Copper Dyshomeostasis in Neurodegenerative Diseases—Therapeutic Implications




Abstract
:1. Introduction
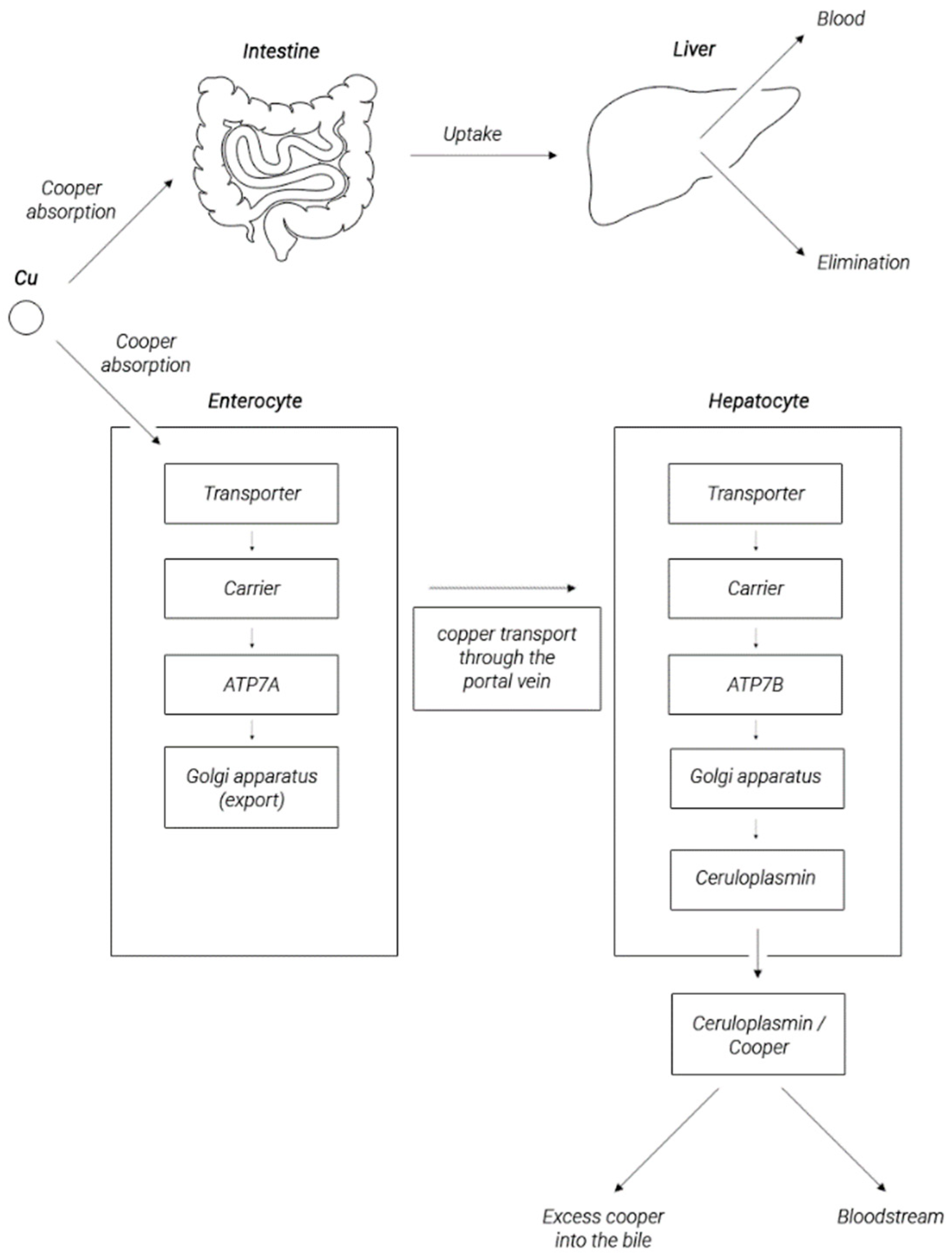
2. Copper Metabolism in the Human Body
3. Copper Content in the Brain
4. Copper Role in the Brain
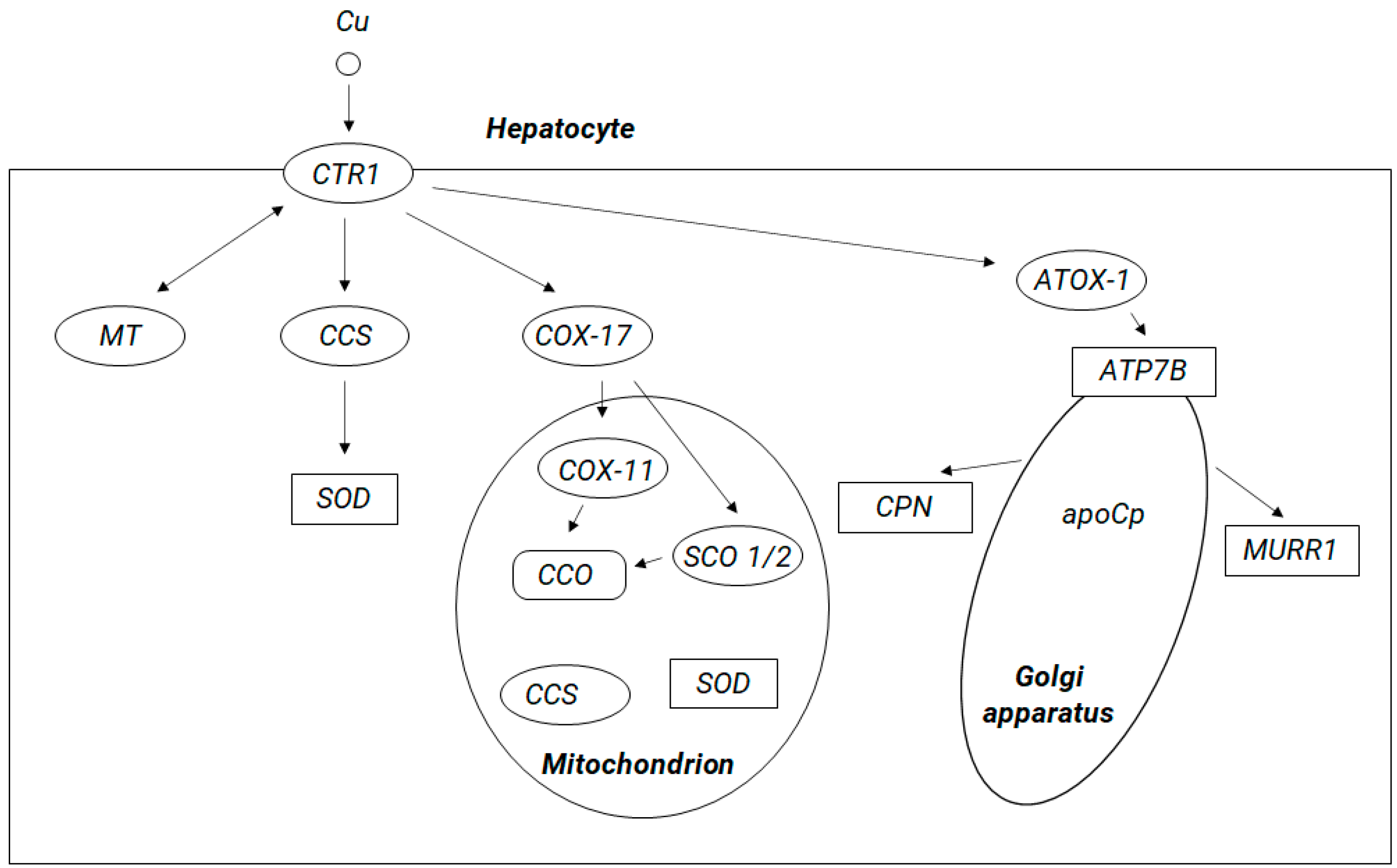
5. Copper Brain Metabolism
6. Abnormal Copper Homeostasis—Neurodegenerative Disease
6.1. Copper Excess—Neurodegeneration: Wilson’s Disease
6.2. Copper Excess—Neurodegeneration: Alzheimer’s Disease
6.3. Copper Excess—Neurodegeneration: Parkinson’s Disease
6.4. Copper Toxicity—Modifying Factors: Lessons from WD
7. Copper Deficiency and Neurodegeneration
7.1. Menkes Disease
7.2. Alzheimer’s Disease
7.3. Parkinson’s Disease
7.4. Copper Deficiency—Neurodegeneration: SOD1
8. Therapeutic Strategies to Reverse Disturbances in Brain Copper Homeostasis
9. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Adams, P.C. Epidemiology and diagnostic testing for hemochromatosis and iron overload. Int. J. Lab. Hematol. 2015, 37 (Suppl. 1), 25–30. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.; Gautam, N.; Mishra, A.; Gupta, R. Heavy metals and living systems: An overview. Indian J. Pharmacol. 2011, 43, 246–253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bisaglia, M.; Bubacco, L. Copper Ions and Parkinson’s Disease: Why Is Homeostasis So Relevant? Biomolecules 2020, 10, 195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aguilar-Martinez, P.; Grandchamp, B.; Cunat, S.; Cadet, E.; Blanc, F.; Nourrit, M.; Lassoued, K.; Schved, J.F.; Rochette, J. Iron overload in HFE C282Y heterozygotes at first genetic testing: A strategy for identifying rare HFE variants. Haematologica 2011, 96, 507–514. [Google Scholar] [CrossRef] [Green Version]
- Aisen, P.; Enns, C.; Wessling-Resnick, M. Chemistry and biology of eukaryotic iron metabolism. Int. J. Biochem. Cell Biol. 2001, 33, 940–959. [Google Scholar] [CrossRef]
- Nagakubo, T.; Kumano, T.; Ohta, T.; Hashimoto, Y.; Kobayashi, M. Copper amine oxidases catalyze the oxidative deamination and hydrolysis of cyclic imines. Nat. Commun. 2019, 10, 413. [Google Scholar] [CrossRef]
- Fukai, T.; Ushio-Fukai, M. Superoxide dismutases: Role in redox signaling, vascular function, and diseases. Antioxid. Redox Signal. 2011, 15, 1583–1606. [Google Scholar] [CrossRef] [Green Version]
- Akil, M.; Schwartz, J.A.; Dutchak, D.; Yuzbasiyan-Gurkan, V.; Brewer, G.J. The psychiatric presentations of Wilson’s disease. J. Neuropsychiatry Clin. Neurosci. 1991, 3, 377–382. [Google Scholar] [CrossRef]
- Vashchenko, G.; MacGillivray, R.T. Multi-copper oxidases and human iron metabolism. Nutrients 2013, 5, 2289–2313. [Google Scholar] [CrossRef] [Green Version]
- Heldin, C.H.; Lu, B.; Evans, R.; Gutkind, J.S. Signals and Receptors. Cold Spring Harb. Perspect. Biol. 2016, 8, a005900. [Google Scholar] [CrossRef] [Green Version]
- Gaier, E.D.; Eipper, B.A.; Mains, R.E. Copper signaling in the mammalian nervous system: Synaptic effects. J. Neurosci. Res. 2013, 91, 2–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allen, K.J.; Buck, N.E.; Cheah, D.M.; Gazeas, S.; Bhathal, P.; Mercer, J.F. Chronological changes in tissue copper, zinc and iron in the toxic milk mouse and effects of copper loading. Biometals 2006, 19, 555–564. [Google Scholar] [CrossRef] [PubMed]
- Kardos, J.; Héja, L.; Simon, Á.; Jablonkai, I.; Kovács, R.; Jemnitz, K. Copper signalling: Causes and consequences. Cell Commun. Signal. 2018, 16, 71, Erratum in 2018, 16, 80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurutas, E.B. The importance of antioxidants which play the role in cellular response against oxidative/nitrosative stress: Current state. Nutr. J. 2016, 15, 71. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Guo, C.; Kong, J. Oxidative stress in neurodegenerative diseases. Neural. Regen. Res. 2012, 7, 376–385. [Google Scholar]
- Telianidis, J.; Hung, Y.H.; Materia, S.; Fontaine, S.L. Role of the P-Type ATPases, ATP7A and ATP7B in brain copper homeostasis. Front. Aging Neurosci. 2013, 5, 44. [Google Scholar] [CrossRef] [Green Version]
- Choo, X.Y.; Alukaidey, L.; White, A.R.; Grubman, A. Neuroinflammation and copper in Alzheimer’s disease. Int. J. Alzheimers Dis. 2013, 2013, 145345. [Google Scholar] [CrossRef] [Green Version]
- Prashanth, L.; Kattapagari, K.K.; Chitturi, R.T.; Baddam, V.R.; Prasad, L.K. A review on role of essential trace elements in health and disease. J. Ntr. Univ. Health Sci. 2015, 4, 75–85. [Google Scholar]
- Schuchardt, J.P.; Hahn, A. Intestinal Absorption and Factors Influencing Bioavailability of Magnesium-An Update. Curr. Nutr. Food Sci. 2017, 13, 260–278. [Google Scholar] [CrossRef]
- Lane, D.J.; Bae, D.H.; Merlot, A.M.; Sahni, S.; Richardson, D.R. Duodenal cytochrome b (DCYTB) in iron metabolism: An update on function and regulation. Nutrients 2015, 7, 2274–2296. [Google Scholar] [CrossRef] [Green Version]
