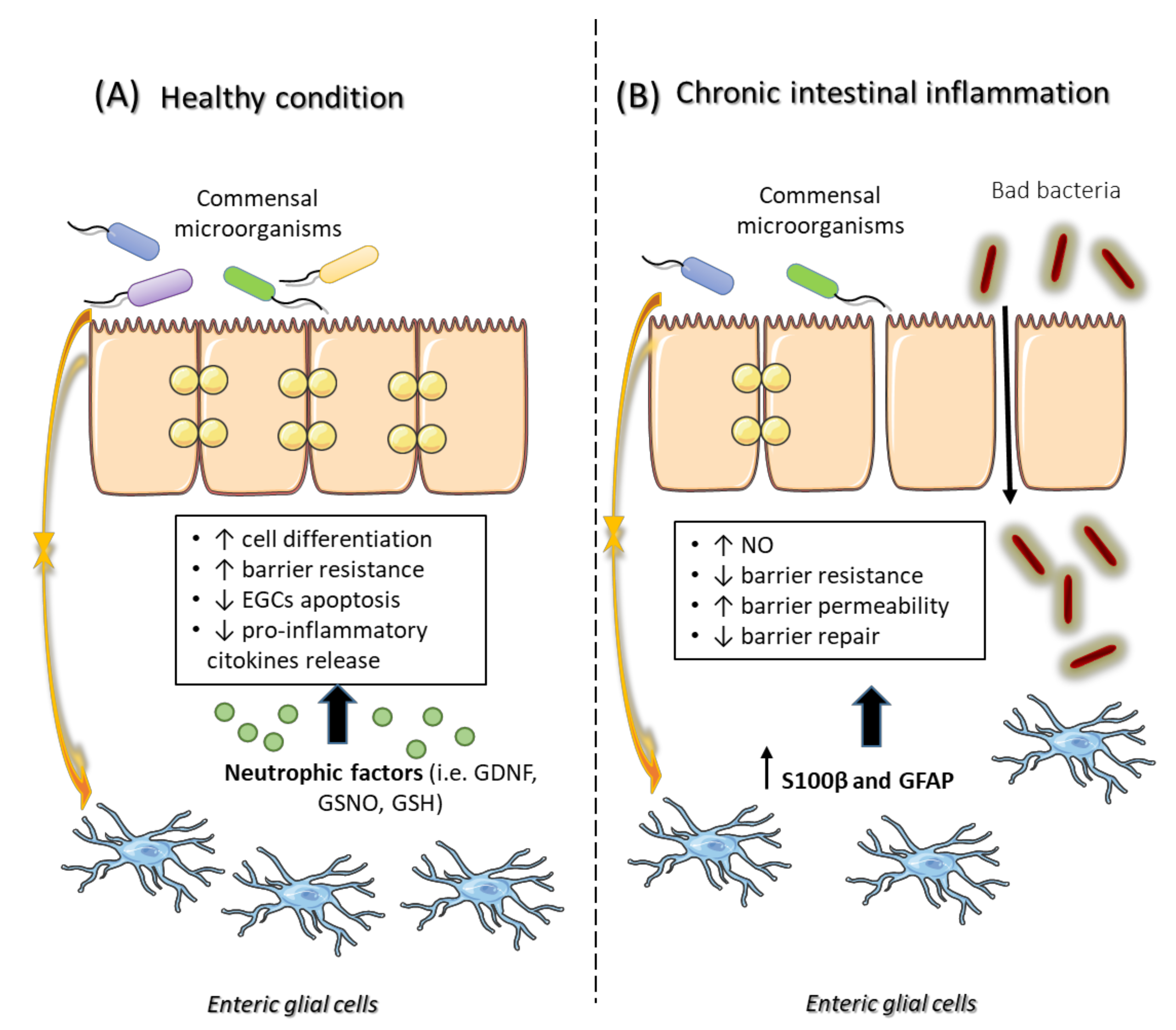
5. The Crosstalk between EGCs, Intestinal Epithelial Barrier and Gut Microbiota
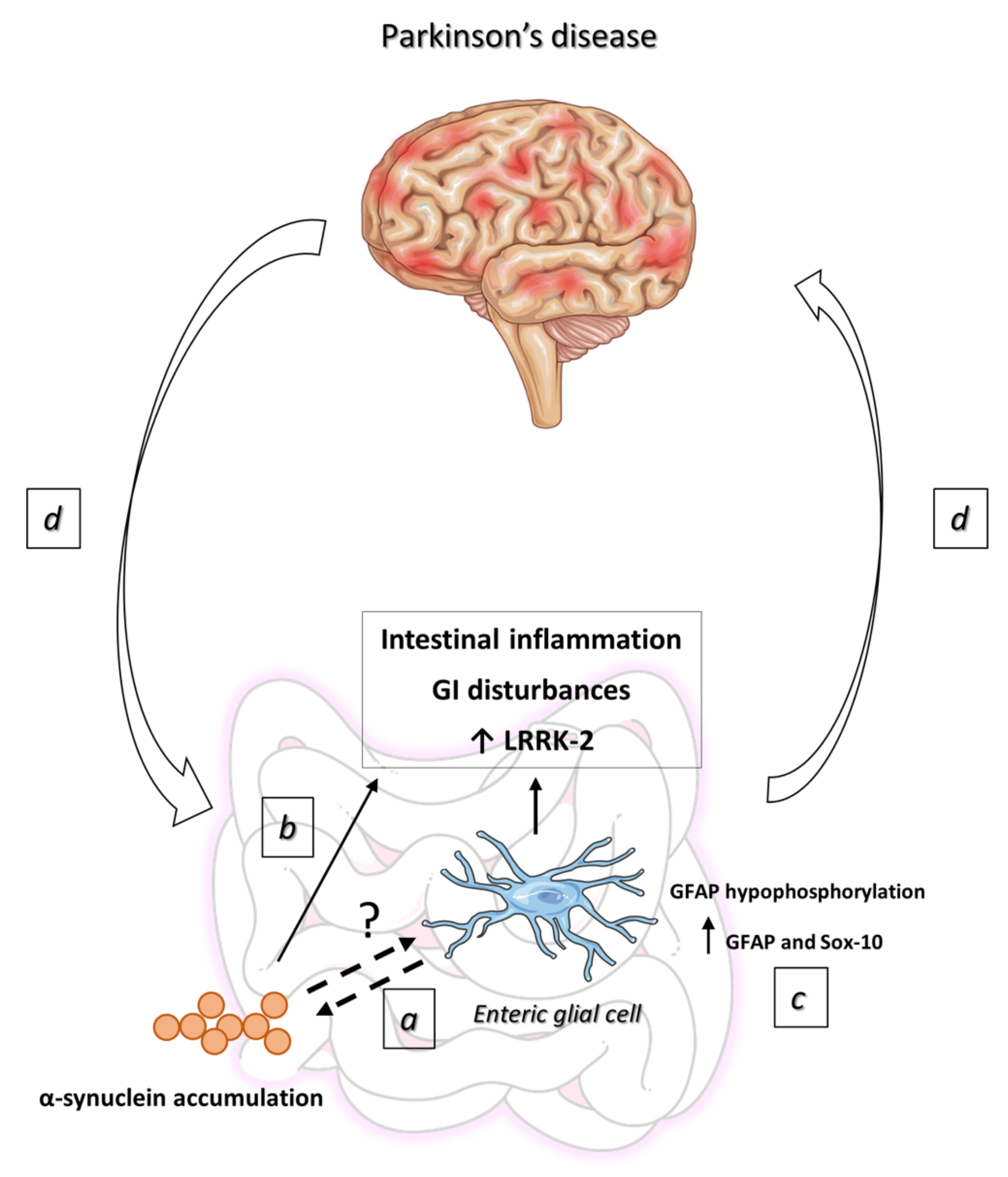
6. Enteric α-Synuclein Accumulation in PD
7. EGCs in the Pathophysiology of PD
8. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- De Virgilio, A.; Greco, A.; Fabbrini, G.; Inghilleri, M.; Rizzo, M.I.; Gallo, A.; Conte, M.; Rosato, C.; Ciniglio Appiani, M.; de Vincentiis, M. Parkinson’s disease: Autoimmunity and neuroinflammation. Autoimmun. Rev. 2016, 15, 1005–1011. [Google Scholar] [CrossRef] [Green Version]
- Obeso, J.A.; Rodrıguez-Oroz, M.C.; Benitez-Temino, B.; Blesa, F.J.; Guridi, J.; Marin, C.; Rodriguez, M. Functional Organization of the Basal Ganglia: Therapeutic Implications for Parkinson’s Disease. Mov. Disord. 2008, 23, S548–S559. [Google Scholar] [CrossRef] [PubMed]
- Davie, C.A. A review of Parkinson’s disease. Br. Med. Bull. 2008, 86, 109–127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Derkinderen, P.; Rouaud, T.; Lebouvier, T.; Bruley des Varannes, S.; Neunlist, M.; De Giorgio, R. Parkinson disease: The enteric nervous system spills its guts. Neurology 2011, 77, 1761–1767. [Google Scholar] [CrossRef] [PubMed]
- Jost, W.H. Gastrointestinal dysfunction in Parkinson’s disease. J. Neurol. Sci. 2010, 289, 69–73. [Google Scholar] [CrossRef] [PubMed]
- Keshavarzian, A.; Green, S.J.; Engen, P.A.; Voigt, R.M.; Naqib, A.; Forsyth, C.B.; Mutlu, E.; Shannon, K.M. Colonic bacterial composition in Parkinson’s disease. Mov. Disord. 2015, 30, 1351–1360. [Google Scholar] [CrossRef] [PubMed]
- Dodiya, H.B.; Forsyth, C.B.; Voigt, R.M.; Engen, A.P.; Patel, J.; Shaikh, M.; Green, S.J.; Naqib, A.; Roy, A.; Kordower, J.H.; et al. Chronic stress-induced gut dysfunction exacerbates Parkinson’s disease phenotype and pathology in a rotenone-induced mouse model of Parkinson’s disease. Neurobiol. Dis. 2018, 135, 104352. [Google Scholar] [CrossRef] [PubMed]
- Manfredsson, F.P.; Luk, K.C.; Benskey, M.J.; Gezer, A.; Garcia, J.; Kuhn, N.C.; Sandoval, I.M.; Patterson, J.R.; O’Mara, A.; Yonkers, R.; et al. Induction of alphasynuclein pathology in the enteric nervous system of the rat and non-human primate results in gastrointestinal dysmotility and transient CNS pathology. Neurobiol. Dis. 2018, 112, 106–118. [Google Scholar] [CrossRef]
- Uemura, N.; Yagi, H.; Uemura, M.T.; Hatanaka, Y.; Yamakado, H.; Takahashi, R. Inoculation of α-synuclein preformed fibrils into the mouse gastrointestinal tract induces Lewy body-like aggregates in the brainstem via the vagus nerve. Mol. Neurodegener. 2018, 13, 21. [Google Scholar] [CrossRef] [Green Version]
- Gulbransen, B.D.; Bashashati, M.; Hirota, S.A.; Gui, X.; Roberts, J.A.; MacDonald, J.A.; Muruve, D.A.; McKay, D.M.; Beck, P.L.; Mawe, G.M.; et al. Activation of neuronal P2X7 receptor-pannexin-1 mediates death of enteric neurons during colitis. Nat. Med. 2012, 18, 600–604. [Google Scholar] [CrossRef] [Green Version]
- Cabarrocas, J.; Savidge, T.C.; Liblau, R.S. Role of enteric glial cells in inflammatory bowel disease. Glia 2003, 41, 81–93. [Google Scholar] [CrossRef] [PubMed]
- Gulbransen, B.D.; Sharkey, K.A. Novel functional roles for enteric glia in the gastrointestinal tract. Nat. Rev. Gastroenterol. Hepatol. 2012, 9, 625–632. [Google Scholar] [CrossRef] [PubMed]
- Sharkey, K.A. Emerging roles for enteric glia in gastrointestinal disorders. J. Clin. Invest. 2015, 125, 918–925. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heanue, T.A.; Pachnis, V. Enteric nervous system development and Hirschsprung’s disease: Advances in genetic and stem cell studies. Nat. Rev. Neurosci. 2007, 8, 466–479. [Google Scholar] [CrossRef]
