Cellular Consequences of Coenzyme Q10 Deficiency in Neurodegeneration of the Retina and Brain

{kind=link}
{kind=link}
Abstract
:1. Introduction
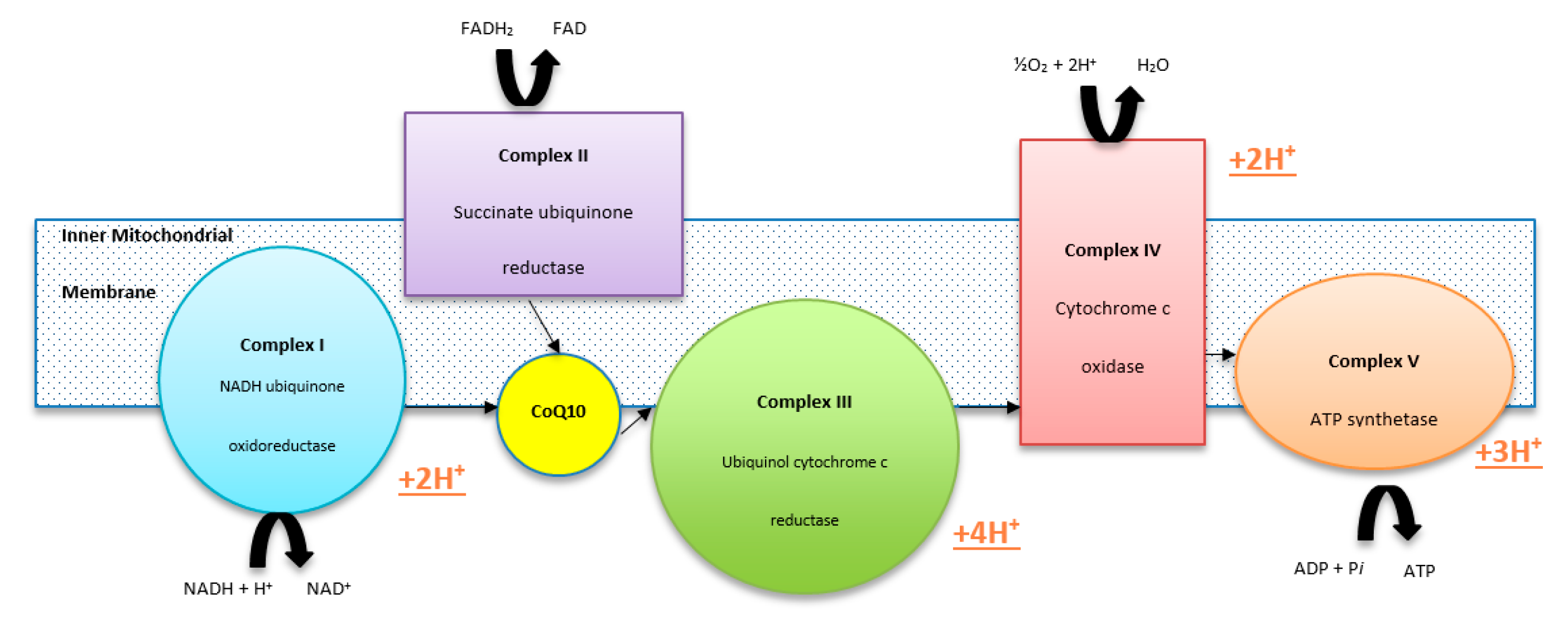
2. Deficiencies of CoQ10
3. Consequences of CoQ10 Deficiency
Cellular Consequences of CoQ10 Deficiency
4. Retina
5. Brain
5.1. Alzheimer’s Disease
5.2. Parkinson’s Disease
5.3. Huntington’s Disease
5.4. Friedrich’s Ataxia
6. Conclusions
Funding
Conflicts of Interest
Abbreviations
| MDPI | Multidisciplinary Digital Publishing Institute |
| DOAJ | Directory of open access journals |
| TLA | Three letter acronym |
| LD | Linear dichroism |
References
- Silverman, H.M.; Romano, J.; Elmer, G. The Vitamin Book; Bantam Books: New York, NY, USA, 1993. [Google Scholar]
- Acosta, M.J.; Fonseca, L.V.; Desbats, M.A.; Cerqua, C.; Zordan, R.; Trevisson, E.; Salviati, L. Coenzyme Q biosynthesis in health and disease. Biochimica Biophysica Acta (BBA)-Bioenerg. 2016, 1857, 1079–1085. [Google Scholar] [CrossRef] [PubMed]
- Nohl, H.; Gille, L. The role of coenzyme Q in lysosomes. In Coenzyme Q: Molecular Mechanisms in Health and Disease; CRC Press: Boca Raton, FL, USA, 2001; pp. 99–106. [Google Scholar]
- Challem, J. Coenzyme Q10: It may just be the miracle vitamin of the 1990s. The Nutritional Reporter, 4 December 1996. [Google Scholar]
- Mancini, A.; Festa, R.; Raimondo, S.; Pontecorvi, A.; Littarru, G.P. Hormonal influence on coenzyme Q10 levels in blood plasma. Int. J. Mol. Sci. 2011, 12, 9216–9225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Mauro, S.; Quinzii, C.M.; Hirano, M. Mutations in coenzyme Q 10 biosynthetic genes. J. Clin. Investig. 2007, 117, 587–589. [Google Scholar] [CrossRef] [PubMed]
- Quinzii, C.M.; Hirano, M. Primary and secondary CoQ10 deficiencies in humans. Biofactors 2011, 37, 361–365. [Google Scholar] [CrossRef]
- Potgieter, M.; Pretorius, E.; Pepper, M.S. Primary and secondary coenzyme Q10 deficiency: The role of therapeutic supplementation. Nutr. Rev. 2013, 71, 180–188. [Google Scholar] [CrossRef]
- Åberg, F.; Appelkvist, E.L.; Dallner, G.; Ernster, L. Distribution and redox state of ubiquinones in rat and human tissues. Arch. Biochem. Biophys. 1992, 295, 230–234. [Google Scholar] [CrossRef]
- Miles, M.V.; Horn, P.S.; Morrison, J.A.; Tang, P.H.; De Grauw, T.; Pesce, A.J. Plasma coenzyme Q10 reference intervals, but not redox status, are affected by gender and race in self-reported healthy adults. Clin. Chim. Acta 2003, 332, 123–132. [Google Scholar] [CrossRef]
- Quinzii, C.M.; López, L.C.; Gilkerson, R.W.; Dorado, B.; Coku, J.; Naini, A.B.; Lagier-Tourenne, C.; Schuelke, M.; Salviati, L.; Carrozzo, R.; et al. Reactive oxygen species, oxidative stress, and cell death correlate with level of CoQ10 deficiency. FASEB J. 2010, 24, 3733–3743. [Google Scholar] [CrossRef] [Green Version]