- Nose, Y.; Wood, L.K.; Kim, B.E.; Prohaska, J.R.; Fry, R.S.; Spears, J.W.; Thiele, D.J. Ctr1 is an apical copper transporter in mammalian intestinal epithelial cells in vivo that is controlled at the level of protein stability. J. Biol. Chem. 2010, 285, 32385–32392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zimnicka, A.M.; Maryon, E.B.; Kaplan, J.H. Human copper transporter hCTR1 mediates basolateral uptake of copper into enterocytes: Implications for copper homeostasis. J. Biol. Chem. 2007, 282, 26471–26480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, A.; Lutsenko, S. Human copper transporters: Mechanism, role in human diseases and therapeutic potential. Future Med. Chem. 2009, 1, 1125–1142. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.; Petris, M.J.; Thiele, D.J. Characterization of mouse embryonic cells deficient in the ctr1 high affinity copper transporter. Identification of a Ctr1-independent copper transport system. J. Biol. Chem. 2002, 277, 40253–40259. [Google Scholar] [CrossRef] [Green Version]
- Arredondo, M.; Muñoz, P.; Mura, C.V.; Nùñez, M.T. DMT1, a physiologically relevant apical Cu1+ transporter of intestinal cells. American journal of physiology. Cell Physiol. 2003, 284, C1525–C1530. [Google Scholar] [CrossRef] [PubMed]
- Zimnicka, A.M.; Ivy, K.; Kaplan, J.H. Acquisition of dietary copper: A role for anion transporters in intestinal apical copper uptake. Am. J. Physiol. Cell Physiol. 2011, 300, C588–C599. [Google Scholar] [CrossRef] [Green Version]
- Pierson, H.; Muchenditsi, A.; Kim, B.E.; Ralle, M.; Zachos, N.; Huster, D.; Lutsenko, S. The Function of ATPase Copper Transporter ATP7B in Intestine. Gastroenterology 2018, 154, 168–180. [Google Scholar] [CrossRef] [Green Version]
- Ravia, J.J.; Stephen, R.M.; Ghishan, F.K.; Collins, J.F. Menkes Copper ATPase (Atp7a) is a novel metal-responsive gene in rat duodenum, and immunoreactive protein is present on brush-border and basolateral membrane domains. J. Biol. Chem. 2005, 280, 36221–36227. [Google Scholar] [CrossRef] [Green Version]
- La Fontaine, S.; Mercer, J.F. Trafficking of the copperATPases, ATP7A and ATP7B: Role in copper homeostasis. Arch. Biochem. Biophys. 2007, 463, 149–167. [Google Scholar] [CrossRef]
- Nyasae, L.; Bustos, R.; Braiterman, L.; Eipper, B.; Hubbard, A. Dynamics of endogenous ATP7A (Menkes protein) in intestinal epithelial cells: Copper-dependent redistribution between two intracellular sites. Am. J. Physiol. Gastrointest. Liver Physiol. 2007, 292, G1181–G1194. [Google Scholar] [CrossRef] [Green Version]
- Lloyd, D.R.; Phillips, D.H. Oxidative DNA damage mediated by copper(II), iron(II) and nickel(II) fenton reactions: Evidence for site-specific mechanisms in the formation of double-strand breaks, 8-hydroxydeoxyguanosine and putative intrastrand cross-links. Mutat. Res. 1999, 424, 23–36. [Google Scholar] [CrossRef]
- Sharp, P. The molecular basis of copper and iron interactions. Proc. Nutr. Soc. 2004, 63, 563–569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Przybyłkowski, A.; Gromadzka, G.; Wawer, A.; Grygorowicz, T.; Cybulska, A.; Członkowska, A. Intestinal expression of metal transporters in Wilson’s disease. Biometals 2013, 26, 925–934. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bauerly, K.A.; Kelleher, S.L.; Lonnerdal, B. Effects of copper supplementation on copper absorption, tissue distribution, and copper transporter expression in an infant rat model. Am. J. Physiol. Gastrointest. Liver Physiol. 2005, 288, G1007–G1014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, I.S.; Chen, H.H.; Aiba, I.; Hossain, A.; Liang, Z.D.; Klomp, L.W.; Kuo, M.T. Transcription factor Sp1 plays an important role in the regulation of copper homeostasis in mammalian cells. Mol. Pharmacol. 2008, 74, 705–713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, Z.D.; Tsai, W.B.; Lee, M.Y.; Savaraj, N.; Kuo, M.T. Specificity protein 1 (sp1) oscillation is involved in copper homeostasis maintenance by regulating human highaffinity copper transporter 1 expression. Mol. Pharm. 2012, 81, 455–464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liwei Xie, J.C. Involvement of Sp1 in transcriptional regulation of Atp7a during hypoxia. FASEB J. 2012, 26, 248. [Google Scholar]
- Skopp, A.; Boyd, S.D.; Ullrich, M.S.; Liu, L.; Winkler, D.D. Copper–zinc superoxide dismutase (Sod1) activation terminates interaction between its copper chaperone (Ccs) and the cytosolic metal-binding domain of the copper importer Ctr1. BioMetals 2019, 32, 695–705. [Google Scholar] [CrossRef] [Green Version]
- Kaplan, J.H.; Maryon, E.B. How Mammalian Cells Acquire Copper: An Essential but Potentially Toxic Metal. Biophys. J. 2016, 110, 7–13. [Google Scholar] [CrossRef] [Green Version]
- Kaplan, J.H.; Lutsenko, S. Copper transport in mammalian cells: Special care for a metal with special needs. J. Biol. Chem. 2009, 284, 25461–25465. [Google Scholar] [CrossRef] [Green Version]
- Collins, J.F.; Prohaska, J.R.; Knutson, M.D. Metabolic crossroads of iron and copper. Nutr. Rev. 2010, 68, 133–147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamza, I.; Gitlin, J.D. Hepatic Copper Transport. In Madame Curie Bioscience Database [Internet]; Landes Bioscience: Austin, TX, USA, 2013. Available online: https://www.ncbi.nlm.nih.gov/books/NBK6381/ (accessed on 3 December 2020).