- Rothman, T.P.; Tennyson, V.M.; Gershon, M.D. Colonization of the bowel by the precursors of enteric glia: Studies of normal and congenitally aganglionic mutant mice. J. Comp. Neurol. 1986, 252, 493–506. [Google Scholar] [CrossRef]
- Kabouridis, P.S.; Lasrado, R.; McCallum, S.; Chng, S.H.; Snippert, H.J.; Clevers, H.; Pettersson, S.; Pachnis, V. Microbiota controls the homeostasis of glial cells in the gut lamina propria. Neuron 2015, 85, 289–295. [Google Scholar] [CrossRef] [Green Version]
- Jessen, K.R.; Saffrey, M.J.; Burnstock, G. The enteric nervous system in tissue culture. I. Cell types and their interactions in explants of the myenteric and submucous plexuses from guinea pig, rabbit and rat. Brain Res. 1983, 262, 17–35. [Google Scholar] [CrossRef]
- Ferri, G.L.; Probert, L.; Cocchia, D.; Michetti, F.; Marangos, P.J.; Polak, J.M. Evidence for the presence of S-100 protein in the glial component of the human enteric nervous system. Nature 1982, 297, 409–410. [Google Scholar] [CrossRef]
- McClain, J.L.; Grubisic, V.; Fried, D.; Gomez-Suarez, R.A.; Leinninger, G.M.; Sevigny, J.; Parpura, V.; Gulbransen, B.D. Ca2+ responses in enteric glia are mediated by connexin-43 hemichannels and modulate colonic transit in mice. Gastroenterology 2014, 146, 497–507. [Google Scholar] [CrossRef] [Green Version]
- Rao, M.; Nelms, B.D.; Dong, L.; Salinas-Rios, V.; Rutlin, M.; Gershon, M.D.; Corfas, G. Enteric glia express proteolipid protein 1 and are a transcriptionally unique population of glia in the mammalian nervous system. Glia 2015, 63, 2040–2057. [Google Scholar] [CrossRef]
- Boesmans, W.; Lasrado, R.; Vanden Berghe, P.; Pachnis, V. Heterogeneity and phenotypic plasticity of glial cells in the mammalian enteric nervous system. Glia 2015, 63, 229–241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoff, S.; Zeller, F.; vonWeyhern, C.W.; Wegner, M.; Schemann, M.; Michel, K.; Ruhl, A. Quantitative assessment of glial cells in the human and guinea pig enteric nervous system with an anti-Sox8/9/10 antibody. J. Comp. Neurol. 2008, 509, 356–371. [Google Scholar] [CrossRef] [PubMed]
- Broadhead, M.J.; Bayguinov, P.O.; Okamoto, T.; Heredia, D.J.; Smith, T.K. Ca2+ transients in myenteric glial cells during the colonic migrating motor complex in the isolated murine large intestine. J. Physiol. 2012, 590, 335–350. [Google Scholar] [CrossRef] [PubMed]
- Von Boyen, G.B.; Steinkamp, M.; Geerling, I.; Reinshagen, M.; Schäfer, K.H.; Adler, G.; Kirsch, J. Proinflammatory cytokines induce neurotrophic factor expression in enteric glia: A key to the regulation of epithelial apoptosis in Crohn’s disease. Inflamm. Bowel Dis. 2006, 12, 346–354. [Google Scholar] [CrossRef] [PubMed]
- Gulbransen, B.D.; Sharkey, K.A. Purinergic neuron-to-glia signaling in the enteric nervous system. Gastroenterology 2009, 136, 1349–1358. [Google Scholar] [CrossRef] [PubMed]
- Neunlist, M.; Rolli-Derkinderen, M.; Latorre, R.; Van Landeghem, L.; Coron, E.; Derkinderen, P.; De Giorgio, R. Enteric glial cells: Recent developments and future directions. Gastroenterology 2014, 147, 1230–1237. [Google Scholar] [CrossRef]
- Gulbransen, B.D.; Bains, J.S.; Sharkey, K.A. Enteric glia are targets of the sympathetic innervation of the myenteric plexus in the guinea pig distal colon. J. Neurosci. 2010, 30, 6801–6809. [Google Scholar] [CrossRef]