- Quinzii, C.M.; Luna-Sanchez, M.; Ziosi, M.; Hidalgo-Gutierrez, A.; Kleiner, G.; Lopez, L.C. The role of sulfide oxidation impairment in the pathogenesis of primary CoQ deficiency. Front. Physiol. 2017, 8, 525. [Google Scholar] [CrossRef]
- Lagendijk, J.; Ubbink, J.B.; Vermaak, W.J. Measurement of the ratio between the reduced and oxidized forms of coenzyme Q10 in human plasma as a possible marker of oxidative stress. J. Lipid Res. 1996, 37, 67–75. [Google Scholar] [PubMed]
- Kühlbrandt, W. Structure and function of mitochondrial membrane protein complexes. BMC Biol. 2015, 13, 89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heaton, R.A.; Heales, S.; Rahman, K.; Sexton, D.W.; Hargreaves, I. The Effect of Cellular Coenzyme Q10 Deficiency on Lysosomal Acidification. J. Clin. Med. 2020, 9, 1923. [Google Scholar] [CrossRef] [PubMed]
- De Barcelos, I.P.D.; Haas, R.H. Coq10 and ageing. Biology 2019, 8, 28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rahman, S.; Hargreaves, I.; Clayton, P.; Heales, S. Neonatal presentation of coenzyme Q10 deficiency. J. Pediatrics 2001, 139, 456–458. [Google Scholar] [CrossRef] [PubMed]
- Gempel, K.; Topaloglu, H.; Talim, B.; Schneiderat, P.; Schoser, B.G.; Hans, V.H.; Pálmafy, B.; Kale, G.; Tokatli, A.; Quinzii, C.; et al. The myopathic form of coenzyme Q10 deficiency is caused by mutations in the electron-transferring-flavoprotein dehydrogenase (ETFDH) gene. Brain 2007, 130, 2037–2044. [Google Scholar] [CrossRef]
- Rötig, A.; Appelkvist, E.L.; Geromel, V.; Chretien, D.; Kadhom, N.; Edery, P.; Lebideau, M.; Dallner, G.; Munnich, A.; Ernster, L.; et al. Quinone-responsive multiple respiratory-chain dysfunction due to widespread coenzyme Q10 deficiency. Lancet 2000, 356, 391–395. [Google Scholar] [CrossRef]
- Mollet, J.; Giurgea, I.; Schlemmer, D.; Dallner, G.; Chretien, D.; Delahodde, A.; Bacq, D.; de Lonlay, P.; Munnich, A.; Rötig, A. Prenyldiphosphate synthase, subunit 1 (PDSS1) and OH-benzoate polyprenyltransferase (COQ2) mutations in ubiquinone deficiency and oxidative phosphorylation disorders. J. Clin. Investig. 2007, 117, 765–772. [Google Scholar] [CrossRef] [Green Version]
- Desbats, M.A.; Morbidoni, V.; Silic-Benussi, M.; Doimo, M.; Ciminale, V.; Cassina, M.; Sacconi, S.; Hirano, M.; Basso, G.; Pierrel, F.; et al. The COQ2 genotype predicts the severity of coenzyme Q10 deficiency. Hum. Mol. Genet. 2016, 25, 4256–4265. [Google Scholar] [CrossRef] [Green Version]
- Qu, J.; Kaufman, Y.; Washington, I. Coenzyme Q10 in the human retina. Investig. Ophthalmol. Vis. Sci. 2009, 50, 1814–1818. [Google Scholar] [CrossRef] [Green Version]
