Dual mTOR/DNA-PK Inhibitor CC-115 Induces Cell Death in Melanoma Cells and Has Radiosensitizing Potential



 ,
, 
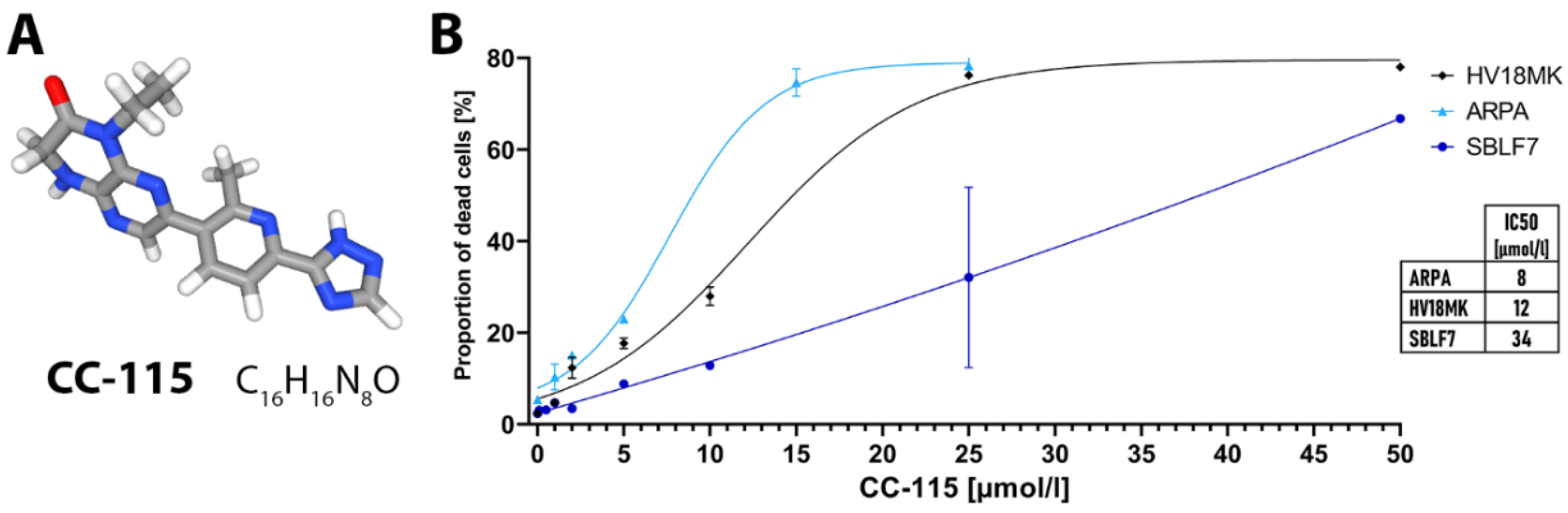{kind=link}
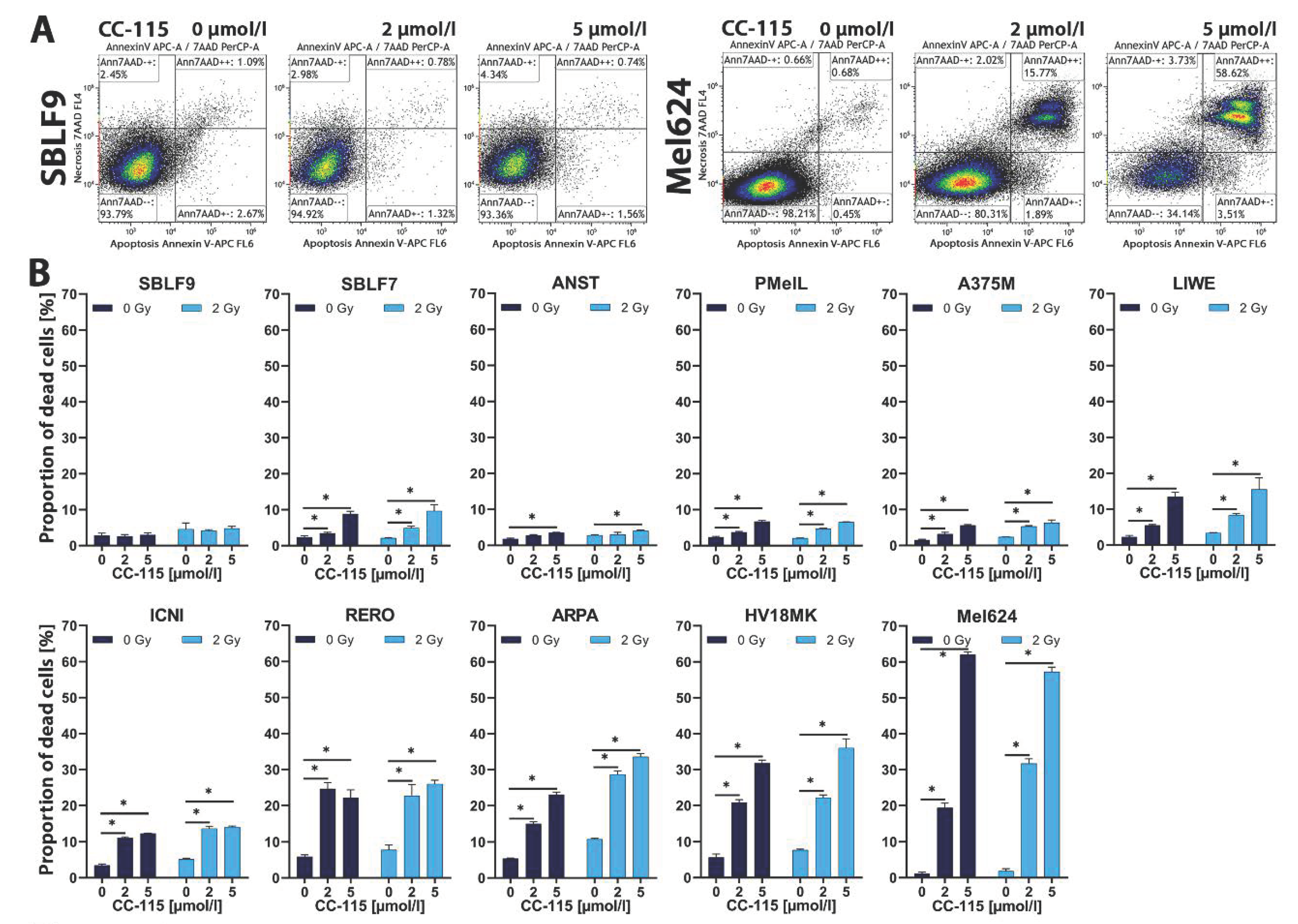{kind=link}
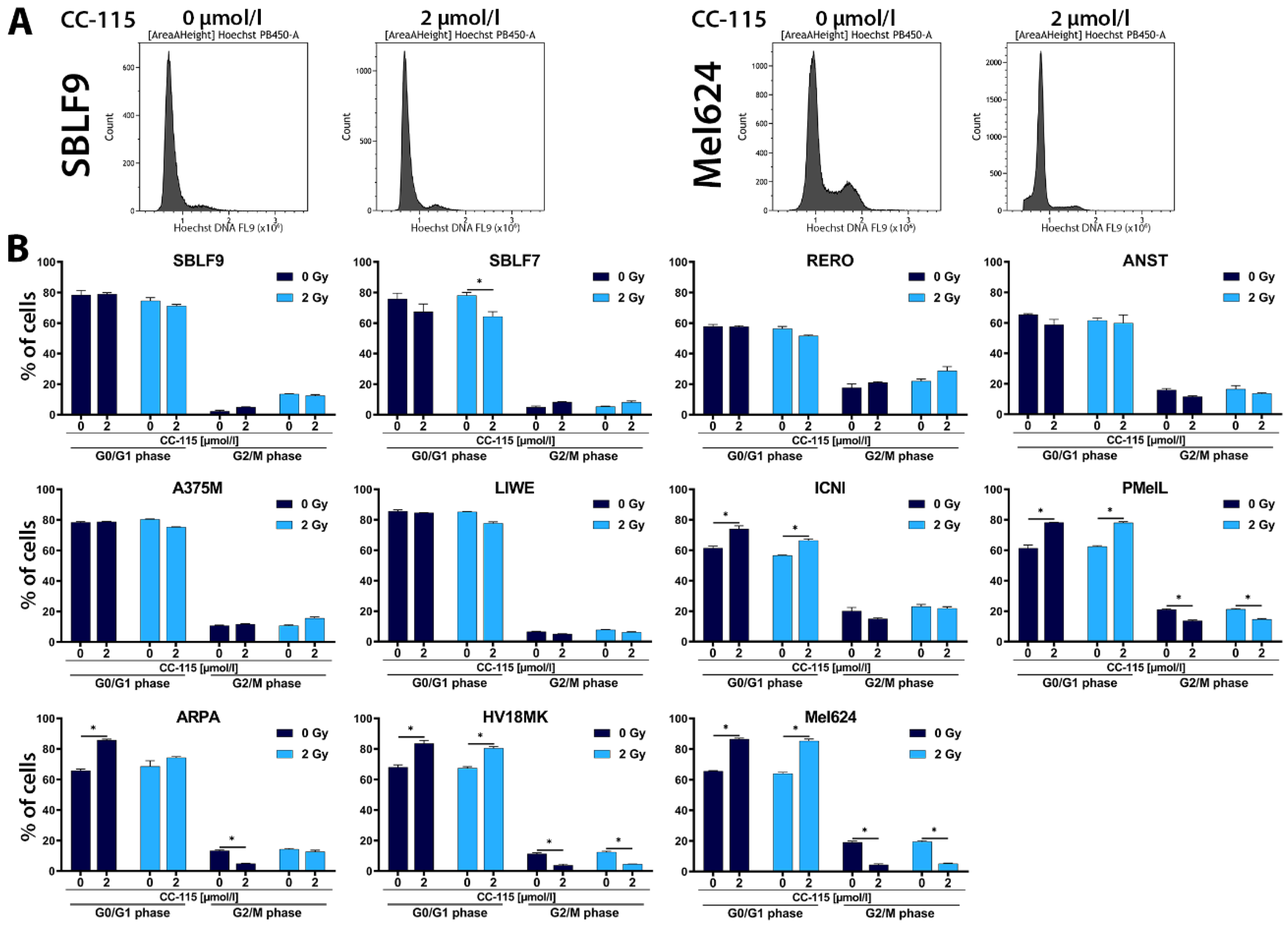{kind=link}
{kind=link}
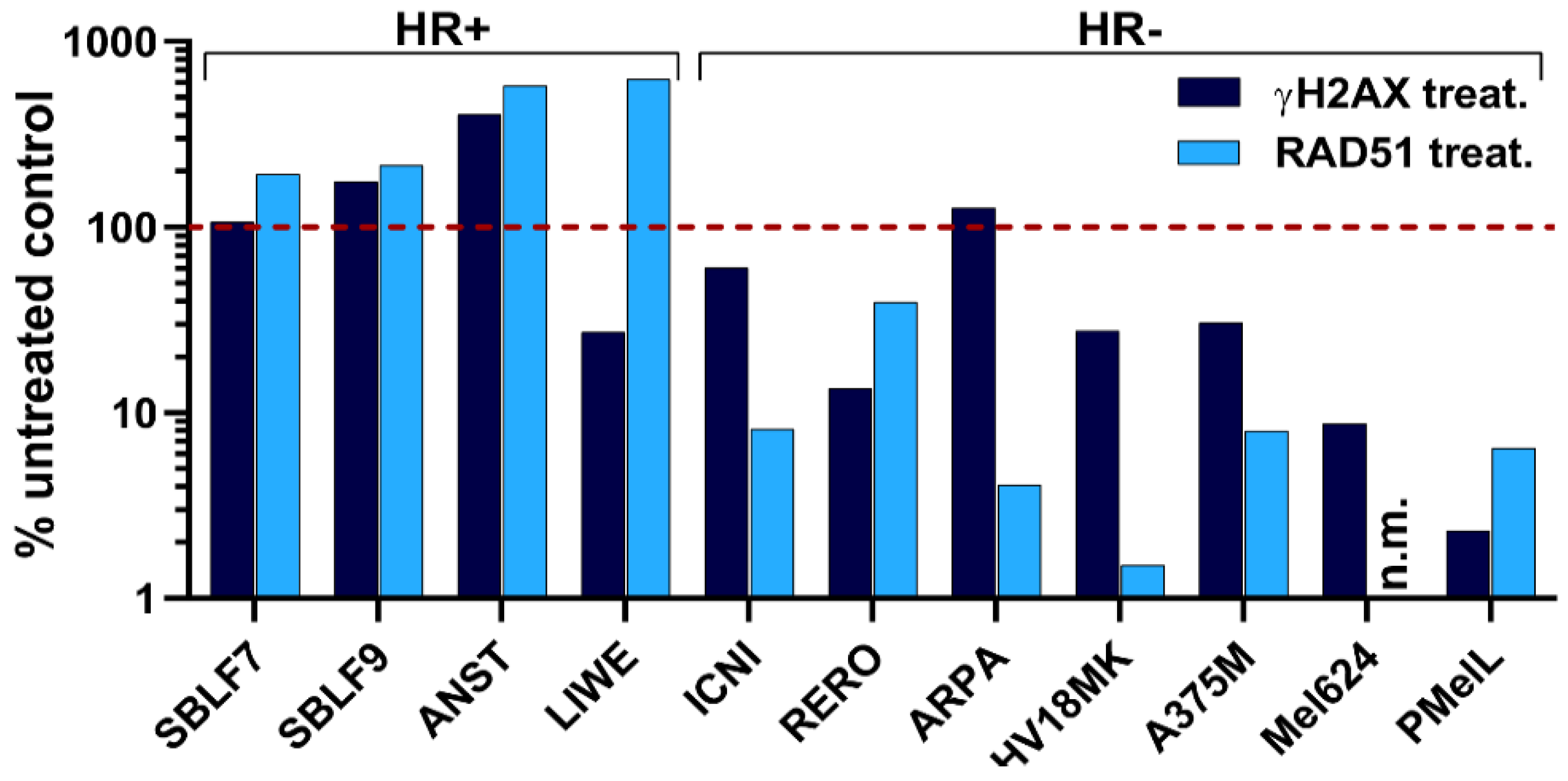{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Apoptosis and Necrosis Analysis
2.2. Cell Cycle Analysis
2.3. Colony Forming Assay
2.4. Homologous Recombination Assay
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Irradiation and Inhibitor
4.3. Flow Cytometry
4.3.1. Apoptosis and Necrosis Analysis
4.3.2. Cell Cycle Analysis
4.4. Colony Forming Assay
4.5. Homologous Recombination Assay (RAD51 Immunofluorescence Staining)
4.6. Statistics
4.7. Biosecurity and Institutional Safety Procedures
4.8. Ethics Approval and Consent to Participate
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| 7-AAD | 7-amino-actinomycin D |
| DDR | DNA-damage response |
| DEF | Dose enhancement factor |
| DNA–PK | DNA–dependent protein kinase |
| DSB | DNA double-strand break |
| FBS | Fetal bovine serum |
| GBM | Glioblastoma multiforme |
| HR | Homologous recombination |
| IR | Ionizing radiation |
| mTOR | Mechanistic target of rapamycin |
| mTORC1 | mTOR complex 1 |
| mTORC2 | mTOR complex 2 |
| NEA | Non-essential amino acid |
References
- Heinen, C.D.; Schmutte, C.; Fishel, R. DNA repair and tumorigenesis: Lessons from hereditary cancer syndromes. Cancer Biol. Ther. 2002, 1, 477–485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rajendran, P.; Ho, E.; Williams, D.E.; Dashwood, R.H. Dietary phytochemicals, HDAC inhibition, and DNA damage/repair defects in cancer cells. Clin. Epigenetics 2011, 3, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barbie, D.; Hahn, W.; Pellman, D. Destabilization of the cancer genome. Cancer Princ. Pract. Oncol. 2008, 8, 35–51. [Google Scholar]
- Dobler, C.; Jost, T.; Hecht, M.; Fietkau, R.; Distel, L. Senescence Induction by Combined Ionizing Radiation and DNA Damage Response Inhibitors in Head and Neck Squamous Cell Carcinoma Cells. Cells 2020, 9, 2012. [Google Scholar] [CrossRef] [PubMed]