- Neunlist, M.; Van Landeghem, L.; Mahe, M.M.; Derkinderen, P.; des Varannes, S.B.; Rolli-Derkinderen, M. The digestive neuronal-glial-epithelial unit: A new actor in gut health and disease. Nat. Rev. Gastroenterol. Hepatol. 2013, 10, 90–100. [Google Scholar] [CrossRef]
- D’Antongiovanni, V.; Pellegrini, C.; Fornai, M.; Colucci, R.; Blandizzi, C.; Antonioli, L.; Bernardini, N. Intestinal epithelial barrier and neuromuscular compartment in health and disease. World J. Gastroenterol. 2020, 26, 1564–1579. [Google Scholar] [CrossRef]
- Ko, H.M.; Lee, S.H.; Kim, K.C.; Joo, S.H.; Choi, W.S.; Shin, C.Y. The role of TLR4 and Fyn interaction on lipopolysaccharide-Stimulated PAI-1 expression in astrocytes. Mol. Neurobiol. 2015, 52, 8–25. [Google Scholar] [CrossRef]
- Viana, S.D.; Valero, J.; Rodrigues-Santos, P. Regulation of striatal astrocytic receptor for advanced glycation end-products variants in an early stage of experimental Parkinson’s disease. J. Neurochem. 2016, 138, 598–609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brkic, M.; Balusu, S.; Libert, C.; Vandenbroucke, R.E. Friends or foes: Matrix metalloproteinases and their multifaceted roles in neurodegenerative diseases. Mediators Inflamm. 2015, 2015, 62058. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Srinivasan, M.; Lahiri, D.K. Significance of NF-κB as a pivotal therapeutic target in the neurodegenerative pathologies of Alzheimer’s disease and multiple sclerosis. Expert Opin. Ther. Targets 2015, 19, 471–487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Antonioli, L.; D’Antongiovanni, V.; Pellegrini, C.; Fornai, M.; Benvenuti, L.; di Carlo, A.; van den Wijngaard, R.; Caputi, V.; Cerantola, S.; Giron, M.C.; et al. Colonic dysmotility associated with high-fat diet-induced obesity: Role of enteric glia. FASEB J. 2020, 34, 5512–5524. [Google Scholar] [CrossRef] [Green Version]
- D’Antongiovanni, V.; Benvenuti, L.; Fornai, M.; Pellegrini, C.; Van Den Wijngaard, R.; Cerantola, S.; Giron, M.C.; Caputi, V.; Colucci, R.; Hasko, G.; et al. Glial A2B Adenosine Receptors Modulate Abnormal Tachykininergic Responses and Prevent Enteric Inflammation Associated with High Fat Diet-Induced Obesity. Cells 2020, 9, 1245. [Google Scholar] [CrossRef]
- Ruhl, A.; Franzke, S.; Collins, S.M.; Stremmel, W. Interleukin-6 expression and regulation in rat enteric glial cells. Am. J. Physiol. Gastrointest. Liver. Physiol. 2001, 280, G1163–G1171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruhl, A.; Nasser, Y.; Sharkey, K.A. Enteric glia. Neurogastroenterol Motil. 2004, 16 (Suppl. S1), 44–49. [Google Scholar] [CrossRef]
- Da Silveira, A.B.M.; De Oliveira, E.C.; Neto, S.G.; Luquetti, A.O.; Fujiwara, R.T.; Oliveira, R.C.; Brehmer, A. Enteroglial cells act as antigen-presenting cells in chagasic megacolon. Hum. Pathol. 2011, 42, 522–532. [Google Scholar] [CrossRef]
- Solleiro-Villavicencio, H.; Rivas-Arancibia, S. Effect of chronic oxidative stress on neuroinflammatory response mediated by CD4+T cells in neurodegenerative diseases. Front. Cell. Neurosci. 2018, 12, 114. [Google Scholar] [CrossRef] [Green Version]
- Frossi, B.; De Carli, M.; Piemonte, M.; Pucillo, C. Oxidative microenvironment exerts an opposite regulatory effect on cytokine production by Th1 and Th2 cells. Mol. Immunol. 2008, 45, 58–64. [Google Scholar] [CrossRef]
- Litteljohn, D.; Mangano, E.; Clarke, M.; Bobyn, J.; Moloney, K.; Hayley, S. Inflammatory mechanisms of neurodegeneration in toxin-based models of Parkinson’s disease. Parkinsons Dis. 2010, 2011, 713517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- González, H.; Pacheco, R. T-cell-mediated regulation of neuroinflammation involved in neurodegenerative diseases. J. Neuroinflamm. 2014, 11, 201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- MacEachern, S.J.; Patel, B.A.; McKay, D.M.; Sharkey, K.A. Nitric oxide regulation of colonic epithelial ion transport: A novel role for enteric glia in the myenteric plexus. J. Physiol. 2011, 589, 3333–3348. [Google Scholar] [CrossRef] [PubMed]