- Söderberg, M.; Edlund, C.; Kristensson, K.; Dallner, G. Lipid compositions of different regions of the human brain during aging. J. Neurochem. 1990, 54, 415–423. [Google Scholar] [CrossRef] [PubMed]
- Edlund, C.; Söderberg, M.; Kristensson, K.; Dallner, G. Ubiquinone, dolichol, and cholesterol metabolism in aging and Alzheimer’s disease. Biochem. Cell Biol. 1992, 70, 422–428. [Google Scholar] [CrossRef] [PubMed]
- Nagase, M.; Yamamoto, Y.; Matsumoto, N.; Arai, Y.; Hirose, N. Increased oxidative stress and coenzyme Q10 deficiency in centenarians. J. Clin. Biochem. Nutr. 2018, 63, 17–124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knekt, P.; Heliövaara, M.; Rissanen, A.; Aromaa, A.; Aaran, R.K. Serum antioxidant vitamins and risk of cataract. BMJ 1992, 305, 1392–1394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dusting, G.J.; Triggle, C. Are we over oxidized? Oxidative stress, cardiovascular disease, and the future of intervention studies with antioxidants. Vasc. Health Risk Manag. 2005, 1, 93–97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nunomura, A.; Moreira, P.I.; Lee, H.G.; Zhu, X.; Castellani, R.J.; Smith, M.A.; Perry, G. Neuronal Death and Survival Under Oxidative Stress in Alzheimer and Parkinson Diseases. CNS Neurol. Disord. Drug Targets 2008, 6, 411–423. [Google Scholar] [CrossRef] [Green Version]
- Lenaz, G. Role of mitochondria in oxidative stress and ageing. Biochim. Biophys. Acta Bioenerg. 1998, 1366, 53–67. [Google Scholar] [CrossRef] [Green Version]
- Blasi, M.A.; Bovina, C.; Carella, G.; Genova, M.L.; Jansen, A.M.; Lenaz, G.; Brancato, R. Does coenzyme Q10 play a role in opposing oxidative stress in patients with age-related macular degeneration? Ophthalmologica 2001, 215, 51–54. [Google Scholar] [CrossRef]
- Beal, M.F.; Oakes, D.; Shoulson, I.; Henchcliffe, C.; Galpern, W.R.; Haas, R.; Juncos, J.L.; Nutt, J.G.; Voss, T.S.; Ravina, B.; et al. A randomized clinical trial of high-dosage coenzyme Q10 in early Parkinson disease: No evidence of benefit. JAMA Neurol. 2014, 71, 543–552. [Google Scholar]
- Nucci, C.; Tartaglione, R.; Cerulli, A.; Mancino, R.; Spano, A.; Cavaliere, F.; Rombola, L.; Bagetta, G.; Corasaniti, M.T.; Morrone, L.A. Retinal damage caused by high intraocular pressure–induced transient ischemia is prevented by coenzyme Q10 in rat. Int. Rev. Neurobiol. 2007, 82, 397–406. [Google Scholar]
- Russo, R.; Cavaliere, F.; Rombolà, L.; Gliozzi, M.; Cerulli, A.; Nucci, C.; Fazzi, E.; Bagetta, G.; Corasaniti, M.T.; Morrone, L.A. Rational basis for the development of coenzyme Q10 as a neurotherapeutic agent for retinal protection. Prog. Brain Res. 2008, 173, 575–582. [Google Scholar] [PubMed]