- Wu, F.; Wang, J.; Pu, C.; Qiao, L.; Jiang, C. Wilson’s disease: A comprehensive review of the molecular mechanisms. Int. J. Mol. Sci. 2015, 16, 6419–6431. [Google Scholar] [CrossRef] [PubMed]
- Kiela, P.R.; Ghishan, F.K. Physiology of Intestinal Absorption and Secretion. Best Pract. Res. Clin. Gastroenterol. 2016, 30, 145–159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, W.; Monnot, A.D. Regulation of brain iron and coppe homeostasis by brain barrier systems: Implication in neurodegenerative diseases. Pharmacol. Ther. 2012, 133, 177–188. [Google Scholar] [CrossRef] [Green Version]
- Song, M.; Zhou, Z.; Chen, T.; Zhang, J.; McClain, C.J. Copper deficiency exacerbates bile duct ligation-induced liver injury and fibrosis in rats. J. Pharmacol. Exp. Ther. 2011, 339, 298–306. [Google Scholar] [CrossRef] [Green Version]
- Tórsdóttir, G.; Kristinsson, J.; Sveinbjörnsdóttir, S.; Snaedal, J.; Jóhannesson, T. Copper, ceruloplasmin, superoxide dismutase and iron parameters in Parkinson’s disease. Pharmacol. Toxicol. 1999, 85, 239–243. [Google Scholar] [CrossRef]
- Moriya, M.; Ho, Y.H.; Grana, A.; Nguyen, L.; Alvarez, A.; Jamil, R.; Ackland, M.L.; Michalczyk, A.; Hamer, P.; Ramos, D.; et al. Copper is taken up efficiently from albumin and alpha2-macroglobulin by cultured human cells by more than one mechanism. Am. J. Physiol. Cell Physiol. 2008, 295, C708–C721. [Google Scholar] [CrossRef] [Green Version]
- Linder, M.C. Ceruloplasmin and other copper binding components of blood plasma and their functions: An update. Metallomics 2016, 8, 887–905. [Google Scholar] [CrossRef]
- Ramos, D.; Mar, D.; Ishida, M.; Vargas, R.; Gaite, M.; Montgomery, A.; Linder, M.C. Mechanism of Copper Uptake from Blood Plasma Ceruloplasmin by Mammalian Cells. PLoS ONE 2016, 11, e0149516. [Google Scholar] [CrossRef] [Green Version]
- Antonucci, L.; Porcu, C.; Iannucci, G.; Balsano, C.; Barbaro, B. Non-Alcoholic Fatty Liver Disease and Nutritional Implications: Special Focus on Copper. Nutrients 2017, 9, 1137. [Google Scholar] [CrossRef] [Green Version]
- Bush, A.I. Metals and neuroscience. Curr. Opin. Chem. Biol. 2000, 4, 184–191. [Google Scholar] [CrossRef]
- Montes, S.; Rivera-Mancia, S.; Diaz-Ruiz, A.; Tristan-Lopez, L.; Rios, C. Copper and copper proteins in Parkinson’s disease. Oxid. Med. Cell Longev. 2014, 2014, 147251. [Google Scholar] [CrossRef] [Green Version]
- Stuerenburg, H.J. CSF copper concentrations, blood-brain barrier function, and coeruloplasmin synthesis during the treatment of Wilson’s disease. J. Neural. Transm. (Vienna) 2000, 107, 321–329. [Google Scholar] [CrossRef] [PubMed]
- Lech, T.; Sadlik, J.K. Contribution to the data on copper concentration in blood and urine in patients with Wilson’s disease and in normal subjects. Biol. Trace Elem. Res. 2007, 118, 16–20. [Google Scholar] [CrossRef] [PubMed]
- Rahil-Khazen, R.; Bolann, B.J.; Myking, A.; Ulvik, R.J. Multi-element analysis of trace element levels in human autopsy tissues by using inductively coupled atomic emission spectrometry technique (ICP-AES). J. Trace Elem. Med. Biol. 2002, 16, 15–25. [Google Scholar] [CrossRef]
- Taylor, A.A.; Tsuji, J.S.; Garry, M.R.; McArdle, M.E.; Goodfellow, W.L.; Adams, W.J., Jr.; Menzie, C.A. Critical Review of Exposure and Effects: Implications for Setting Regulatory Health Criteria for Ingested Copper. Environ. Manag. 2020, 65, 131–159. [Google Scholar] [CrossRef] [Green Version]
- Schlief, M.L.; Gitlin, J.D. Copper homeostasis in the CNS: A novel link between the NMDA receptor and copper homeostasis in the hippocampus. Mol. Neurobiol. 2006, 33, 81–90. [Google Scholar] [CrossRef]
- DiDonato, M.; Narindrasorasak, S.; Forbes, J.R.; Cox, D.W.; Sarkar, B. Expression, purification, and metal binding properties of the N-terminal domain from the Wilson disease putative copper-transporting ATPase (ATP7B). J. Biol. Chem. 1997, 272, 33279–33282. [Google Scholar] [CrossRef] [Green Version]
- Lönnerdal, B. Intestinal regulation of copper homeostasis: A developmental perspective. Am. J. Clin. Nutr. 2008, 88, 846S–850S. [Google Scholar] [CrossRef] [Green Version]
- Trombley, P.Q.; Horning, M.S.; Blakemore, L.J. Interactions between carnosine and zinc and copper: Implications for neuromodulation and neuroprotection. Biochemistry (Mosc.) 2000, 65, 807–816. [Google Scholar]
- Blanke, M.L.; VanDongen, A.M.J. Chapter 13: Activation Mechanisms of the NMDA Receptor. In Biology of the NMDA Receptor; Van Dongen, A.M., Ed.; CRC Press/Taylor & Francis: Boca Raton, FL, USA, 2009. Available online: https://www.ncbi.nlm.nih.gov/books/NBK5274/ (accessed on 3 December 2020).
- Johnson, K.A.; Conn, P.J.; Niswender, C.M. Glutamate receptors as therapeutic targets for Parkinson’s disease. CNS Neurol. Disord. Drug Targets 2009, 8, 475–491. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Chang, L.; Song, Y.; Li, H.; Wu, Y. The Role of NMDA Receptors in Alzheimer’s Disease. Front. Neurosci. 2019, 13, 43. [Google Scholar] [CrossRef] [Green Version]
- D’Ambrosi, N.; Rossi, L. Copper at synapse: Release, binding and modulation of neurotransmission. Neurochem. Int. 2015, 90, 36–45. [Google Scholar] [CrossRef] [PubMed]
- Que, E.L.; Domaille, D.W.; Chang, C.J. Metals in neurobiology: Probing their chemistry and biology with molecular imaging. Chem. Rev. 2008, 108, 1517–1549, Erratum in 2008, 108, 4328. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.J. Searching for harmony in transition-metal signaling. Nat. Chem. Biol. 2015, 11, 744–747. [Google Scholar] [CrossRef] [PubMed]
- Araque, A.; Navarrete, M. Glial cells in neuronal network function. Philos. Trans. R Soc. Lond. B Biol. Sci. 2010, 365, 2375–2381. [Google Scholar] [CrossRef] [PubMed]
- Barber, C.N.; Raben, D.M. Lipid Metabolism Crosstalk in the Brain: Glia and Neurons. Front. Cell Neurosci. 2019, 13, 212. [Google Scholar] [CrossRef] [Green Version]
- Travaglia, A.; La Mendola, D.; Magrì, A.; Nicoletti, V.G.; Pietropaolo, A.; Rizzarelli, E. Copper, BDNF and Its N-terminal domain: Inorganic features and biological perspetives. Chemistry 2012, 18, 15618–15631. [Google Scholar] [CrossRef]
- Naletova, I.; Satriano, C.; Pietropaolo, A.; Gianì, F.; Pandini, G.; Triaca, V.; Amadoro, G.; Latina, V.; Calissano, P.; Travaglia, A.; et al. The Copper (II)-Assisted Connection between NGF and BDNF by Means of Nerve Growth Factor-Mimicking Short Peptides. Cells 2019, 8, 301. [Google Scholar] [CrossRef] [Green Version]
- Chao, M.V. Neurotrophins and their receptors: A convergence point for many signalling pathways. Nat. Rev. Neurosci. 2003, 4, 299–309. [Google Scholar] [CrossRef]
- Ross, G.M.; Shamovsky, I.L.; Woo, S.B.; Post, J.I.; Vrkljan, P.N.; Lawrance, G.; Solc, M.; Dostaler, S.M.; Neet, K.E.; Riopelle, R.J. The binding of zinc and copper ions to nerve growth factor is differentially affected by pH: Implications for cerebral acidosis. J. Neurochem. 2001, 78, 515–523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, W.; Post, J.I.; Dow, K.E.; Shin, S.H.; Riopelle, R.J.; Ross, G.M. Zinc and copper inhibit nerve growth factor-mediated protection from oxidative stress-induced apoptosis. Neurosci. Lett. 1999, 259, 115–118. [Google Scholar] [CrossRef]
- Birkaya, B.; Aletta, J.M. NGF promotes copper accumulation required for optimum neurite outgrowth and protein methylation. J. Neurobiol. 2005, 63, 49–61. [Google Scholar] [CrossRef] [PubMed]
- Li, G.J.; Zheng, W. Regulation of neuroactive metals by the choroid plexus. In The Blood-Cerebrospinal Barrier; Zheng, W., Chodobski, A., Eds.; CRC Press: New York, NY, USA, 2005; pp. 211–239. [Google Scholar]
- Sofroniew, M.V. Astrocyte barriers to neurotoxic inflammation. Nat. Rev. Neurosci. 2015, 16, 249–263. [Google Scholar] [CrossRef] [Green Version]
- Bylicky, M.A.; Mueller, G.P.; Day, R.M. Mechanisms of Endogenous Neuroprotective Effects of Astrocytes in Brain Injury. Oxid. Med. Cell Longev. 2018, 2018, 6501031. [Google Scholar] [CrossRef]
- Dringen, R.; Scheiber, I.F.; Mercer, J.F. Copper metabolism of astrocytes. Front. Aging Neurosci. 2013, 5, 9. [Google Scholar] [CrossRef] [Green Version]
- Skjørringe, T.; Burkhart, A.; Johnsen, K.B.; Moos, T. Divalent metal transporter 1 (DMT1) in the brain: Implications for a role in iron transport at the blood-brain barrier, and neuronal and glial pathology. Front. Mol. Neurosci. 2015, 8, 19. [Google Scholar]
- Howell, S.B.; Safaei, R.; Larson, C.A.; Sailor, M.J. Copper transporters and the cellular pharmacology of the platinum-containing cancer drugs. Mol. Pharmacol. 2010, 77, 887–894. [Google Scholar] [CrossRef] [Green Version]
- Garza-Lombó, C.; Posadas, Y.; Quintanar, L.; Gonsebatt, M.E.; Franco, R. Neurotoxicity Linked to Dysfunctional Metal Ion Homeostasis and Xenobiotic Metal Exposure: Redox Signaling and Oxidative Stress. Antioxid. Redox Signal. 2018, 28, 1669–1703. [Google Scholar] [CrossRef] [Green Version]
- Del Rio, R.; Quintanilla, R.A.; Orellana, J.A.; Retamal, M.A. Neuron-Glia Crosstalk in the Autonomic Nervous System and Its Possible Role in the Progression of Metabolic Syndrome: A New Hypothesis. Front. Physiol. 2015, 6, 350. [Google Scholar] [CrossRef] [Green Version]