- Mangerich, A.; Bürkle, A. How to kill tumor cells with inhibitors of poly (ADP-ribosyl) ation. Int. J. Cancer 2011, 128, 251–265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rassool, F.V.; Tomkinson, A.E. Targeting abnormal DNA double strand break repair in cancer. Cell. Mol. Life Sci. 2010, 67, 3699–3710. [Google Scholar] [CrossRef] [Green Version]
- Vignard, J.; Mirey, G.; Salles, B. Ionizing-radiation induced DNA double-strand breaks: A direct and indirect lighting up. Radiother. Oncol. 2013, 108, 362–369. [Google Scholar] [CrossRef] [Green Version]
- Khanna, K.K.; Jackson, S.P. DNA double-strand breaks: Signaling, repair and the cancer connection. Nat. Genet. 2001, 27, 247–254. [Google Scholar] [CrossRef]
- Barton, M.B.; Jacob, S.; Shafiq, J.; Wong, K.; Thompson, S.R.; Hanna, T.P.; Delaney, G.P. Estimating the demand for radiotherapy from the evidence: A review of changes from 2003 to 2012. Radiother. Oncol. 2014, 112, 140–144. [Google Scholar] [CrossRef]
- Delaney, G.; Jacob, S.; Featherstone, C.; Barton, M. The role of radiotherapy in cancer treatment: Estimating optimal utilization from a review of evidence-based clinical guidelines. Cancer 2005, 104, 1129–1137. [Google Scholar] [CrossRef]
- Pospisilova, M.; Seifrtova, M.; Rezacova, M. Small molecule inhibitors of DNA-PK for tumor sensitization to anticancer therapy. J. Physiol. Pharmacol. 2017, 68, 337–344. [Google Scholar]
- Uziel, T.; Lerenthal, Y.; Moyal, L.; Andegeko, Y.; Mittelman, L.; Shiloh, Y. Requirement of the MRN complex for ATM activation by DNA damage. EMBO J. 2003, 22, 5612–5621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karanam, K.; Kafri, R.; Loewer, A.; Lahav, G. Quantitative live cell imaging reveals a gradual shift between DNA repair mechanisms and a maximal use of HR in mid S phase. Mol. Cell 2012, 47, 320–329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davis, A.J.; Chen, D.J. DNA double strand break repair via non-homologous end-joining. Transl. Cancer Res. 2013, 2, 130–143. [Google Scholar] [CrossRef] [PubMed]