- Lee, D.; Shim, M.S.; Kim, K.Y.; Noh, Y.H.; Kim, H.; Kim, S.Y.; Weinreb, R.N.; Ju, W.K. Coenzyme Q10 inhibits glutamate excitotoxicity and oxidative stress–mediated mitochondrial alteration in a mouse model of glaucoma. Investig. Ophthalmol. Vis. Sci. 2014, 55, 993–1005. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhagavan, H.N.; Chopra, R.K. Coenzyme Q10: Absorption, tissue uptake, metabolism and pharmacokinetics. Free Radic. Res. 2006, 40, 445–453. [Google Scholar] [CrossRef] [PubMed]
- Nucci, C.; Russo, R.; Martucci, A.; Giannini, C.; Garaci, F.; Floris, R.; Bagetta, G.; Morrone, L.A. New strategies for neuroprotection in glaucoma, a disease that affects the central nervous system. Eur. J. Pharmacol. 2016, 787, 119–126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mancino, R.; Cesareo, M.; Martucci, A.; Di Carlo, E.; Ciuffoletti, E.; Giannini, C.; Morrone, L.A.; Nucci, C.; Garaci, F. Neurodegenerative process linking the eye and the brain. Curr. Med. Chem. 2019, 26, 3754–3763. [Google Scholar] [CrossRef] [PubMed]
- Lulli, M.; Witort, E.; Papucci, L.; Torre, E.; Schipani, C.; Bergamini, C.; Dal Monte, M.; Capaccioli, S. Coenzyme Q10 instilled as eye drops on the cornea reaches the retina and protects retinal layers from apoptosis in a mouse model of kainate-induced retinal damage. Investig. Ophthalmol. Vis. Sci. 2012, 53, 8295–8302. [Google Scholar] [CrossRef]
- Davis, B.M.; Tian, K.; Pahlitzsch, M.; Brenton, J.; Ravindran, N.; Butt, G.; Malaguarnera, G.; Normando, E.M.; Guo, L.; Cordeiro, M.F. Topical Coenzyme Q10 demonstrates mitochondrial-mediated neuroprotection in a rodent model of ocular hypertension. Mitochondrion 2017, 36, 114–123. [Google Scholar] [CrossRef]
- Ozates, S.; Elgin, K.U.; Yilmaz, N.S.; Demirel, O.O.; Sen, E.; Yilmazbas, P. Evaluation of oxidative stress in pseudo-exfoliative glaucoma patients treated with and without topical coenzyme Q10 and vitamin E. Eur. J. Ophthalmol. 2019, 29, 196–201. [Google Scholar] [CrossRef]
- Quaranta, L.; Riva, I.; Biagioli, E.; Rulli, E.; Rulli, E.; Poli, D.; CoQun® Study Group. Evaluating the effects of an ophthalmic solution of coenzyme Q10 and vitamin E in open-angle glaucoma patients: A study protocol. Adv. Ther. 2019, 36, 2506–2514. [Google Scholar] [CrossRef]
- Yamagishi, K.; Ikeda, A.; Moriyama, Y.; Chei, C.L.; Noda, H.; Umesawa, M.; Cui, R.; Nagao, M.; Kitamura, A.; Yamamoto, Y.; et al. Serum coenzyme Q10 and risk of disabling dementia: The Circulatory Risk in Communities Study (CIRCS). Atherosclerosis 2014, 237, 400–403. [Google Scholar] [CrossRef] [Green Version]