- Kielian, T. Glial connexins and gap junctions in CNS inflammation and disease. J. Neurochem. 2008, 106, 1000–1016. [Google Scholar] [CrossRef] [PubMed]
- Millhauser, G.L. Copper binding in the prion protein. Acc. Chem. Res. 2004, 37, 79–85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, L.R.; Harris, D.A. Copper and zinc cause delivery of the prion protein from the plasma membrane to a subset of early endosomes and the Golgi. J. Neurochem. 2003, 87, 353–363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Varela-Nallar, L.; Toledo, E.M.; Chacón, M.A.; Inestrosa, N.C. The functional links between prion protein and copper. Biol. Res. 2006, 39, 39–44. [Google Scholar] [CrossRef] [PubMed]
- Fukai, T.; Ushio-Fukai, M.; Kaplan, J.H. Copper transporters and copper chaperones: Roles in cardiovascular physiology and disease. Am. J. Physiol Cell Physiol. 2018, 315, C186–C201. [Google Scholar] [CrossRef]
- Bhattacharjee, A.; Chakraborty, K.; Shukla, A. Cellular copper homeostasis: Current concepts on its interplay with glutathione homeostasis and its implication in physiology and human diseases. Metallomics 2017, 9, 1376–1388. [Google Scholar] [CrossRef]
- Hatori, Y.; Lutsenko, S. The Role of Copper Chaperone Atox1 in Coupling Redox Homeostasis to Intracellular Copper Distribution. Antioxidants 2016, 5, 25. [Google Scholar] [CrossRef] [Green Version]
- Polishchuk, R.; Lutsenko, S. Golgi in copper homeostasis: A view from the membrane trafficking field. Histochem. Cell Biol. 2013, 140, 285–295. [Google Scholar] [CrossRef] [Green Version]
- Kaler, S.G. ATP7A-related copper transport diseases-emerging concepts and future trends. Nat. Rev. Neurol. 2011, 7, 15–29. [Google Scholar] [CrossRef] [Green Version]
- Chang, I.J.; Hahn, S.H. The genetics of Wilson disease. Handb. Clin. Neurol. 2017, 142, 19–34. [Google Scholar]
- De Bie, P.; van de Sluis, B.; Burstein, E.; van de Berghe, P.V.; Muller, P.; Berger, R.; Gitlin, J.D.; Wijmenga, C.; Klomp, L.W. Distinct Wilson’s disease mutations in ATP7B areassociated with enhanced binding to COMMD1 and reduced stability of ATP7B. Gastroenterology 2007, 133, 1316–1326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, C.H.; Yang, N.; Bothe, J.; Tonelli, M.; Nokhrin, S.; Dolgova, N.V.; Braiterman, L.; Lutsenko, S.; Dmitriev, O.Y. The metal chaperone Atox1 regulates the activity of the human copper transporter ATP7B by modulating domain dynamics. J. Biol. Chem. 2017, 292, 18169–18177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shanmugavel, K.P.; Wittung-Stafshede, P. Copper relay path through the N-terminus of Wilson disease protein, ATP7B. Metallomics 2019, 11, 1472–1480. [Google Scholar] [CrossRef] [Green Version]
- Forbes, J.R.; His, G.; Cox, D.W. Role of the copper-binding domain in the copper transport function of ATP7B, the P-type ATPase defective in Wilson disease. J. Biol. Chem. 1999, 274, 12408–12413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goodyer, I.D.; Jones, E.E.; Monaco, A.P.; Francis, M.J. Characterization of the Menkes protein copper-binding domains and their role in copper-induced protein relocalization. Hum. Mol. Genet. 1999, 8, 1473–1478. [Google Scholar] [CrossRef] [Green Version]
- Banci, L.; Bertini, I.; Cantini, F.; Rosenzweig, A.C.; Yatsunyk, L.A. Metal binding domains 3 and 4 of the Wilson disease protein: Solution structure and interaction with the copper(I) chaperone HAH1. Biochemistry 2008, 47, 7423–7429. [Google Scholar] [CrossRef] [Green Version]
- Mohr, I.; Weiss, K.H. Biochemical Markers for the Diagnosis and Monitoring of Wilson Disease. Clin. Biochem. Rev. 2019, 40, 59–77. [Google Scholar]
- Osredkar, J.; Sustar, N. Copper and Zinc, Biological Role and Significance of Copper/Zinc Imbalance. J. Clin. Toxicol. 2011, S3, 001. [Google Scholar] [CrossRef] [Green Version]
- Ruttkay-Nedecky, B.; Nejdl, L.; Gumulec, J.; Zitka, O.; Masarik, M.; Eckschlager, T.; Stiborova, M.; Adam, V.; Kizek, R. The role of metallothionein in oxidative stress. Int. J. Mol. Sci. 2013, 14, 6044–6066. [Google Scholar] [CrossRef] [Green Version]
- Tassabehji, N.M.; VanLandingham, J.W.; Levenson, C.W. Copper alters the conformation and transcriptional activity of the tumor suppressor protein p53 in human Hep G2 cells. Exp. Biol. Med. (Maywood) 2005, 230, 699–708. [Google Scholar] [CrossRef]
- Holley, A.K.; St Clair, D.K. Watching the watcher: Regulation of p53 by mitochondria. Future Oncol. 2009, 5, 117–130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lang, P.A.; Schenck, M.; Nicolay, J.P.; Becker, J.U.; Kempe, D.S.; Lupescu, A.; Koka, S.; Eisele, K.; Klarl, B.A.; Rübben, H.; et al. Liver cell death and anemia in Wilson disease involve acid sphingomyelinase and ceramide. Nat. Med. 2007, 13, 164–170. [Google Scholar] [CrossRef] [PubMed]
- Li, P.L.; Zhang, Y. Cross talk between ceramide and redox signaling: Implications for endothelial dysfunction and renal disease. Handb. Exp. Pharmacol. 2013, 216, 171–197. [Google Scholar]
- Sian, J.; Youdim, M.B.H.; Riederer, P.; Gerlach, M. Hepatolenticular Degeneration: Wilson’s Disease. In Basic Neurochemistry: Molecular, Cellular and Medical Aspects, 6th ed.; Elsevier: Philadelphia, PA, USA, 1999. [Google Scholar]
- Lange, S.C.; Bak, L.K.; Waagepetersen, H.S.; Schousboe, A.; Norenberg, M.D. Primary cultures of astrocytes: Their value in understanding astrocytes in health and disease. Neurochem. Res. 2012, 37, 2569–2588. [Google Scholar] [CrossRef] [Green Version]
- Suárez, I.; Bodega, G.; Fernández, B. Glutamine synthetase in brain: Effect of ammonia. Neurochem. Int. 2002, 41, 123–142. [Google Scholar] [CrossRef]
- Bertrand, E.; Lewandowska, E.; Szpak, G.M.; Hoogenraad, T. Neuropathological analysis of pathological forms of astroglia in Wilson’s disease. Folia Neuropathol. 2001, 39, 73–79. [Google Scholar]
- Xie, J.J.; Wu, Z.Y. Wilson’s Disease in China. Neurosci. Bull. 2017, 33, 323–330. [Google Scholar] [CrossRef]
- Dusek, P.; Litwin, T.; Członkowska, A. Neurologic impairment in Wilson disease. Ann. Transl. Med. 2019, 7 (Suppl. 2), S64. [Google Scholar] [CrossRef]
- Ma, K.C. Alzheimer-type I astrogliopathy (AIA) and its implications for dynamic plasticity of astroglia: A historical review of the significance of AIA. J. Neuropathol. Exp. Neurol. 2001, 60, 121–131. [Google Scholar] [CrossRef] [Green Version]
- Butterworth, R.F. Hepatic encephalopathy—A serious complication of alcoholic liver disease. Alcohol. Res. Health 2003, 27, 143–145. [Google Scholar]
- Verkhratsky, A.; Parpura, V. Recent advances in (patho)physiology of astroglia. Acta Pharm. Sin. 2010, 31, 1044–1054. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agarwal, A.N.; Mais, D.D. Sensitivity and Specificity of Alzheimer Type II Astrocytes in Hepatic Encephalopathy. Arch. Pathol. Lab. Med. 2019, 143, 1256–1258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gomes, F.C.; Paulin, D.; Moura Neto, V. Glial fibrillary acidic protein (GFAP): Modulation by growth factors and its implication in astrocyte differentiation. Braz. J. Med. Biol. Res. 1999, 32, 619–631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sosunov, A.A.; McKhann, G.M., 2nd; Goldman, J.E. The origin of Rosenthal fibers and their contributions to astrocyte pathology in Alexander disease. Acta Neuropathol. Commun. 2017, 5, 27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, D.C.; Morrison, E.H. Histology, Astrocytes. In StatPearls [Internet]; StatPearls Publishing: Treasure Island, FL, USA, 2020. Available online: https://www.ncbi.nlm.nih.gov/books/NBK545142/ (accessed on 3 December 2020).
- Schiweck, J.; Eickholt, B.J.; Murk, K. Important Shapeshifter: Mechanisms Allowing Astrocytes to Respond to the Changing Nervous System during Development, Injury and Disease. Front. Cell Neurosci. 2018, 12, 261. [Google Scholar] [CrossRef] [Green Version]
- Matias, I.; Morgado, J.; Gomes, F.C.A. Astrocyte Heterogeneity: Impact to Brain Aging and Disease. Front. Aging Neurosci. 2019, 11, 59. [Google Scholar] [CrossRef] [Green Version]
- Mossakowski, M.J.; Weinrauder, H. Natural history of Opalski cells. Neuropat. Pol. 1984, 22, 471–481. [Google Scholar]
- Newell, K.L.; Kleinschmidt-De Masters, B.K. Central pontine myelinolysis at autopsy; a twelve year retrospective analysis. J. Neurol. Sci. 1996, 142, 134–139. [Google Scholar] [CrossRef]
- Brewer, G.J.; Dick, R.D.; Yuzbasiyan-Gurkin, V.; Tankanow, R.; Young, A.B.; Kluin, K.J. Initial therapy of patients with Wilson’s disease with tetrathiomolybdate. Arch. Neurol. 1991, 48, 42–47. [Google Scholar] [CrossRef]
- Starzl, T.E.; Iwatsuki, S.; Van Thiel, D.H. Evolution of liver transplantation. Hepatology 1982, 2, 614–636. [Google Scholar] [CrossRef] [Green Version]
- Członkowska, A.; Rodo, M.; Gromadzka, G. Late onset Wilson’s disease: Therapeutic implications. Mov. Disord. 2008, 23, 896–898. [Google Scholar] [CrossRef] [PubMed]
- Ala, A.; Walker, A.P.; Ashkan, K.; Dooley, J.S.; Schilsky, M.L. Wilson’s disease. Lancet 2007, 369, 397–408. [Google Scholar] [CrossRef]
- Sinha, S.; Taly, A.B.; Ravishankar, S.; Prashanth, L.K.; Venugopal, K.S.; Arunodaya, G.R.; Vasudev, M.K.; Swamy, H.S. Wilson’s disease: Cranial MRI observations and clinical correlation. Neuroradiology 2006, 48, 613–621. [Google Scholar] [CrossRef] [PubMed]