- MacEachern, S.J.; Patel, B.A.; Keenan, C.M.; Dicay, M.; Chapman, K.; McCafferty, D.M.; Savidge, T.C.; Beck, P.L.; MacNaughton, W.K.; Sharkey, K.A. Inhibiting inducible nitric oxide synthase in enteric glia restores electrogenic ion transport in mice with colitis. Gastroenterology 2015, 149, 445–455. [Google Scholar] [CrossRef] [Green Version]
- Brown, I.A.M.; McClain, J.L.; Watson, R.E.; Patel, B.A.; Gulbransen, B.D. Enteric glia mediate neuron death in colitis through purinergic pathways that require Connexin-43 and nitric oxide. Cell. Mol. Gastroenterol. Hepatol. 2015, 2, 77–91. [Google Scholar] [CrossRef] [Green Version]
- Fasano, A.; Shea-Donohue, T. Mechanisms of disease: The role of intestinal barrier function in the pathogenesis of gastrointestinal autoimmune diseases. Nat. Clin. Pract. Gastroenterol. Hepatol. 2005, 2, 416–422. [Google Scholar] [CrossRef]
- Fornai, M.; van den Wijngaard, R.M.; Antonioli, L.; Pellegrini, C.; Blandizzi, C.; de Jonge, W.J. Neuronal regulation of intestinal immune functions in health and disease. Neurogastroenterol. Motil. 2018, 30, e13406. [Google Scholar] [CrossRef]
- Van Landeghem, L.; Chevalier, J.; Mahé, M.M.; Wedel, T.; Urvil, P.; Derkinderen, P.; Savidge, T.; Neunlist, M. Enteric glia promote intestinal mucosal healing via activation of focal adhesion kinase and release of proEGF. Am. J. Physiol. Gastrointest. Liver Physiol. 2011, 300, G976–G987. [Google Scholar] [CrossRef] [Green Version]
- Aube, A.C.; Cabarrocas, J.; Bauer, J.; Philippe, D.; Aubert, P.; Doulay, F.; Liblau, R.; Galmiche, J.P.; Neunlist, M. Changes in enteric neurone phenotype and intestinal functions in a transgenic mouse model of enteric glia disruption. Gut 2006, 55, 630–637. [Google Scholar] [CrossRef] [Green Version]
- Neunlist, M.; Aubert, P.; Bonnaud, S.; Van Landeghem, L.; Coron, E.; Wedel, T.; Naveilhan, P.; Ruhl, A.; Lardeux, B.; Savidge, T.; et al. Enteric glia inhibit intestinal epithelial cell proliferation partly through a TGF-beta1-dependent pathway. Am. J. Physiol. Gastrointest. Liver Physiol. 2007, 292, G231–G241. [Google Scholar] [CrossRef] [Green Version]
- Van Landeghem, L.; Mahé, M.M.; Teusan, R.; Léger, J.; Guisle, I.; Houlgatte, R.; Neunlist, M. Regulation of intestinal epithelial cells transcriptome by enteric glial cells: Impact on intestinal epithelial barrier functions. BMC Genomics 2009, 10, 507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steinkamp, M.; Geerling, I.; Seufferlein, T.; von Boyen, G.; Egger, B.; Grossmann, J.; Ludwig, L.; Adler, G.; Reinshagen, M. Glial-derived neurotrophic factor regulates apoptosis in colonic epithelial cells. Gastroenterology 2003, 124, 1748–1757. [Google Scholar] [CrossRef]
- Tanaka, F.; Tominaga, K.; Fujikawa, Y.; Nagami, Y.; Kamata, N.; Yamagami, H.; Tanigawa, T.; Shiba, M.; Watanabe, T.; Fujiwara, Y.; et al. Concentration of glial cell line-derived neurotrophic factor positively correlates with symptoms in functional dyspepsia. Dig. Dis. Sci. 2016, 61, 3478–3485. [Google Scholar] [CrossRef] [PubMed]
- Cirillo, C.; Bessissow, T.; Desmet, A.S.; Vanheel, H.; Tack, J.; Vanden Berghe, P. Evidence for neuronal and structural changes in submucous ganglia of patients with functional dyspepsia. Am. J. Gastroenterol. 2015, 110, 1205–1215. [Google Scholar] [CrossRef]