- Choi, H.; Park, H.H.; Koh, S.H.; Choi, N.Y.; Yu, H.J.; Park, J.; Lee, Y.J.; Lee, K.Y. Coenzyme Q10 protects against amyloid beta-induced neuronal cell death by inhibiting oxidative stress and activating the P13K pathway. Neurotoxicology 2012, 33, 85–90. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Yang, Y.; Li, G.; Wang, J.; Yang, E.S. Coenzyme Q10 attenuates β-amyloid pathology in the aged transgenic mice with Alzheimer presenilin 1 mutation. J. Mol. Neurosci. 2008, 34, 165–171. [Google Scholar] [CrossRef] [PubMed]
- Dumont, M.; Kipiani, K.; Yu, F.; Wille, E.; Katz, M.; Calingasan, N.Y.; Gouras, G.K.; Lin, M.T.; Beal, M.F. Coenzyme Q10 decreases amyloid pathology and improves behavior in a transgenic mouse model of Alzheimer’s disease. J. Alzheimer’s Dis. 2011, 27, 211–223. [Google Scholar] [CrossRef] [PubMed]
- Kawamoto, E.M.; Munhoz, C.D.; Glezer, I.; Bahia, V.S.; Caramelli, P.; Nitrini, R.; Marcourakis, T. Oxidative state in platelets and erythrocytes in aging and Alzheimer’s disease. Neurobiol. Aging 2005, 26, 857–864. [Google Scholar] [CrossRef] [PubMed]
- Beal, M.F.; Henshaw, D.R.; Jenkins, B.G.; Rosen, B.R.; Schulz, J.B. Coenzyme Q10 and nicotinamide block striatal lesions produced by the mitochondrial toxin malonate. Ann. Neurol. Off. J. Am. Neurol. Assoc. Child Neurol. Soc. 1994, 36, 882–888. [Google Scholar] [CrossRef]
- Galasko, D.R.; Peskind, E.; Clark, C.M.; Quinn, J.F.; Ringman, J.M.; Jicha, G.A.; Cotman, C.; Cottrell, B.; Montine, T.J.; Thomas, R.G.; et al. Antioxidants for Alzheimer disease: A randomized clinical trial with cerebrospinal fluid biomarker measures. Arch. Neurol. 2012, 69, 836–841. [Google Scholar] [CrossRef] [Green Version]
- Senin, U.; Parnetti, L.; Barbagallo-Sangiorgi, G.; Bartorelli, L.; Bocola, V.; Capurso, A.; Fioravanti, M. Idebenone in senile dementia of Alzheimer type: A multicentre study. Arch. Gerontol. Geriatr. 1992, 15, 249–260. [Google Scholar] [CrossRef]
- Bergamasco, B.; Scarzella, L.; La Commare, P. Idebenone, a new drug in the treatment of cognitive impairment in patients with dementia of the Alzheimer type. Funct. Neurol. 1994, 9, 161–168. [Google Scholar]
- Calne, D.B.; Snow, B.J.; Lee, C. Criteria for diagnosing Parkinson’s disease. Ann. Neurol. Off. J. Am. Neurol. Assoc. Child Neurol. Soc. 1992, 32, S125–S127. [Google Scholar] [CrossRef]
- Braak, H.; Braak, E. Pathoanatomy of Parkinson’s disease. J. Neurol. 2000, 247, II3–II10. [Google Scholar] [CrossRef]
- Forno, L.S. The Neuropathology Of Parkinson’S Disease. In Progress in Parkinson Research; Springer: Boston, MA, USA, 1988; pp. 11–21. [Google Scholar]
- Vaughan, J.R.; Davis, M.B.; Wood, N.W. Genetics of Parkinsonism: A review. Ann. Hum. Genet. 2001, 65, 111–126. [Google Scholar] [CrossRef] [PubMed]
- Sherer, T. Pathogenesis of Parkinson’s Disease. 2001. Available online: https://europepmc.org/article/med/11569943 (accessed on 6 November 2020).