- Burma, S.; Chen, B.P.; Chen, D.J. Role of non-homologous end joining (NHEJ) in maintaining genomic integrity. DNA Repair 2006, 5, 1042–1048. [Google Scholar] [CrossRef] [PubMed]
- Weterings, E.; Chen, D.J. The endless tale of non-homologous end-joining. Cell Res. 2008, 18, 114–124. [Google Scholar] [CrossRef] [Green Version]
- Roos, W.P.; Thomas, A.D.; Kaina, B. DNA damage and the balance between survival and death in cancer biology. Nat. Rev. Cancer 2016, 16, 20–33. [Google Scholar] [CrossRef]
- Surucu, B.; Bozulic, L.; Hynx, D.; Parcellier, A.; Hemmings, B.A. In vivo analysis of protein kinase B (PKB)/Akt regulation in DNA-PKcs-null mice reveals a role for PKB/Akt in DNA damage response and tumorigenesis. J. Biol. Chem. 2008, 283, 30025–30033. [Google Scholar] [CrossRef] [Green Version]
- Meek, K.; Dang, V.; Lees-Miller, S.P. DNA-PK: The means to justify the ends? Adv. Immunol. 2008, 99, 33–58. [Google Scholar] [CrossRef]
- Neal, J.A.; Meek, K. Choosing the right path: Does DNA-PK help make the decision? Mutat. Res./Fundam. Mol. Mech. Mutagenesis 2011, 711, 73–86. [Google Scholar] [CrossRef] [Green Version]
- Cary, R.B.; Peterson, S.R.; Wang, J.; Bear, D.G.; Bradbury, E.M.; Chen, D.J. DNA looping by Ku and the DNA-dependent protein kinase. Proc. Natl. Acad. Sci. USA 1997, 94, 4267–4272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohiuddin, I.S.; Kang, M.H. DNA-PK as an Emerging Therapeutic Target in Cancer. Front. Oncol. 2019, 9. [Google Scholar] [CrossRef] [PubMed]
- Damia, G. Targeting DNA-PK in cancer. Mutat. Res. /Fundam. Mol. Mech. Mutagenesis 2020, 821, 111692. [Google Scholar] [CrossRef] [PubMed]
- Fok, J.H.L.; Ramos-Montoya, A.; Vazquez-Chantada, M.; Wijnhoven, P.W.G.; Follia, V.; James, N.; Farrington, P.M.; Karmokar, A.; Willis, S.E.; Cairns, J.; et al. AZD7648 is a potent and selective DNA-PK inhibitor that enhances radiation, chemotherapy and olaparib activity. Nat. Commun. 2019, 10, 5065. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carr, M.I.; Zimmermann, A.; Chiu, L.-Y.; Zenke, F.T.; Blaukat, A.; Vassilev, L.T. DNA-PK Inhibitor, M3814, as a New Combination Partner of Mylotarg in the Treatment of Acute Myeloid Leukemia. Front. Oncol. 2020, 10. [Google Scholar] [CrossRef] [PubMed]