- Xiao, W.; Wang, W.; Chen, W.; Sun, L.; Li, X.; Zhang, C.; Yang, H. GDNF is involved in the barrier-inducing effect of enteric glial cells on intestinal epithelial cells under acute ischemia reperfusion stimulation. Mol. Neurobiol. 2014, 50, 274–289. [Google Scholar] [CrossRef]
- Meir, M.; Flemming, S.; Burkard, N.; Bergauer, L.; Metzger, M.; Germer, C.T.; Schlegel, N. Glial cell line-derived neurotrophic factor promotes barrier maturation and wound healing in intestinal epithelial cells in vitro. Am. J. Physiol. Gastrointest. Liver Physiol. 2015, 309, G613–G624. [Google Scholar] [CrossRef] [Green Version]
- Bauman, B.D.; Meng, J.; Zhang, L.; Louiselle, A.; Zheng, E.; Banerjee, S.; Roy, S.; Segura, B.J. Enteric glial-mediated enhancement of intestinal barrier integrity is compromised by morphine. J. Surg. Res. 2017, 219, 214–221. [Google Scholar] [CrossRef]
- Cirillo, C.; Sarnelli, G.; Turco, F.; Mango, A.; Grosso, M.; Aprea, G.; Masone, S.; Cuomo, R. Proinflammatory stimuli activates human-derived enteroglial cells and induces autocrine nitric oxide production. Neurogastroenterol. Motil. 2011, 23, e372–e382. [Google Scholar] [CrossRef]
- Esposito, G.; Cirillo, C.; Sarnelli, G.; De Filippis, D.; D’Armiento, F.P.; Rocco, A.; Nardone, G.; Petruzzelli, R.; Grosso, M.; Izzo, P.; et al. Enteric glial-derived S100B protein stimulates nitric oxide production in celiac disease. Gastroenterology 2007, 133, 918–925. [Google Scholar] [CrossRef]
- Turco, F.; Sarnelli, G.; Cirillo, C.; Palumbo, I.; De Giorgi, F.; D’Alessandro, A.; Cammarota, M.; Giuliano, M.; Cuomo, R. Enteroglial-derived S100B protein integrates bacteria-induced Toll-like receptor signalling in human enteric glial cells. Gut 2014, 63, 105–115. [Google Scholar] [CrossRef]
- Esposito, G.; Capoccia, E.; Gigli, S.; Pesce, M.; Bruzzese, E.; D’Alessandro, A.; Cirillo, C.; Di Cerbo, A.; Cuomo, R.; Seguella, L.; et al. HIV-1 Tat-induced diarrhea evokes an enteric glia-dependent neuroinflammatory response in the central nervous system. Sci. Rep. 2017, 7, 7735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cirillo, C.; Sarnelli, G.; Esposito, G.; Turco, F.; Steardo, L.; Cuomo, R. S100B protein in the gut: The evidence for enteroglial-sustained intestinal inflammation. World J. Gastroenterol. 2011, 17, 1261–1266. [Google Scholar] [CrossRef] [PubMed]
- Xiao, W.D.; Chen, W.; Sun, L.H.; Wang, W.S.; Zhou, S.W.; Yang, H. The protective effect of enteric glial cells on intestinal epithelial barrier function is enhanced by inhibiting inducible nitric oxide synthase activity under lipopolysaccharide stimulation. Mol. Cell. Neurosci. 2011, 46, 527–534. [Google Scholar] [CrossRef]
- Rao, M.; Rastelli, D.; Dong, L.; Chiu, S.; Setlik, W.; Gershon, M.D.; Corfas, G. Enteric Glia Regulate Gastrointestinal Motility but Are Not Required for Maintenance of the Epithelium in Mice. Gastroenterology. 2017, 153, 1068–1081. [Google Scholar] [CrossRef] [PubMed]
- Grubišić, V.; Gulbransen, B.D. Enteric glial activity regulates secretomotor function in the mouse colon but does not acutely affect gut permeability. J. Physiol. 2017, 595, 3409–3424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.A.; Chung, Y.C.; Pan, S.T.; Shen, M.Y.; Hou, Y.C.; Peng, S.J.; Pasricha, P.J.; Tang, S.C. 3-D imaging, illustration, and quantitation of enteric glial network in transparent human colon mucosa. Neurogastroenterol. Motil. 2013, 25, e324–e338. [Google Scholar] [CrossRef] [PubMed]