- Olanow, C.W.; Tatton, W.G. Etiology and pathogenesis of Parkinson’s disease. Annu. Rev. Neurosci. 1999, 22, 123–144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schapira, A.H. Mitochondria in the aetiology and pathogenesis of Parkinson’s disease. Lancet Neurol. 2008, 7, 97–109. [Google Scholar] [CrossRef]
- Jenner, P. Oxidative stress in Parkinson’s disease and other neurodegenerative disorders. Pathol. Biol. 1996, 44, 57–64. [Google Scholar] [PubMed]
- Sian, J.; Dexter, D.T.; Lees, A.J.; Daniel, S.; Agid, Y.; Javoy-Agid, F.; Jenner, P.; Marsden, C.D. Alterations in glutathione levels in Parkinson’s disease and other neurodegenerative disorders affecting basal ganglia. Ann. Neurol. 1994, 36, 348–355. [Google Scholar] [CrossRef] [PubMed]
- Parker, W.D.; Boyson, S.J.; Parks, J.K. Abnormalities of the electron transport chain in idiopathic parkinson’s disease. Ann. Neurol. 1989, 26, 719–723. [Google Scholar] [CrossRef] [PubMed]
- Turner, C.; Schapira, A.H.V. Mitochondrial dysfunction in neurodegenerative disorders and ageing. In Advances in Experimental Medicine and Biology; Springer: Boston, MA, USA, 2001; pp. 229–251. [Google Scholar]
- Krige, D.; Carroll, M.T.; Cooper, J.M.; Marsden, C.D.; Schapira, A.H.V. Platelet mitochondria function in Parkinson’s disease. Ann. Neurol. 1992, 32, 782–788. [Google Scholar] [CrossRef]
- Przedborski, S.; Jackson-Lewis, V. Mechanisms of MPTP toxicity. Mov. Disord. 1998, 13 (Suppl. 1), 35–38. [Google Scholar]
- William Langston, J.; Ballard, P.; Tetrud, J.W.; Irwin, I. Chronic parkinsonism in humans due to a product of meperidine-analog synthesis. Science 1983, 219, 979–980. [Google Scholar] [CrossRef] [Green Version]
- Fahn, S. Levodopa in the treatment of Parkinson’s disease. In Oxidative Stress and Neuroprotection; Springer: Vienna, Austria, 2006; pp. 1–15. [Google Scholar]
- Shults, C.W.; Nasirian, F.; Ward, D.M.; Nakano, K.; Pay, M.; Hill, L.R. Carbidopa/levodopa and selegiline do not affect platelet mitochondrial function in early parkinsonism. Neurology 1995, 45, 344–348. [Google Scholar] [CrossRef]
- Shults, C.W.; Haas, R.H.; Passov, D.; Beal, M.F. Coenzyme Q10 levels correlate with the activities of complexes I and II/III in mitochondria from Parkinsonian and nonparkinsonian subjects. Ann. Neurol. 1997, 42, 261–264. [Google Scholar] [CrossRef] [PubMed]
- Beal, M.F.; Matthews, R.T.; Tieleman, A.; Shults, C.W. Coenzyme Q10 attenuates the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) induced loss of striatal dopamine and dopaminergic axons in aged mice. Brain Res. 1998, 783, 109–114. [Google Scholar] [CrossRef]
- Shults, C.W.; Flint Beal, M.; Fontaine, D.; Nakano, K.; Haas, R.H. Absorption, tolerability, and effects on mitochondrial activity of oral coenzyme Q10 in parkinsonian patients. Neurology 1998, 50, 793–795. [Google Scholar] [CrossRef] [PubMed]
- Shults, C.W.; Oakes, D.; Kieburtz, K.; Flint Beal, M.; Haas, R.; Plumb, S. Effects of coenzyme Q 10 in early Parkinson disease: Evidence of slowing of the functional decline. Arch Neurol. 2002, 59, 1541–1550. [Google Scholar] [CrossRef] [PubMed]