- Laplante, M.; Sabatini, D.M. mTOR Signaling in Growth Control and Disease. Cell 2012, 149, 274–293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hua, H.; Kong, Q.; Zhang, H.; Wang, J.; Luo, T.; Jiang, Y. Targeting mTOR for cancer therapy. J. Hematol. Oncol. 2019, 12, 71. [Google Scholar] [CrossRef]
- Morita, M.; Gravel, S.P.; Hulea, L.; Larsson, O.; Pollak, M.; St-Pierre, J.; Topisirovic, I. mTOR coordinates protein synthesis, mitochondrial activity and proliferation. Cell Cycle 2015, 14, 473–480. [Google Scholar] [CrossRef] [Green Version]
- Yin, Y.; Hua, H.; Li, M.; Liu, S.; Kong, Q.; Shao, T.; Wang, J.; Luo, Y.; Wang, Q.; Luo, T.; et al. mTORC2 promotes type I insulin-like growth factor receptor and insulin receptor activation through the tyrosine kinase activity of mTOR. Cell Res. 2016, 26, 46–65. [Google Scholar] [CrossRef] [Green Version]
- Soria, J.C.; Shepherd, F.A.; Douillard, J.Y.; Wolf, J.; Giaccone, G.; Crino, L.; Cappuzzo, F.; Sharma, S.; Gross, S.H.; Dimitrijevic, S.; et al. Efficacy of everolimus (RAD001) in patients with advanced NSCLC previously treated with chemotherapy alone or with chemotherapy and EGFR inhibitors. Ann. Oncol. 2009, 20, 1674–1681. [Google Scholar] [CrossRef]
- Ellard, S.L.; Clemons, M.; Gelmon, K.A.; Norris, B.; Kennecke, H.; Chia, S.; Pritchard, K.; Eisen, A.; Vandenberg, T.; Taylor, M.; et al. Randomized phase II study comparing two schedules of everolimus in patients with recurrent/metastatic breast cancer: NCIC Clinical Trials Group IND.163. J. Clin. Oncol. 2009, 27, 4536–4541. [Google Scholar] [CrossRef] [PubMed]