- Kabouridis, P.S.; Pachnis, V. Emerging roles of gut microbiota and the immune system in the development of the enteric nervous system. J. Clin. Invest. 2015, 125, 956–964. [Google Scholar] [CrossRef] [Green Version]
- Barajon, I.; Serrao, G.; Arnaboldi, F.; Opizzi, E.; Ripamonti, G.; Balsari, A.; Rumio, C. Toll-like receptors 3, 4, and 7 are expressed in the enteric nervous system and dorsal root ganglia. J. Histochem. Cytochem. 2009, 57, 1013–1023. [Google Scholar] [CrossRef]
- Abreu, M.T. Toll-like receptor signalling in the intestinal epithelium: How bacterial recognition shapes intestinal function. Nat. Rev. Immunol. 2010, 10, 131–144. [Google Scholar] [CrossRef]
- Round, J.L.; Mazmanian, S.K. The gut microbiota shapes intestinal immune responses during health and disease. Nat. Rev. Immunol. 2009, 9, 313–323. [Google Scholar] [CrossRef]
- Muller, P.A.; Koscso, B.; Rajani, G.M.; Stevanovic, K.; Berres, M.L.; Hashimoto, D.; Mortha, A.; Leboeuf, M.; Li, X.M.; Mucida, D.; et al. Crosstalk betweenmuscularis macrophages and enteric neurons regulates gastrointestinal motility. Cell 2014, 158, 300–313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sampson, T.R.; Debelius, J.W.; Thron, T.; Janssen, S.; Shastri, G.G.; Ilhan, Z.E.; Challis, C.; Schretter, C.E.; Rocha, S.; Gradinaru, V.; et al. Gut microbiota regulate motor deficits and neuroinflammation in a model of Parkinson’s disease. Cell 2016, 167, 1469–1480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clairembault, T.; Leclair-Visonneau, L.; Neunlist, M.; Derkinderen, P. Enteric glial cells: New players in Parkinson’s disease? Mov. Disord. 2015, 30, 494–498. [Google Scholar] [CrossRef] [PubMed]
- Perez-Pardo, P.; Dodiya, H.B.; Engen, P.A.; Forsyth, C.B.; Huschens, A.M.; Shaikh, M.; Voigt, R.M.; Naqib, A.; Green, S.J.; Kordower, J.H.; et al. Role of TLR4 in the gut-brain axis in Parkinson’s disease: A translational study from men to mice. Gut 2019, 68, 829–843. [Google Scholar] [CrossRef] [PubMed]
- Spillantini, M.G.; Schmidt, M.L.; Lee, V.M.; Trojanowski, J.Q.; Jakes, R.; Goedert, M. Alpha-synuclein in Lewy bodies. Nature 1997, 388, 839–840. [Google Scholar] [CrossRef]
- Gelpi, E.; Navarro-Otano, J.; Tolosa, E.; Gaig, C.; Compta, Y.; Rey, M.J.; Martí, M.J.; Hernández, I.; Valldeoriola, F.; Reñé, R.; et al. Multiple organ involvement by alpha-synuclein pathology in Lewy body disorders. Mov. Disord. 2014, 29, 1010–1018. [Google Scholar] [CrossRef]
- Beach, T.G.; Adler, C.H.; Sue, L.I.; Vedders, L.; Lue, L.; White, C.L., III; Akiyama, H.; Caviness, J.N.; Shill, H.A.; Sabbagh, M.N.; et al. Multi-organ distribution of phosphorylated alpha-synuclein histopathology in subjects with Lewy body disorders. Acta Neuropathol. 2009, 119, 689–702. [Google Scholar] [CrossRef] [Green Version]
- Rota, L.; Pellegrini, C.; Benvenuti, L.; Antonioli, L.; Fornai, M.; Blandizzi, C.; Cattaneo, A.; Colla, E. Constipation, deficit in colon contractions, and alpha-synuclein inclusions within the colon precede motor abnormalities and neurodegeneration in the central nervous system in a mouse model of alpha-synucleinopathy. Transl. Neurod. 2019, 8, 5. [Google Scholar] [CrossRef] [Green Version]
- Yan, F.; Chen, Y.; Li, M.; Wang, Y.; Zhang, W.; Chen, X.; Ye, Q. Gastrointestinal nervous system α-synuclein as a potential biomarker of Parkinson disease. Medicine 2018, 97, e11337. [Google Scholar] [CrossRef]
- Feng, Y.K.; Wu, Q.L.; Peng, Y.W.; Liang, F.Y.; You, H.J.; Feng, Y.W.; Li, G.; Li, X.J.; Liu, S.H.; Li, Y.C.; et al. Gingivalis impairs gut permeability and mediates immune responses associated with neurodegeneration in LRRK2 R1441G mice. J. Neuroinflamm. 2020, 17, 347. [Google Scholar] [CrossRef]