- Popovic, N.; Brundin, P. Therapeutic potential of controlled drug delivery systems in neurodegenerative diseases. Int. J. Pharm. 2006, 314, 120–126. [Google Scholar] [CrossRef]
- Park, H.W.; Park, C.G.; Park, M.; Lee, S.H.; Park, H.R.; Lim, J.; Park, S.H.; Choy, Y.B. Intrastriatal administration of coenzyme Q10 enhances neuroprotection in a Parkinson’s disease rat model. Sci. Rep. 2020, 10, 1–2. [Google Scholar] [CrossRef]
- Feigin, A.; Kieburtz, K.; Como, P.; Hickey, C.; Abwender, D.; Zimmerman, C.; Shoulson, I. Assessment of coenzyme Q10 tolerability in Huntington’s disease. Mov. Disord. Off. J. Mov. Disord. Soc. 1996, 11, 321–323. [Google Scholar] [CrossRef]
- Ferrante, R.J.; Andreassen, O.A.; Dedeoglu, A.; Ferrante, K.L.; Jenkins, B.G.; Hersch, S.M.; Beal, M.F. Therapeutic effects of coenzyme Q10 and remacemide in transgenic mouse models of Huntington’s disease. J. Neurosci. 2002, 22, 1592–1599. [Google Scholar] [CrossRef] [Green Version]
- Koroshetz, W.J.; Jenkins, B.G.; Rosen, B.R.; Beal, M.F. Energy metabolism defects in Huntington’s disease and effects of coenzyme Q10. Ann. Neurol. Off. J. Am. Neurol. Assoc. Child Neurol. Soc. 1997, 41, 160–165. [Google Scholar] [CrossRef]
- Huntington Study Group. A randomized, placebo-controlled trial of coenzyme Q10 and remacemide in Huntington’s disease. Neurology 2001, 57, 397–404. [Google Scholar]
- Bresolin, N.; Doriguzzi, C.; Ponzetto, C.; Angelini, C.; Moroni, I.; Castelli, E.; Piccolo, G. Ubidecarenone in the treatment of mitochondrial myopathies: A multi-center double-blind trial. J. Neurol. Sci. 1990, 100, 70–78. [Google Scholar] [CrossRef]
- Harding, A.E. Friedreich’s ataxia: A clinical and genetic study of 90 families with an analysis of early diagnostic criteria and intrafamilial clustering of clinical features. Brain 1981, 104, 589–620. [Google Scholar] [CrossRef] [PubMed]
- Dürr, A.; Cossee, M.; Agid, Y.; Campuzano, V.; Mignard, C.; Penet, C. Clinical and Genetic Abnormalities in Patients with Friedreich’s Ataxia. N. Engl. J. Med. 1996, 335, 1169–1175. [Google Scholar] [CrossRef] [PubMed]
- Bürk, K. Friedreich Ataxia: Current status and future prospects. Cerebellum Ataxias 2017, 4, 4. [Google Scholar] [CrossRef] [Green Version]
- Campuzano, V.; Montermini, L.; Moltò, M.D.; Pianese, L.; Cossée, M.; Cavalcanti, F. Friedreich’s ataxia: Autosomal recessive disease caused by an intronic GAA triplet repeat expansion. Science 1996, 271, 1423–1427. [Google Scholar] [CrossRef]
- Campuzano, V.; Montermini, L.; Lutz, Y.; Cova, L.; Hindelang, C.; Jiralerspong, S. Frataxin is reduced in Friedreich ataxia patients and is associated with mitochondrial membranes. Hum. Mol. Genet. 1997, 6, 1771–1780. [Google Scholar] [CrossRef] [Green Version]
- Wong, A.; Yang, J.; Cavadini, P.; Gellera, C.; Lonnerdal, B.; Taroni, F. The Friedreich’s ataxia mutation confers cellular sensitivity to oxidant stress which is rescued by chelators of iron and calcium and inhibitors of apoptosis. Hum. Mol. Genet. 1999, 8, 425–430. [Google Scholar] [CrossRef] [Green Version]