- Magalhaes, A.; Caramelli, P.; Menezes, J. Wilson’s disease: MRI with clinical correlation. Neuroradiology 1994, 36, 97–100. [Google Scholar] [CrossRef] [PubMed]
- Prayer, L.; Wimberger, D.; Kramer, J. Cranial MRI in Wilson’s disease. Neuroradiology 1990, 32, 211–214. [Google Scholar] [CrossRef] [PubMed]
- Nazer, H.; Brismar, J.; Al-Kawi, M. Magnetic resonance imaging of the brain in Wilson’s disease. Neuroradiology 1993, 35, 130–133. [Google Scholar] [CrossRef]
- Oder, W.; Prayer, L.; Grimm, G. Wilson’s diasease: Evidence of subgroups derived from clinical findings and brain lesions. Neurology 2004, 43, 120–124. [Google Scholar] [CrossRef]
- Kim, T.J.; Kim, I.O.; Kim, W.S.; Cheon, J.E.; Moon, S.G.; Kwon, J.W.; Seo, J.K.; Yeon, K.M. MR imaging of the brain in Wilson disease of childhood: Findings before and after treatment with clinical correlation. AJNR Am. J. Neuroradiol. 2006, 27, 1373–1378. [Google Scholar]
- Cumings, J.N. The effects of B.A.L. in hepatolenticular degeneration. Brain 1951, 74, 10–22. [Google Scholar] [CrossRef]
- Svetel, M.; Mijajlović, M.; Tomić, A.; Kresojević, N.; Pekmezović, T.; Kostić, V.S. Transcranial sonography in Wilson’s disease. Parkinsonism. Rel. Dis. 2012, 18, 234–238. [Google Scholar] [CrossRef]
- Walter, U.; Skowrońska, M.; Litwin, T.; Szpak, G.M.; Jabłonka-Salach, K.; Skoloudík, D.; Bulska, E.; Członkowska, A. Lenticular nucleus hyperechogenicity in Wilson’s disease reflects local copper, but not iron accumulation. J. Neural. Transm. 2014, 121, 1273–1279. [Google Scholar] [CrossRef] [PubMed]
- Bruehlmeier, M.; Leenders, K.L.; Vontobel, P.; Calonder, C.; Antonini, A.; Weindl, A. Increased cerebral iron uptake in Wilson’s disease: A 52Fe-citrate PET study. J. Nucl. Med. 2000, 41, 778–781. [Google Scholar]
- Danielsen, E.; Ross, B. Magnetic Resonance Spectroscopy Diagnosis of Neurological Diseases; Marcel Dekker: New York, NY, USA, 1999. [Google Scholar]
- Tarnacka, B.; Szeszkowski, W.; Gołębiowski, M.; Członkowska, A. Metabolic changes in 37 newly diagnosed Wilson’s disease patients assessed by Magnetic Resonance Spectroscopy. Park Rel. Dis. 2009, 15, 582–586. [Google Scholar] [CrossRef] [PubMed]
- Van der Hart, M.; Czeh, B.; Biurrun, G.; Michaelis, T.; Watanabe, T.; Natt, O.; Frahm, J.; Fuchs, E. Substance P receptor antagonist an cloimipramine prevent stressinduced alternations in cerebral metabolites, cytogenesis in the dentate gyrus and hippocampal volume. Mol. Psychiatry 2002, 7, 933–941. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tarnacka, B.; Szeszkowski, W.; Gołębiowski, M.; Członkowska, A. MR spectroscopy in monitoring the treatment of Wilson’s disease patients. Mov. Disord. 2008, 23, 1456–1560. [Google Scholar] [CrossRef] [PubMed]
- Rudkin, T.; Arnold, D. Proton magnetic resonance spectroscopy for the diagnosis and management of cerebral disorders. Arch. Neurol. 1999, 56, 919–926. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arciello, M.; Rotilio, G.; Rossi, L. Copper-dependent toxicity in SH-SY5Y neuroblastoma cells involves mitochondrial damage. Biochem. Biophys. Res. Commun. 2005, 327, 454–459. [Google Scholar] [CrossRef]
- Van Cauter, S.; Severino, M.; Ammendola, R.; Van Berkel, B.; Vavro, H.; Van den Hauwe, L.; Rumboldt, Z. Bilateral lesions of the basal ganglia and thalami (central grey matter)-pictorial review. Neuroradiology 2020, 62, 1565–1605. [Google Scholar] [CrossRef]
- Meenakshi-Sundaram, S.; Mahadevan, A.; Taly, A.B.; Arunodaya, G.R.; Swamy, H.S.; Shankar, S.K. Wilson’s disease: A clinico-neuropathological autopsy study. J. Clin. Neurosci. 2008, 15, 409–417. [Google Scholar] [CrossRef]
- Choi, B.S.; Zheng, W. Copper transport to the brain by the blood-brain barrier and blood-CSF barrier. Brain Res. 2009, 1248, 14–21. [Google Scholar] [CrossRef] [Green Version]
- Svetel, M.; Potrebić, A.; Pekmezović, T.; Tomić, A.; Kresojević, N.; Jesić, R.; Dragasević, N.; Kostić, V.S. Neuropsychiatric aspects of treated Wilson’s disease. Parkinsonism Relat. Disord. 2009, 15, 772–775. [Google Scholar] [CrossRef] [PubMed]
- Seniów, J.; Bak, T.; Gajda, J.; Poniatowska, R.; Czlonkowska, A. Cognitive functioning in neurologically symptomatic and asymptomatic forms of Wilson’s disease. Mov. Disord. 2002, 17, 1077–1083. [Google Scholar] [CrossRef] [PubMed]
- Steinberg, H.; Sternlieb, I. Wilson’s Disease. Major Problems in Internal Medicine; Lloyd, H.S., Ed.; W.B. Saunders: Philadelphia, PA, USA, 1984; Volume XXIII. [Google Scholar]
- Bearn, A.G. Agenetical analysis of thirty families with Wilson’s disease (hepatolenticular degeneration). Ann. Hum. Genet. 1960, 24, 33–43. [Google Scholar] [CrossRef] [PubMed]
- Li, D.D.; Zhang, W.; Wang, Z.Y.; Zhao, P. Serum Copper, Zinc, and Iron Levels in Patients with Alzheimer’s Disease: A Meta-Analysis of Case-Control Studies. Front. Aging Neurosci. 2017, 9, 300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bagheri, S.; Squitti, R.; Haertlé, T.; Siotto, M.; Saboury, A.A. Role of Copper in the Onset of Alzheimer’s Disease Compared to Other Metals. Front. Aging Neurosci. 2018, 9, 446. [Google Scholar] [CrossRef] [PubMed]
- Hordyjewska, A.; Popiołek, Ł.; Kocot, J. The many “faces” of copper in medicine and treatment. Biometals 2014, 27, 611–621. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Zhao, B. Oxidative stress and the pathogenesis of Alzheimer’s disease. Oxid. Med. Cell Longev. 2013, 2013, 316523. [Google Scholar] [CrossRef] [Green Version]
- Singh, S.K.; Srivastav, S.; Yadav, A.K.; Srikrishna, S.; Perry, G. Overview of Alzheimer’s Disease and Some Therapeutic Approaches Targeting Aβ by Using Several Synthetic and Herbal Compounds. Oxid. Med. Cell Longev. 2016, 2016, 7361613. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.L.; Fan, Y.G.; Yang, Z.S.; Wang, Z.Y.; Guo, C. Iron and Alzheimer’s Disease: From Pathogenesis to Therapeutic Implications. Front. Neurosci. 2018, 12, 632. [Google Scholar] [CrossRef] [Green Version]
- Maynard, C.J.; Bush, A.I.; Masters, C.L.; Cappai, R.; Li, Q.X. Metals and amyloid-beta in Alzheimer’s disease. Int. J. Exp. Pathol. 2005, 86, 147–159. [Google Scholar] [CrossRef]
- Kim, A.C.; Lim, S.; Kim, Y.K. Metal Ion Effects on Aβ and Tau Aggregation. Int. J. Mol. Sci. 2018, 19, 128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, D.; Zhang, L.; Grant, G.P.; Dudzik, C.G.; Chen, S.; Patel, S.; Hao, Y.; Millhauser, G.L.; Zhou, F. The elevated copper binding strength of amyloid-β aggregates allows the sequestration of copper from albumin: A pathway to accumulation of copper in senile plaques. Biochemistry 2013, 52, 547–556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gu, M.; Bode, D.C.; Viles, J.H. Copper Redox Cycling Inhibits Aβ Fibre Formation and promotes Fibre Fragmentation, while Generating a Dityrosine Aβ Dimer. Sci. Rep. 2018, 8, 16190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.P.; Falangola, M.F.; Nixon, R.A.; Duff, K.; Helpern, J.A. Visualization of beta-amyloid plaques in a transgenic mouse model of Alzheimer’s disease using MR microscopy without contrast reagents. Magn. Reson. Med. 2004, 52, 538–544. [Google Scholar] [CrossRef] [Green Version]
- James, S.A.; Churches, Q.I.; de Jonge, M.D.; Birchall, I.E.; Streltsov, V.; McColl, G.; Adlard, P.A.; Hare, D.J. Iron, Copper, and Zinc Concentration in Aβ Plaques in the APP/PS1 Mouse Model of Alzheimer’s Disease Correlates with Metal Levels in the Surrounding Neuropil. ACS Chem. Neurosci. 2017, 8, 629–637. [Google Scholar] [CrossRef]
- Cheignon, C.; Tomas, M.; Bonnefont-Rousselot, D.; Faller, P.; Hureau, C.; Collin, F. Oxidative stress and the amyloid beta peptide in Alzheimer’s disease. Redox. Biol. 2018, 14, 450–464. [Google Scholar] [CrossRef]
- Cole, S.L.; Vassar, R. The Basic Biology of BACE1: A Key Therapeutic Target for Alzheimer’s Disease. Curr. Genom. 2007, 8, 509–530. [Google Scholar]
- Kaden, D.; Bush, A.I.; Danzeisen, R.; Bayer, T.A.; Multhaup, G. Disturbed copper bioavailability in Alzheimer’s disease. Int. J. Alzheimers Dis. 2011, 2011, 345614. [Google Scholar] [CrossRef] [Green Version]
- Zheng, Z.; White, C.; Lee, J.; Peterson, T.S.; Bush, A.I.; Sun, G.Y.; Weisman, G.A.; Petris, M.J. Altered microglial copper homeostasis in a mouse model of Alzheimer’s disease. J. Neurochem. 2010, 114, 1630–1638. [Google Scholar] [CrossRef] [Green Version]
- Acevedo, K.M.; Hung, Y.H.; Dalziel, A.H.; Li, Q.X.; Laughton, K.; Wikhe, K.; Rembach, A.; Roberts, B.; Masters, C.L.; Bush, A.I.; et al. Copper promotes the trafficking of the amyloid precursor protein. J. Biol. Chem. 2011, 286, 8252–8262. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Zhu, M.; Hjorth, E.; Cortés-Toro, V.; Eyjolfsdottir, H.; Graff, C.; Nennesmo, I.; Palmblad, J.; Eriksdotter, M.; Sambamurti, K.; et al. Resolution of inflammation is altered in Alzheimer’s disease. Alzheimers Dement. 2015, 11, 40–50. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.S.; Kim, H.; Brahim, J.S.; Rowan, J.; Lee, G.; Dionne, R.A. Acetaminophen selectively suppresses peripheral prostaglandin E2 release and increases COX-2 gene expression in a clinical model of acute inflammation. Pain 2007, 129, 279–286. [Google Scholar] [CrossRef]