- Houghton, P.J. Everolimus. Clin. Cancer Res. 2010, 16, 1368–1372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hudes, G.; Carducci, M.; Tomczak, P.; Dutcher, J.; Figlin, R.; Kapoor, A.; Staroslawska, E.; Sosman, J.; McDermott, D.; Bodrogi, I. Temsirolimus, interferon alfa, or both for advanced renal-cell carcinoma. N. Engl. J. Med. 2007, 356, 2271–2281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thijssen, R.; Ter Burg, J.; Garrick, B.; van Bochove, G.G.; Brown, J.R.; Fernandes, S.M.; Rodriguez, M.S.; Michot, J.M.; Hallek, M.; Eichhorst, B.; et al. Dual TORK/DNA-PK inhibition blocks critical signaling pathways in chronic lymphocytic leukemia. Blood 2016, 128, 574–583. [Google Scholar] [CrossRef]
- Tsuji, T.; Sapinoso, L.M.; Tran, T.; Gaffney, B.; Wong, L.; Sankar, S.; Raymon, H.K.; Mortensen, D.S.; Xu, S. CC-115, a dual inhibitor of mTOR kinase and DNA-PK, blocks DNA damage repair pathways and selectively inhibits ATM-deficient cell growth in vitro. Oncotarget 2017, 8, 74688–74702. [Google Scholar] [CrossRef] [Green Version]
- Mortensen, D.S.; Perrin-Ninkovic, S.M.; Shevlin, G.; Elsner, J.; Zhao, J.; Whitefield, B.; Tehrani, L.; Sapienza, J.; Riggs, J.R.; Parnes, J.S.; et al. Optimization of a Series of Triazole Containing Mammalian Target of Rapamycin (mTOR) Kinase Inhibitors and the Discovery of CC-115. J. Med. Chem. 2015, 58, 5599–5608. [Google Scholar] [CrossRef]
- Munster, P.; Mita, M.; Mahipal, A.; Nemunaitis, J.; Massard, C.; Mikkelsen, T.; Cruz, C.; Paz-Ares, L.; Hidalgo, M.; Rathkopf, D.; et al. First-In-Human Phase I Study of a Dual mTOR Kinase And DNA-PK Inhibitor (CC-115) In Advanced Malignancy. Cancer Manag. Res. 2019, 11, 10463–10476. [Google Scholar] [CrossRef] [Green Version]
- Dzwierzynski, W.W. Managing malignant melanoma. Plastic Reconstr. Surg. 2013, 132, 446e–460e. [Google Scholar] [CrossRef]
- Holmes, D. The cancer that rises with the Sun. Nature 2014, 515, S110–S111. [Google Scholar] [CrossRef] [Green Version]
- Bibault, J.-E.; Dewas, S.; Mirabel, X.; Mortier, L.; Penel, N.; Vanseymortier, L.; Lartigau, E. Adjuvant radiation therapy in metastatic lymph nodes from melanoma. Radiat. Oncol. 2011, 6, 12. [Google Scholar] [CrossRef] [Green Version]