- Kelly, L.P.; Carvey, P.M.; Keshavarzian, A.; Shannon, K.M.; Shaikh, M.; Bakay, R.A.; Kordower, J.H. Progression of intestinal permeability changes and alpha-synuclein expression in a mouse model of Parkinson’s disease. Mov. Disord. 2014, 29, 999–1009. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Qian, Y.; Xu, S.; Song, Y.; Xiao, Q. Longitudinal Analysis of Fecal Microbiome and Pathologic Processes in a Rotenone Induced Mice Model of Parkinson’s Disease. Front. Aging Neurosci. 2018, 9, 441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holmqvist, S.; Chutna, O.; Bousset, L.; Aldrin-Kirk, P.; Li, W.; Björklund, T.; Wang, Z.Y.; Roybon, L.; Melki, R.; Li, J.Y. Direct evidence of Parkinson pathology spread from the gastrointestinal tract to the brain in rats. Acta Neuropathol. 2014, 128, 805–820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan-Montojo, F.; Schwarz, M.; Winkler, C.; Arnhold, M.; O’Sullivan, G.A.; Pal, A.; Said, J.; Marsico, G.; Verbavatz, J.M.; Rodrigo-Angulo, M.; et al. Environmental toxins trigger PD-like progression via increased alpha-synuclein release from enteric neurons in mice. Sci. Rep. 2012, 2, 898. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.; Kwon, S.H.; Kam, T.I.; Panicker, N.; Karuppagounder, S.S.; Lee, S.; Lee, J.H.; Kim, W.R.; Kook, M.; Foss, C.A.; et al. Transneuronal Propagation of Pathologic α-Synuclein from the Gut to the Brain Models Parkinson’s Disease. Neuron 2019, 103, 627–641. [Google Scholar] [CrossRef] [PubMed]
- Svensson, E.; Horvath-Puho, E.; Thomsen, R.W.; Djurhuus, J.C.; Pedersen, L.; Borghammer, P.; Sorensen, H.T. Vagotomy and Subsequent Risk of Parkinson’s Disease. Ann. Neurol. 2015, 78, 522–529. [Google Scholar] [CrossRef]
- Liu, B.; Fang, F.; Pedersen, N.L.; Tillander, A.; Ludvigsson, J.F.; Ekbom, A.; Svenningsson, P.; Chen, H.; Wirdefeldt, K. Vagotomy and Parkinson disease: A Swedish register-based matched-cohort study. Neurology 2017, 88, 1996–2002. [Google Scholar] [CrossRef] [Green Version]
- Shannon, K.M.; Keshavarzian, A.; Mutlu, E.; Dodiya, H.B.; Daian, D.; Jaglin, J.A.; Kordower, J.H. Alpha-synuclein in colonic submucosa in early untreated Parkinson’s disease. Mov. Disord. 2012, 27, 709–715. [Google Scholar] [CrossRef]
- Anderson, G.; Seo, M.; Berk, M.; Carvalho, A.F.; Maes, M. Gut permeability and microbiota in Parkinson’s disease: Role of depression, tryptophan catabolites, oxidative and nitrosative stress and melatonergic pathways. Curr. Pharm. Des. 2016, 22, 6142–6151. [Google Scholar] [CrossRef]
- Devos, D.; Lebouvier, T.; Lardeux, B.; Biraud, M.; Rouaud, T.; Pouclet, H.; Coron, E.; Varannes, S.B.D.; Naveilhan, P.; Nguyen, J.-M.; et al. Colonic inflammation in Parkinson’s disease. Neurobiol. Dis. 2013, 50, 42–48. [Google Scholar] [CrossRef]
- Takemura, M.; Gomi, H.; Colucci-Guyon, E.; Itohara, S. Protective role of phosphorylation in turnover of glial fibrillary acidic protein in mice. J. Neurosci. 2002, 22, 6972–6979. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, S.M.; Sullivan, R.K.; Miller, S.M.; Ireland, Z.; Björkman, S.T.; Pow, D.V.; Colditz, P.B. Phosphorylation of GFAP is associated with injury in the neonatal pig hypoxicischemic brain. Neurochem. Res. 2012, 37, 2364–2378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korolainen, M.A.; Auriola, S.; Nyman, T.A.; Alafuzoff, I.; Pirttilä, T. Proteomic analysis of glial fibrillary acidic protein in Alzheimer’s disease and aging brain. Neurobiol. Dis. 2005, 20, 858–870. [Google Scholar] [CrossRef] [PubMed]