- Osaki, S.; Johnson, D.A.; Frieden, E. The possible significance of the ferrous oxidase activity of ceruloplasmin in normal human serum. J. Biol. Chem. 1966, 241, 2746–2751. [Google Scholar]
- Nemeth, E.; Ganz, T. The role of hepcidin in iron metabolism. Acta Haematol. 2009, 122, 78–86. [Google Scholar] [CrossRef] [Green Version]
- Venti, A.; Giordano, T.; Eder, P.; Bush, A.I.; Lahiri, D.K.; Greig, N.H.; Rogers, J.T. The integrated role of desferrioxamine and phenserine targeted to an iron-responsive element in the APP-mRNA 5’-untranslated region. Ann. N. Y. Acad. Sci. 2004, 1035, 34–48. [Google Scholar] [CrossRef]
- Myhre, O.; Utkilen, H.; Duale, N.; Brunborg, G.; Hofer, T. Metal dyshomeostasis and inflammation in Alzheimer’s and Parkinson’s diseases: Possible impact of environmental exposures. Oxid. Med. Cell. Longev. 2013, 726954. [Google Scholar] [CrossRef] [Green Version]
- Wei, H.; Frei, B.; Beckman, J.S.; Zhang, W.J. Copper chelation by tetrathiomolybdate inhibits lipopolysaccharide-induced inflammatory responses in vivo. Am. J. Physiol. Heart Circ. Physiol. 2011, 301, H712–H720. [Google Scholar] [CrossRef]
- Wang, C.Y.; Xie, J.W.; Xu, Y.; Wang, T.; Cai, J.H.; Wang, X.; Zhao, B.L.; An, L.; Wang, Z.Y. Trientine reduces BACE1 activity and mitigates amyloidosis via the AGE/RAGE/NF-κB pathway in a transgenic mouse model of Alzheimer’s disease. Antioxid. Redox. Signal. 2013, 19, 2024–2039. [Google Scholar] [CrossRef] [Green Version]
- White, A.R.; Multhaup, G.; Maher, F.; Bellingham, S.; Camakaris, J.; Zheng, H.; Bush, A.I.; Beyreuther, K.; Masters, C.L.; Cappai, R. The Alzheimer’s disease amyloid precursor protein modulates copper-induced toxicity and oxidative stress in primary neuronal cultures. J. Neurosci. 1999, 19, 9170–9179. [Google Scholar] [CrossRef] [Green Version]
- Squitti, R.; Rossini, P.M.; Cassetta, E.; Moffa, F.; Pasqualetti, P.; Cortesi, M.; Colloca, A.; Rossi, L.; Finazzi-Agró, A. D-penicillamine reduces serum oxidative stress in Alzheimer’s disease patients. Eur. J. Clin. Investig. 2002, 32, 51–59. [Google Scholar] [CrossRef]
- Wojsiat, J.; Zoltowska, K.M.; Laskowska-Kaszub, K.; Wojda, U. Oxidant/Antioxidant Imbalance in Alzheimer’s Disease: Therapeutic and Diagnostic Prospects. Oxid. Med. Cell Longev. 2018, 2018, 6435861. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uversky, V.N.; Eliezer, D. Biophysics of Parkinson’s disease: Structure and aggregation of alpha-synuclein. Curr. Protein Pept. Sci. 2009, 10, 483–499. [Google Scholar] [CrossRef] [PubMed]
- Stefanis, L. α-Synuclein in Parkinson’s disease. Cold Spring Harb. Perspect. Med. 2012, 2, a009399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hijaz, B.A.; Volpicelli-Daley, L.A. Initiation and propagation of α-synuclein aggregation in the nervous system. Mol. Neurodegener. 2020, 15, 19. [Google Scholar] [CrossRef]
- Spillantini, M.G.; Schmidt, M.L.; Lee, V.M.; Trojanowski, J.Q.; Jakes, R.; Goedert, M. Alpha-synuclein in Lewy bodies. Nature 1997, 388, 839–840. [Google Scholar] [CrossRef]
- Sung, Y.H.; Rospigliosi, C.; Eliezer, D. Nmr mapping of copper binding sites in alpha-synuclein. Biochim. Biophys. Acta 2006, 1764, 5–12. [Google Scholar] [CrossRef]
- McDowall, J.S.; Brown, D.R. Alpha-synuclein: Relating metals to structure, function and inhibition. Metallomics 2016, 8, 385–397. [Google Scholar] [CrossRef]
- Rasia, R.M.; Bertoncini, C.W.; Marsh, D.; Hoyer, W.; Cherny, D.; Zweckstetter, M.; Griesinger, C.; Jovin, T.M.; Fernandez, C.O. Structural characterization of copper(ii) binding to alpha-synuclein: Insights into the bioinorganic chemistry of parkinson’s disease. Proc. Natl. Acad. Sci. USA 2005, 102, 4294–4299. [Google Scholar] [CrossRef] [Green Version]
- Gaetke, L.M.; Chow-Johnson, H.S.; Chow, C.K. Copper: Toxicological relevance and mechanisms. Arch. Toxicol. 2014, 88, 1929–1938. [Google Scholar] [CrossRef]
- Filograna, R.; Beltramini, M.; Bubacco, L.; Bisaglia, M. Anti-Oxidants in Parkinson’s Disease Therapy: A Critical Point of View. Curr. Neuropharmacol. 2016, 14, 260–271. [Google Scholar] [CrossRef] [Green Version]
- Aracena, P.; Aguirre, P.; Muñoz, P.; Núñez, M.T. Iron and glutathione at the crossroad of redox metabolism in neurons. Biol. Res. 2006, 39, 157–165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smeyne, M.; Smeyne, R.J. Glutathione metabolism and Parkinson’s disease. Free Radic. Biol. Med. 2013, 62, 13–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dodani, S.C.; Domaille, D.W.; Nam, C.I.; Miller, E.W.; Finney, L.A.; Vogt, S.; Chang, C.J. Calcium-dependent copper redistributions in neuronal cells revealed by a fluorescent copper sensor and X-ray fluorescence microscopy. Proc. Natl. Acad. Sci. USA 2011, 108, 5980–5985. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monzani, E.; Nicolis, S.; Dell’Acqua, S.; Capucciati, A.; Bacchella, C.; Zucca, F.A.; Mosharov, E.V.; Sulzer, D.; Zecca, L.; Casella, L. Dopamine, Oxidative Stress and Protein-Quinone Modifications in Parkinson’s and Other Neurodegenerative Diseases. Angew. Chem. Int. Ed. Engl. 2019, 58, 6512–6527. [Google Scholar] [CrossRef] [PubMed]
- Deas, E.; Cremades, N.; Angelova, P.R.; Ludtmann, M.H.; Yao, Z.; Chen, S.; Horrocks, M.H.; Banushi, B.; Little, D.; Devine, M.J.; et al. Alpha-synuclein oligomers interact with metal ions to induce oxidative stress and neuronal death in parkinson’s disease. Antioxid. Redon. Signal. 2016, 24, 376–391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dias, V.; Junn, E.; Mouradian, M.M. The role of oxidative stress in Parkinson’s disease. J. Parkinsons Dis. 2013, 3, 461–491. [Google Scholar] [CrossRef] [Green Version]
- Bisaglia, M.; Filograna, R.; Beltramini, M.; Bubacco, L. Are dopamine derivatives implicated in the pathogenesis of Parkinson’s disease? Ageing Res. Rev. 2014, 13, 107–114. [Google Scholar] [CrossRef]
- Mosharov, E.V.; Borgkvist, A.; Sulzer, D. Presynaptic effects of levodopa and their possible role in dyskinesia. Mov. Disord. 2015, 30, 45–53. [Google Scholar] [CrossRef] [Green Version]
- Palumbo, A.; d’Ischia, M.; Misuraca, G.; Prota, G. Effect of metal ions on the rearrangement of dopachrome. Biochim. Biophys. Acta 1987, 925, 203–209. [Google Scholar] [CrossRef]
- Pham, A.N.; Waite, T.D. Cu(ii)-catalyzed oxidation of dopamine in aqueous solutions: Mechanism and kinetics. J. Inorg. Biochem. 2014, 137, 74–84. [Google Scholar] [CrossRef]
- Bhattacharyya, A.; Chattopadhyay, R.; Mitra, S.; Crowe, S.E. Oxidative stress: An essential factor in the pathogenesis of gastrointestinal mucosal diseases. Physiol. Rev. 2014, 94, 329–354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gromadzka, G.; Członkowska, A. Influence of IL-1RN intron 2 variable number of tandem repeats (VNTR) polymorphism on the age at onset of neuropsychiatric symptoms in Wilson’s disease. Int. J. Neurosci. 2011, 121, 8–15. [Google Scholar] [CrossRef] [PubMed]
- Oldreive, C.E.; Doherty, G.H. Neurotoxic effects of homocysteine on cerebellar Purkinje neurons in vitro. Neurosci. Lett. 2007, 413, 52–57. [Google Scholar] [CrossRef] [PubMed]
- Carrasco-Pozo, C.; Alvarez-Lueje, A.; Olea-Azar, C.; López-Alarcón, C.; Speisky, H. In vitro interaction between homocysteine and copper ions: Potential redox implications. Exp. Biol. Med. (Maywood) 2006, 231, 1569–1575. [Google Scholar] [CrossRef]
- White, A.R.; Huang, X.; Jobling, M.F.; Barrow, C.J.; Beyreuther, K.; Masters, C.L.; Bush, A.I.; Cappai, R. Homocysteine potentiates copper-and amyloid beta peptide-mediated toxicity in primary neuronal cultures: Possible risk factors in the Alzheimer’s-type neurodegenerative pathways. J. Neurochem. 2001, 76, 1509–1520. [Google Scholar] [CrossRef] [Green Version]
- Linnebank, M.; Lutz, H.; Jarre, E.; Vielhaber, S.; Noelker, C.; Struys, E.; Jakobs, C.; Klockgether, T.; Evert, B.O.; Kunz, W.S.; et al. Binding of copper is a mechanism of homocysteine toxicity leading to COX deficiency and apoptosis in primary neurons, PC12 and SHSY-5Y cells. Neurobiol. Dis. 2006, 23, 725–730. [Google Scholar] [CrossRef]
- Van der Put, N.M.; Gabreëls, F.; Stevens, E.M.; Smeitink, J.A.; Trijbels, F.J.; Eskes, T.K.; Van den Heuvel, L.P.; Blom, H.J. A second common mutation in the methylenetetrahydrofolate reductase gene: An additional risk factor for neural-tube defects? Am. J. Hum. Genet. 1998, 62, 1044–1051. [Google Scholar] [CrossRef] [Green Version]
- Ravin, H.A. An improved colorimetric enzymatic assay of ceruloplasmin. J. Lab. Clin. Med. 1961, 58, 161–168. [Google Scholar]
- Gromadzka, G.; Rudnicka, M.; Chabik, G.; Przybyłkowski, A.; Członkowska, A. Genetic variability in the methylenetetrahydrofolate reductase gene (MTHFR) affects clinical expression of Wilson’s disease. J. Hepatol. 2011, 55, 913–919. [Google Scholar] [CrossRef]
- Litwin, T.; Gromadzka, G.; Członkowska, A. Apolipoprotein E gene (APOE) genotype in Wilson’s disease: Impact on clinical presentation. Parkinsonism Relat. Disord. 2012, 18, 367–369. [Google Scholar] [CrossRef]