- Cagney, D.N.; Martin, A.M.; Catalano, P.J.; Redig, A.J.; Lin, N.U.; Lee, E.Q.; Wen, P.Y.; Dunn, I.F.; Bi, W.L.; Weiss, S.E.; et al. Incidence and prognosis of patients with brain metastases at diagnosis of systemic malignancy: A population-based study. Neuro-Oncology 2017, 19, 1511–1521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glitza, I.C.; Heimberger, A.B.; Sulman, E.P.; Davies, M.A. Prognostic factors for survival in melanoma patients with brain metastases. In Brain Metastases from Primary Tumors; Hayat, M.A., Ed.; Elsevier: Amsterdam, Netherlands, 2016; Volume 3, pp. 267–297. [Google Scholar]
- Farshad, A.; Burg, G.; Panizzon, R.; Dummer, R. A retrospective study of 150 patients with lentigo maligna and lentigo maligna melanoma and the efficacy of radiotherapy using Grenz or soft X-rays. Br. J. Dermatol. 2002, 146, 1042–1046. [Google Scholar] [CrossRef] [PubMed]
- Sause, W.T.; Cooper, J.S.; Rush, S.; Ago, C.T.; Cosmatos, D.; Coughlin, C.T.; JanJan, N.; Lipsett, J. Fraction size in external beam radiation therapy in the treatment of melanoma. Int. J. Radiat. Oncol. Biol. Phys. 1991, 20, 429–432. [Google Scholar] [CrossRef]
- Deutsch, M.; Parsons, J.A.; Mercado, R., Jr. Radiotherapy for intracranial metastases. Cancer 1974, 34, 1607–1611. [Google Scholar] [CrossRef]
- Linskey, M.E.; Andrews, D.W.; Asher, A.L.; Burri, S.H.; Kondziolka, D.; Robinson, P.D.; Ammirati, M.; Cobbs, C.S.; Gaspar, L.E.; Loeffler, J.S. The role of stereotactic radiosurgery in the management of patients with newly diagnosed brain metastases: A systematic review and evidence-based clinical practice guideline. J. Neuro-Oncol. 2010, 96, 45–68. [Google Scholar] [CrossRef] [Green Version]
- Mazzola, R.; Jereczek-Fossa, B.A.; Franceschini, D.; Tubin, S.; Filippi, A.R.; Tolia, M.; Lancia, A.; Minniti, G.; Corradini, S.; Arcangeli, S.; et al. Oligometastasis and local ablation in the era of systemic targeted and immunotherapy. Radiat. Oncol. 2020, 15, 92. [Google Scholar] [CrossRef]