- Herskowitz, J.H.; Seyfried, N.T.; Duong, D.M.; Xia, Q.; Rees, H.D.; Gearing, M.; Peng, J.; Lah, J.J.; Levey, A.I. Phosphoproteomic analysis reveals site-specific changes in GFAP and NDRG2 phosphorylation in frontotemporal lobar degeneration. J. Proteome Res. 2010, 9, 6368–6379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clairembault, T.; Kamphuis, W.; Leclair-Visonneau, L.; Rolli-Derkinderen, M.; Coron, E.; Neunlist, M.; Hol, E.M.; Derkinderen, P. Enteric GFAP expression and phosphorylation in Parkinson’s disease. J. Neurochem. 2014, 130, 805–815. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burda, J.E.; Sofroniew, M.V. Reactive gliosis and the multicellular response to CNS damage and disease. Neuron 2014, 81, 229–248. [Google Scholar] [CrossRef] [Green Version]
- Gerhard, A.; Pavese, N.; Hotton, G.; Turkheimer, F.; Es, M.; Hammers, A.; Eggert, K.; Oertel, W.; Banati, R.B.; Brooks, D.J. In vivo imaging of microglial activation with [11C](R)-PK11195 PET in idiopathic Parkinson’s disease. Neurobiol. Dis. 2006, 21, 404–412. [Google Scholar] [CrossRef]
- Pellegrini, C.; Fornai, M.; Colucci, R.; Tirotta, E.; Blandini, F.; Levandis, G.; Cerri, S.; Segnani, C.; Ippolito, C.; Bernardini, N.; et al. Alteration of colonic excitatory tachykininergic motility and enteric inflammation following dopaminergic nigrostriatal neurodegeneration. J. Neuroinflamm. 2016, 13, 146. [Google Scholar] [CrossRef] [Green Version]
- Pellegrini, C.; Ippolito, C.; Segnani, C.; Dolfi, A.; Errede, M.; Virgintino, D.; Fornai, M.; Antonioli, L.; Garelli, F.; Nericcio, A.; et al. Pathological remodelling of colonic wall following dopaminergic nigrostriatal neurodegeneration. Neurobiol. Dis. 2020, 139, 104821. [Google Scholar] [CrossRef]
- Schwiertz, A.; Spiegel, J.; Dillmann, U.; Grundmann, D.; Bürmann, J.; Faßbender, K.; Schäfer, K.H.; Unger, M.M. Fecal markers of intestinal inflammation and intestinal permeability are elevated in Parkinson’s disease. Parkinsonism Relat. Disord. 2018, 50, 104–107. [Google Scholar] [CrossRef]
- Braak, H.; Del Tredici, K.; Rüb, U.; de Vos, R.A.; Jansen Steur, E.N.; Braak, E. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol. Aging 2003, 24, 197–211. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Benvenuti, L.; D'Antongiovanni, V.; Pellegrini, C.; Antonioli, L.; Bernardini, N.; Blandizzi, C.; Fornai, M. Enteric Glia at the Crossroads between Intestinal Immune System and Epithelial Barrier: Implications for Parkinson Disease. Int. J. Mol. Sci. 2020, 21, 9199. https://doi.org/10.3390/ijms21239199
Benvenuti L, D'Antongiovanni V, Pellegrini C, Antonioli L, Bernardini N, Blandizzi C, Fornai M. Enteric Glia at the Crossroads between Intestinal Immune System and Epithelial Barrier: Implications for Parkinson Disease. International Journal of Molecular Sciences. 2020; 21(23):9199. https://doi.org/10.3390/ijms21239199
Chicago/Turabian StyleBenvenuti, Laura, Vanessa D'Antongiovanni, Carolina Pellegrini, Luca Antonioli, Nunzia Bernardini, Corrado Blandizzi, and Matteo Fornai. 2020. "Enteric Glia at the Crossroads between Intestinal Immune System and Epithelial Barrier: Implications for Parkinson Disease" International Journal of Molecular Sciences 21, no. 23: 9199. https://doi.org/10.3390/ijms21239199
APA StyleBenvenuti, L., D'Antongiovanni, V., Pellegrini, C., Antonioli, L., Bernardini, N., Blandizzi, C., & Fornai, M. (2020). Enteric Glia at the Crossroads between Intestinal Immune System and Epithelial Barrier: Implications for Parkinson Disease. International Journal of Molecular Sciences, 21(23), 9199. https://doi.org/10.3390/ijms21239199