- Gromadzka, G.; Karpińska, A.; Przybyłkowski, A.; Litwin, T.; Wierzchowska-Ciok, A.; Dzieżyc, K.; Chabik, G.; Członkowska, A. Treatment with D-penicillamine or zinc sulphate affects copper metabolism and improves but not normalizes antioxidant capacity parameters in Wilson disease. Biometals 2014, 27, 207–215, Erratum in 2014, 27, 217–215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gromadzka, G.; Kruszyńska, M.; Wierzbicka, D.; Litwin, T.; Dzieżyc, K.; Wierzchowska-Ciok, A.; Chabik, G.; Członkowska, A. Gene variants encoding proteins involved in antioxidant defense system and the clinical expression of Wilson disease. Liver Int. 2015, 35, 215–222. [Google Scholar] [CrossRef] [PubMed]
- Gromadzka, G.; Wierzbicka, D.; Litwin, T.; Przybylkowski, A. Effect of homeostatic iron regulator protein gene mutation on Wilson’s disease clinical manifestation: Original data and literature review. Int. J. Neurosci. 2020, 1–11. [Google Scholar] [CrossRef]
- Rivera-Mancía, S.; Pérez-Neri, I.; Ríos, C.; Tristán-López, L.; Rivera-Espinosa, L.; Montes, S. The transition metals copper and iron in neurodegenerative diseases. Chem. Biol. Interact. 2010, 186, 184–199. [Google Scholar] [CrossRef]
- Kodama, H.; Fujisawa, C.; Bhadhprasit, W. Inherited Copper Transport Disorders: Biochemical Mechanisms, Diagnosis, and Treatment. Curr. Drug Metab. 2012, 13, 237–250. [Google Scholar] [CrossRef] [Green Version]
- Scheiber, I.F.; Schmidt, M.M.; Dringen, R. Copper export from cultured astrocytes. Neurochem. Int. 2012, 60, 292–300. [Google Scholar] [CrossRef]
- Tümer, Z.; Møller, L.B. Menkes disease. Eur. J. Hum. Genet. 2010, 18, 511–518. [Google Scholar] [CrossRef] [Green Version]
- Bellingham, S.A.; Ciccotosto, G.D.; Needham, B.E.; Fodero, L.R.; White, A.R.; Masters, C.L.; Cappai, R.; Camakaris, J. Gene knockout of amyloid precursor protein and amyloid precursor-like protein-2 increases cellular copper levels in primary mouse cortical neurons and embryonic fibroblasts. J. Neurochem. 2004, 91, 423–428. [Google Scholar] [CrossRef]
- Kessler, H.; Pajonk, F.G.; Meisser, P.; Schneider-Axmann, T.; Hoffmann, K.H.; Supprian, T.; Herrmann, W.; Obeid, R.; Multhaup, G.; Falkai, P.; et al. Cerebrospinal fluid diagnostic markers correlate with lower plasma copper and ceruloplasmin in patients with Alzheimer’s disease. J. Neural. Transm (Vienna) 2006, 113, 1763–1769. [Google Scholar] [CrossRef]
- Roberti Mdo, R.; Borges Filho, H.M.; Gonçalves, C.H.; Lima, F.L. Aceruloplasminemia: A rare disease—Diagnosis and treatment of two cases. Rev. Bras. Hematol. Hemoter. 2011, 33, 389–392. [Google Scholar] [CrossRef] [Green Version]
- Nishiyama, K.; Murayama, S.; Shimizu, J.; Ohya, Y.; Kwak, S.; Asayama, K.; Kanazawa, I. Cu/zn superoxide dismutase-like immunoreactivity is present in lewy bodies from parkinson disease: A light and electron microscopic immunocytochemical study. Acta Neuropathol. 1995, 89, 471–474. [Google Scholar] [CrossRef] [PubMed]
- Trist, B.G.; Davies, K.M.; Cottam, V.; Genoud, S.; Ortega, R.; Roudeau, S.; Carmona, A.; De Silva, K.; Wasinger, V.; Lewis, S.J.G.; et al. Amyotrophic lateral sclerosis-like superoxide dismutase 1 proteinopathy is associated with neuronal loss in parkinson’s disease brain. Acta Neuropathol. 2017, 134, 113–127. [Google Scholar] [CrossRef] [PubMed]
- Roudeau, S.; Chevreux, S.; Carmona, A.; Ortega, R. Reduced net charge and heterogeneity of pi isoforms in familial amyotrophic lateral sclerosis mutants of copper/zinc superoxide dismutase. Electrophoresis 2015, 36, 2482–2488. [Google Scholar] [CrossRef] [PubMed]
- Lu, J. Triethylenetetramine pharmacology and its clinical applications. Mol. Cancer 2010, 9, 2458–2467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tisato, F.; Marzano, C.; Porchia, M.; Pellei, M.; Santini, C. Copper in diseases and treatments, and copper-based anticancer strategies. Med. Res. Rev. 2010, 30, 708–749. [Google Scholar] [CrossRef]
- Janssen, R.; de Brouwer, B.; von der Thüsen, J.H.; Wouters, E.F.M. Copper as the most likely pathogenic divergence factor between lung fibrosis and emphysema. Med. Hypotheses 2018, 120, 49–54. [Google Scholar] [CrossRef]
- Gil-Bea, F.J.; Aldanondo, G.; Lasa-Fernández, H.; de Munain, A.L.; Vallejo-Illarramendi, A. Insights into the mechanisms of copper dyshomeostasis in amyotrophic lateral sclerosis. Expert. Rev. Mol. Med. 2017, 19, e7. [Google Scholar] [CrossRef] [Green Version]
- Lowe, J.; Taveira-da-Silva, R.; Hilário-Souza, E. Dissecting copper homeostasis in diabetes mellitus. IUBMB Life 2017, 69, 255–262. [Google Scholar] [CrossRef] [Green Version]
- Van Horssen, J.; Witte, M.E.; Schreibelt, G.; De Vries, H.E. Radical changes in multiple sclerosis pathogenesis. Biochim. Biophys. Acta 2011, 1812, 141–150. [Google Scholar] [CrossRef] [Green Version]
- Tosato, M.; Di Marco, V. Metal Chelation Therapy and Parkinson’s Disease: A Critical Review on the Thermodynamics of Complex Formation between Relevant Metal Ions and Promising or Established Drugs. Biomolecules 2019, 9, 269. [Google Scholar] [CrossRef] [Green Version]
- Ding, W.Q.; Lind, S.E. Metal ionophores—An emerging class of anticancer drugs. IUBMB Life 2009, 61, 1013–1018. [Google Scholar] [CrossRef] [PubMed]
- Denoyer, D.; Pearson, H.B.; Clatworthy, S.A.; Smith, Z.M.; Francis, P.S.; Llanos, R.M.; Volitakis, I.; Phillips, W.A.; Meggyesy, P.M.; Masaldan, S.; et al. Copper as a target for prostate cancer therapeutics: Copper-ionophore pharmacology and altering systemic copper distribution. Oncotarget 2016, 7, 37064. [Google Scholar] [CrossRef] [Green Version]
- Yu, Y.; Suryo Rahmanto, Y.; Richardson, D.R. Bp44mT: An orally active iron chelator of the thiosemicarbazone class with potent anti-tumour efficacy. Br. J. Pharmacol. 2012, 165, 148–166. [Google Scholar] [CrossRef] [Green Version]
- Park, K.C.; Fouani, L.; Jansson, P.J.; Wooi, D.; Sahni, S.; Lane, D.J.; Palanimuthu, D.; Lok, H.C.; Kovacevic, Z.; Huang, M.L.; et al. Copper and conquer: Copper complexes of di-2-pyridylketone thiosemicarbazones as novel anti-cancer therapeutics. Metallomics 2016, 8, 874–886. [Google Scholar] [CrossRef] [PubMed]
- McCance, R.A.; Widdowson, E.M. Observations on the administration of BAL-Intrav to man. Nature 1946, 157, 837. [Google Scholar] [CrossRef]
- Walshe, J. Penicillamine a new oral therapy for Wilson’s disease. Am. J. Med. 1956, 21, 487–495. [Google Scholar] [CrossRef]
- Brewer, G.; Askari, F.; Lorincz, M.; Carlson, M.; Schilsky, M.; Kluin, K.; Hedera, P.; Moretti, P.; Fink, J.; Tankanow, R.; et al. Treatment of Wilson Disease with Ammonium Tetrathiomolybdate: IV. Comparison of Tetrathiomolybdate and Trientine in a Double-Blind Study of Treatment of the Neurologic Presentation of Wilson Disease. Arch. Neurol. 2006; 63, 521–527. [Google Scholar]
- Brewer, G.J.; Terry, C.A.; Aisen, A.M.; Hill, G.M. Worsening of neurologic syndrome in patients with Wilson’s disease with initial penicillamine therapy. Arch. Neurol. 1987, 44, 490–493. [Google Scholar] [CrossRef]
- Chen, D.B.; Feng, L.; Lin, X.P.; Zhang, W.; Li, F.R.; Liang, X.L.; Li, X.H. Penicillamine Increases Free Copper and Enhances Oxidative Stress in the Brain of Toxic Milk Mice. PLoS ONE 2012, 7, e37709. [Google Scholar]
- Weiss, K.H.; Członkowska, A.; Hedera, P.; Ferenci, P. WTX101–An investigational drug for the treatment of Wilson disease. Expert Opin. Investig. Drugs 2018, 27, 561–567. [Google Scholar] [CrossRef]
- Sensi, S.L.; Granzotto, A.; Siotto, M.; Squitti, R. Copper and zinc dysregulation in Alzheimer’s disease. Trends Pharm. Sci. 2018, 39, 1049–1063. [Google Scholar] [CrossRef]
- Lovell, M.A.; Robertson, J.D.; Teesdale, W.J.; Campbell, J.L.; Markesbery, W.R. Copper, iron and zinc in Alzheimer’s disease senile plaques. J. Neurol. Sci. 1998, 158, 47–52. [Google Scholar] [CrossRef]
- Ribaric, S. Peptides as Potential Therapeutics for Alzheimer’s Disease. Molecules 2018, 23, 283. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Wang, Y.; Li, W.; Mao, F.; Sun, Y.; Huang, L.; Li, X. Design, Synthesis, and Evaluation of Multitarget-Directed Selenium-Containing Clioquinol Derivatives for the Treatment of Alzheimer’s Disease. ACS Chem. Neurosci. 2014, 5, 952–962. [Google Scholar] [CrossRef]
- Ejaz, H.; Wang, W.; Lang, M. Copper Toxicity Links to Pathogenesis of Alzheimer’s Disease and Therapeutics Approaches. Int. J. Mol. Sci. 2020, 21, 7660. [Google Scholar] [CrossRef]
- Bush, A.I. Drug development based on the metals hypothesis of Alzheimer’s disease. J. Alzheimers Dis. 2008, 15, 223–240. [Google Scholar] [CrossRef] [Green Version]
- Adlard, P.A.; Cherny, R.A.; Finkelstein, D.I.; Gautier, E.; Robb, E.; Cortes, M.; Volitakis, I.; Liu, X.; Smith, J.P.; Perez, K.; et al. Rapid Restoration of Cognition in Alzheimer’s Transgenic Mice with 8-Hydroxy Quinoline Analogs Is Associated with Decreased Interstitial Aβ. Neuron 2008, 59, 43–55. [Google Scholar] [CrossRef] [Green Version]
- Hegde, M.L.; Bharathi, P.; Suram, A.; Venugopal, C.; Jagannathan, R.; Poddar, P.; Srinivas, P.; Sambamurti, K.; Rao, K.J.; Scancar, J.; et al. Challenges Associated with Metal. Chelation Therapy in Alzheimer’s Disease. J. Alzheimers Dis. 2009, 17, 457–468. [Google Scholar]
- Drew, S.C. The Case for Abandoning Therapeutic Chelation of Copper Ions in Alzheimer’s Disease. Front. Neurosci. 2017, 11, 317. [Google Scholar] [CrossRef] [Green Version]
- Jenagaratnam, L.; McShane, R. Clioquinol for the treatment of Alzheimer’s Disease. Cochrane Database Syst. Rev. 2006, CD005380. [Google Scholar]