- Ugurel, S.; Röhmel, J.; Ascierto, P.A.; Flaherty, K.T.; Grob, J.J.; Hauschild, A.; Larkin, J.; Long, G.V.; Lorigan, P.; McArthur, G.A. Survival of patients with advanced metastatic melanoma: The impact of novel therapies. Eur. J. Cancer 2016, 53, 125–134. [Google Scholar] [CrossRef] [Green Version]
- Davies, M.A.; Saiag, P.; Robert, C.; Grob, J.-J.; Flaherty, K.T.; Arance, A.; Chiarion-Sileni, V.; Thomas, L.; Lesimple, T.; Mortier, L. Dabrafenib plus trametinib in patients with BRAFV600-mutant melanoma brain metastases (COMBI-MB): A multicentre, multicohort, open-label, phase 2 trial. Lancet Oncol. 2017, 18, 863–873. [Google Scholar] [CrossRef]
- Hecht, M.; Zimmer, L.; Loquai, C.; Weishaupt, C.; Gutzmer, R.; Schuster, B.; Gleisner, S.; Schulze, B.; Goldinger, S.; Berking, C. Radiosensitization by BRAF inhibitor therapy—Mechanism and frequency of toxicity in melanoma patients. Ann. Oncol. 2015, 26, 1238–1244. [Google Scholar] [CrossRef]
- Hecht, M.; Meier, F.; Zimmer, L.; Polat, B.; Loquai, C.; Weishaupt, C.; Forschner, A.; Gutzmer, R.; Utikal, J.S.; Goldinger, S.M.; et al. Clinical outcome of concomitant vs interrupted BRAF inhibitor therapy during radiotherapy in melanoma patients. Br. J. Cancer 2018, 118, 785–792. [Google Scholar] [CrossRef] [Green Version]
- Shannan, B.; Matschke, J.; Chauvistré, H.; Vogel, F.; Klein, D.; Meier, F.; Westphal, D.; Bruns, J.; Rauschenberg, R.; Utikal, J.; et al. Sequence-dependent cross-resistance of combined radiotherapy plus BRAFV600E inhibition in melanoma. Eur. J. Cancer 2019, 109, 137–153. [Google Scholar] [CrossRef] [PubMed]
- Knispel, S.; Stang, A.; Zimmer, L.; Lax, H.; Gutzmer, R.; Heinzerling, L.; Weishaupt, C.; Pföhler, C.; Gesierich, A.; Herbst, R.; et al. Impact of a preceding radiotherapy on the outcome of immune checkpoint inhibition in metastatic melanoma: A multicenter retrospective cohort study of the DeCOG. J. Immunother. Cancer 2020, 8, e000395. [Google Scholar] [CrossRef] [PubMed]
- National Center for Biotechnology Information. PubChem Database. Cc-115, CID=58298318. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/Cc-115 (accessed on 18 May 2020).