- Bradley, J.L.; Blake, J.C.; Chamberlain, S.; Thomas, P.K.; Cooper, J.M.; Schapira, A.H.V. Clinical, biochemical and molecular genetic correlations in Friedreich’s ataxia. Hum. Mol. Genet. 2000, 9, 275–282. [Google Scholar] [CrossRef] [Green Version]
- Koutnikova, H.; Campuzano, V.; Foury, F.; Dollé, P.; Cazzalini, O.; Koenig, M. Studies of human, mouse and yeast homologues indicate a mitochondrial function for frataxin. Nat. Genet. 1997, 16, 345–351. [Google Scholar] [CrossRef]
- Babcock, M.; De Silva, D.; Oaks, R.; Davis-Kaplan, S.; Jiralerspong, S.; Montermini, L. Regulation of mitochondrial iron accumulation by Yfh1p, a putative homolog of frataxin. Science 1997, 276, 1709–1712. [Google Scholar] [CrossRef]
- Foury, F.; Cazzalini, O. Deletion of the yeast homologue of the human gene associated with Friedreich’s ataxia elicits iron accumulation in mitochondria. FEBS Lett. 1997, 411, 373–377. [Google Scholar] [CrossRef] [Green Version]
- Al-Mahdawi, S.; Pinto, R.M.; Varshney, D.; Lawrence, L.; Lowrie, M.B.; Hughes, S. GAA repeat expansion mutation mouse models of Friedreich ataxia exhibit oxidative stress leading to progressive neuronal and cardiac pathology. Genomics 2006, 88, 580–590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lodi, R.; Hart, P.E.; Rajagopalan, B.; Taylor, D.J.; Crilley, J.G.; Bradley, J.L. Antioxidant treatment improves in vivo cardiac and skeletal muscle bioenergetics in patients with Friedreich’s ataxia. Ann. Neurol. 2001, 49, 590–596. [Google Scholar] [CrossRef] [PubMed]
- Hart, P.E.; Lodi, R.; Rajagopalan, B.; Bradley, J.L.; Crilley, J.G.; Turner, C. Antioxidant treatment of patients with Friedreich ataxia: Four-year follow-up. Arch Neurol. 2005, 62, 621–626. [Google Scholar] [CrossRef] [PubMed]
- Cooper, J.M.; Korlipara, L.V.P.; Hart, P.E.; Bradley, J.L.; Schapira, A.H.V. Coenzyme Q10 and vitamin e deficiency in Friedreich’s ataxia: Predictor of efficacy of vitamin e and coenzyme Q10 therapy. Eur. J. Neurol. 2008, 15, 1371–1379. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Manzar, H.; Abdulhussein, D.; Yap, T.E.; Cordeiro, M.F. Cellular Consequences of Coenzyme Q10 Deficiency in Neurodegeneration of the Retina and Brain. Int. J. Mol. Sci. 2020, 21, 9299. https://doi.org/10.3390/ijms21239299
Manzar H, Abdulhussein D, Yap TE, Cordeiro MF. Cellular Consequences of Coenzyme Q10 Deficiency in Neurodegeneration of the Retina and Brain. International Journal of Molecular Sciences. 2020; 21(23):9299. https://doi.org/10.3390/ijms21239299
Chicago/Turabian StyleManzar, Haider, Dalia Abdulhussein, Timothy E. Yap, and M. Francesca Cordeiro. 2020. "Cellular Consequences of Coenzyme Q10 Deficiency in Neurodegeneration of the Retina and Brain" International Journal of Molecular Sciences 21, no. 23: 9299. https://doi.org/10.3390/ijms21239299
APA StyleManzar, H., Abdulhussein, D., Yap, T. E., & Cordeiro, M. F. (2020). Cellular Consequences of Coenzyme Q10 Deficiency in Neurodegeneration of the Retina and Brain. International Journal of Molecular Sciences, 21(23), 9299. https://doi.org/10.3390/ijms21239299