- Sampson, E.L.; Jenagaratnam, L.; McShane, R. Metal protein attenuating compounds for the treatment of Alzheimer’s disease. Cochrane Database Syst. Rev. 2008, CD005380. [Google Scholar]
- Sampson, E.L.; Jenagaratnam, L.; McShane, R. Metal protein attenuating compounds for the treatment of Alzheimer’s dementia. Cochrane Database Syst. Rev. 2014, CD005380. [Google Scholar] [CrossRef]
- Ryan, T.M.; Roberts, B.R.; McColl, G.; Hare, D.J.; Doble, P.A.; Li, Q.X.; Lind, M.; Roberts, A.M.; Mertens, H.D.T.; Kirby, N.; et al. Stabilization of Nontoxic Aβ-Oligomers: Insights into the Mechanism of Action of Hydroxyquinolines in Alzheimer’s Disease. J. Neurosci. 2015, 35, 2871–2884. [Google Scholar] [CrossRef] [Green Version]
- Barnham, K.J.; Bush, A.I. Biological metals and metal-targeting compounds in major neurodegenerative diseases. Chem. Soc. Rev. 2014, 43, 6727–6749. [Google Scholar] [CrossRef] [Green Version]
- Kim, W.S.; Kågedal, K.; Halliday, G.M. Alpha-synuclein biology in Lewy body diseases. Alzheimers Res. 2014, 6, 73. [Google Scholar] [CrossRef] [Green Version]
- Miotto, M.C.; Rodriguez, E.E.; Valiente-Gabioud, A.A.; Torres-Monserrat, V.; Binolfi, A.; Quintanar, L.; Zweckstetter, M.; Griesinger, C.; Fernández, C.O. Site-specific copper-catalyzed oxidation of α-synuclein:Tightening the link between metal binding and protein oxidative damage in Parkinson’s disease. Inorg. Chem. 2014, 53, 4350–4358. [Google Scholar] [CrossRef]
- Ajsuvakova, O.P.; Tinkov, A.A.; Willkommen, D.; Skalnaya, A.A.; Danilov, A.B.; Pilipovich, A.A.; Aschner, M.; Skalny, A.V.; Michalke, B.; Skalnaya, M.G. Assessment of copper, iron, zinc and manganese status and speciation in patients with Parkinson’s disease: A pilot study. J. Trace Elem. Med. Biol. 2019, 59, 126423. [Google Scholar] [CrossRef]
- Davies, K.M.; Bohic, S.; Carmona, A.; Ortega, R.; Cottam, V.; Hare, D.J.; Finberg, J.P.M.; Reyes, S.; Halliday, G.M.; Mercer, J.F.B.; et al. Copper pathology in vulnerable brain regions in Parkinson’s disease. Neurobiol. Aging 2014, 35, 858–866. [Google Scholar] [CrossRef] [Green Version]
- Maher, P.; Kontoghiorghes, G.J. Characterization of the neuroprotective potential of derivatives of the iron chelating drug deferiprone. Neurochem. Res. 2015, 40, 609–620. [Google Scholar] [CrossRef]
- Timoshnikov, V.A.; Kobzeva, T.; Selyutina, O.Y.; Polyakov, N.E.; Kontoghiorghes, G.J. Effective inhibition of copper-catalyzed production of hydroxyl radicals by deferiprone. J. Biol. Inorg. Chem. 2019, 24, 331–341. [Google Scholar] [CrossRef]
- Cherny, R.A.; Atwood, C.S.; Xilinas, M.E.; Gray, D.N.; Jones, W.D.; McLean, C.A.; Barnham, K.J.; Volitakis, I.; Fraser, F.W.; Kim, Y.; et al. Treatment with a copper-zinc chelator markedly and rapidly inhibits β-amyloid accumulation in Alzheimer’s disease transgenic mice. Neuron 2001, 30, 665–676. [Google Scholar] [CrossRef] [Green Version]
- Torkildsen, Ø.; Brunborg, L.; Myhr, K.M.; Bø, L. The cuprizone model for demyelination. Acta Neurol. Scand. 2008, 117, 72–76. [Google Scholar] [CrossRef]
- Pashalidis, I.; Kontoghiorghes, G.J. Molecular factors affecting the complex formation between deferiprone (L1) and Cu (II). Possible implications on efficacy and toxicity. Arzneimittelforschung 2001, 51, 998–1003. [Google Scholar]
- Leung, M.H.; Harada, T.; Kee, T.W. Delivery of curcumin and medicinal effects of the copper (II)–curcumin complexes. Curr. Pharm. Des. 2013, 19, 2070–2083. [Google Scholar]
- Grubman, A.; White, A.R. Copper as a key regulator of cell signalling pathways. Expert Rev. Mol. Med. 2014, 16, e11. [Google Scholar] [CrossRef]
- Donsante, A.; Yi, L.; Zerfas, P.; Brinster, L.R.; Sullivan, P.; Goldstein, D.S.; Prohaska, J.; Centeno, J.A.; Rushing, E.; Kaler, S.G. ATP7A gene addition to the choroid plexus results in long-term rescue of the lethal copper transport defect in a Menkes disease mouse model. Mol. Ther. 2011, 19, 2114–2123. [Google Scholar] [CrossRef] [Green Version]
- McAllum, E.J.; Lim, N.K.; Hickey, J.L.; Paterson, B.M.; Donnelly, P.S.; Li, Q.X.; Liddell, J.R.; Barnham, K.J.; White, A.R.; Crouch, P.J. Therapeutic effects of CuII (atsm) in the SOD1-G37R mouse model of amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. Front. Degener. 2013, 14, 586–590. [Google Scholar] [CrossRef]
- Williams, J.R.; Trias, E.; Beilby, P.R.; Lopez, N.I.; Labut, E.M.; Bradford, C.S.; Roberts, B.R.; McAllum, E.J.; Crouch, P.J.; Rhoads, T.W.; et al. Copper delivery to the CNS by CuATSM effectively treats motor neuron disease in SOD G93A mice co-expressing the Copper-Chaperone-for-SOD. Neurobiol. Dis. 2016, 89, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Tokuda, E.; Ono, S.; Ishige, K.; Watanabe, S.; Okawa, E.; Ito, Y.; Suzuki, T. Ammonium tetrathiomolybdate delays onset, prolongs survival, and slows progression of disease in a mouse model for amyotrophic lateral sclerosis. Exp. Neurol. 2008, 213, 122–128. [Google Scholar] [CrossRef]
- Hottinger, A.F.; Fine, E.G.; Gurney, M.E.; Zurn, A.D.; Aebischer, P. The copper chelator D- penicillamine delays onset of disease and extends survival in a transgenic mouse model of familial amyotrophic lateral sclerosis. Eur. J. Neurosci. 1997, 9, 1548–1551. [Google Scholar] [CrossRef]
- Andreassen, O.A.; Dedeoglu, A.; Friedlich, A.; Ferrante, K.L.; Hughes, D.; Szabo, C.; Beal, M.F. Effects of an inhibitor of poly (ADP-ribose) polymerase, desmethylselegiline, trientine, and lipoic acid in transgenic ALS mice. Exp. Neurol. 2001, 168, 419–424. [Google Scholar] [CrossRef]
- Azzouz, M. Prevention of mutant SOD1 motoneuron degeneration by copper chelators in vitro. J. Neurobiol. 2000, 42, 49–55. [Google Scholar] [CrossRef]
- Kessler, H.; Pajonk, F.-G.; Bach, D.; Schneider-Axmann, T.; Falkai, P.; Herrmann, W.; Multhaup, G.; Wiltfang, J.; Schäfer, S.; Wirths, O.; et al. Effect of copper intake on CSF parameters in patients with mild Alzheimer’s disease: A pilot phase 2 clinical trial. J. Neural Transm. 2008, 115, 1181–1187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Faux, N.G.; Ritchie, C.W.; Gunn, A.; Rembach, A.; Tsatsanis, A.; Bedo, J.; Harrison, J.; Lannfelt, L.; Blennow, K.; Zetterberg, H.; et al. PBT2 rapidly improves cognition in Alzheimer’s Disease: Additional phase II analyses. J. Alzheimers Dis. 2010, 20, 509–516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lannfelt, L.; Blennow, K.; Zetterberg, H.; Batsman, S.; Ames, D.; Harrison, J.; Masters, C.L.; Targum, S.; Bush, A.I.; Murdoch, R.; et al. Safety, efficacy, and biomarker findings of PBT2 in targeting Abeta as a modifying therapy for Alzheimer’s disease: A phase IIa, double-blind, randomised, placebo-controlled trial. Lancet Neurol. 2008, 7, 779–786. [Google Scholar] [CrossRef]
- Vargas, M.R.; Pehar, M.; Díaz-Amarilla, P.J.; Beckman, J.S.; Barbeito, L. Transcriptional profile of primary astrocytes expressing ALS-linked mutant SOD1. J. Neurosci. Res. 2008, 86, 3515–3525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allaman, I.; Gavillet, M.; Bélanger, M.; Laroche, T.; Viertl, D.; Lashuel, H.A.; Magistretti, P.J. Amyloid-beta aggregates cause alterations of astrocytic metabolic phenotype: Impact on neuronal viability. J. Neurosci. 2010, 30, 3326–3338. [Google Scholar] [CrossRef] [Green Version]
- Scheiber, I.F.; Dringen, R. Astrocyte functions in the copper homeostasis of the brain. Neurochem. Int. 2013, 62, 556–565. [Google Scholar] [CrossRef]
- Halliwell, B.; Gutteridge, J.M.C. Free Radicals in Biology and Medicine, 4th ed.; Oxford University Press: Oxford, UK, 2007. [Google Scholar]



{kind=link}
{kind=link}
| Organ | Average Concentration of Copper FAAS µg/g Wet Tissue | |
|---|---|---|
| Sumio et al. (1975) | Lech and Sadik (2007) | |
| Liver | 9.9 | 3.47 |
| Brain | 5.1 | 3.32 |
| Heart | 3.3 | 3.26 |
| Kidney | 2.6 | 2.15 |
| Intestines | 2.1 | 1.54 |
| Lung | 1.3 | 1.91 |
| Spleen | 1.2 | 1.23 |
| Brain Area | Average Concentration of Copper in Descending Order FAAS (µg/g Dry Tissue) |
|---|---|
| Olfactory bulb | 27.92 |
| Caudate nucleus (tail) | 23.12 |
| Calcarine cortex | 23.07 |
| Occipital pole | 21.69 |
| Mammillary bodies | 19.65 |
| Frontal pole | 18.95 |
| Postcentral gyrus | 18.83 |
| Caudate nucleus (body) | 18.46 |
| Inferior colliculus | 17.92 |
| Optic nerve | 17.79 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gromadzka, G.; Tarnacka, B.; Flaga, A.; Adamczyk, A. Copper Dyshomeostasis in Neurodegenerative Diseases—Therapeutic Implications. Int. J. Mol. Sci. 2020, 21, 9259. https://doi.org/10.3390/ijms21239259
Gromadzka G, Tarnacka B, Flaga A, Adamczyk A. Copper Dyshomeostasis in Neurodegenerative Diseases—Therapeutic Implications. International Journal of Molecular Sciences. 2020; 21(23):9259. https://doi.org/10.3390/ijms21239259
Chicago/Turabian StyleGromadzka, Grażyna, Beata Tarnacka, Anna Flaga, and Agata Adamczyk. 2020. "Copper Dyshomeostasis in Neurodegenerative Diseases—Therapeutic Implications" International Journal of Molecular Sciences 21, no. 23: 9259. https://doi.org/10.3390/ijms21239259
APA StyleGromadzka, G., Tarnacka, B., Flaga, A., & Adamczyk, A. (2020). Copper Dyshomeostasis in Neurodegenerative Diseases—Therapeutic Implications. International Journal of Molecular Sciences, 21(23), 9259. https://doi.org/10.3390/ijms21239259