- Lamm, N.; Rogers, S.; Cesare, A.J. The mTOR pathway: Implications for DNA replication. Prog. Biophys. Mol. Biol. 2019, 147, 17–25. [Google Scholar] [CrossRef] [PubMed]
- Sinclair, W.K. Cyclic X-Ray Responses in Mammalian Cells in Vitro. Radiat. Res. 1968, 33, 620–643. [Google Scholar] [CrossRef]
- Franken, N.A.P.; Rodermond, H.M.; Stap, J.; Haveman, J.; van Bree, C. Clonogenic assay of cells in vitro. Nat. Protoc. 2006, 1, 2315–2319. [Google Scholar] [CrossRef]
- Puck, T.T.; Marcus, P.I. Action of X-rays on Mammalian Cells. J. Exp. Med. 1956, 103, 653–666. [Google Scholar] [CrossRef]
- Akashi, Y.; Okamoto, I.; Iwasa, T.; Yoshida, T.; Suzuki, M.; Hatashita, E.; Yamada, Y.; Satoh, T.; Fukuoka, M.; Ono, K.; et al. Enhancement of the antitumor activity of ionising radiation by nimotuzumab, a humanised monoclonal antibody to the epidermal growth factor receptor, in non-small cell lung cancer cell lines of differing epidermal growth factor receptor status. Br. J. Cancer 2008, 98, 749–755. [Google Scholar] [CrossRef] [Green Version]
- Rahman, W.N.; Bishara, N.; Ackerly, T.; He, C.F.; Jackson, P.; Wong, C.; Davidson, R.; Geso, M. Enhancement of radiation effects by gold nanoparticles for superficial radiation therapy. Nanomed. Nanotechnol. Biol. Med. 2009, 5, 136–142. [Google Scholar] [CrossRef]
- Leith, J.T.; Lee, E.S.; Vayer, A.J., Jr.; Dexter, D.L.; Glicksman, A.S. Enhancement of the responses of human colon adenocarcinoma cells to X-irradiation and cis-platinum by N-methylformamide (NMF). Int. J. Radiat. Oncol. Biol. Phys. 1985, 11, 1971–1976. [Google Scholar] [CrossRef]
- Godin, S.K.; Sullivan, M.R.; Bernstein, K.A. Novel insights into RAD51 activity and regulation during homologous recombination and DNA replication. Biochem. Cell Biol. 2016, 94, 407–418. [Google Scholar] [CrossRef] [Green Version]
- Mukhopadhyay, A.; Elattar, A.; Cerbinskaite, A.; Wilkinson, S.J.; Drew, Y.; Kyle, S.; Los, G.; Hostomsky, Z.; Edmondson, R.J.; Curtin, N.J. Development of a Functional Assay for Homologous Recombination Status in Primary Cultures of Epithelial Ovarian Tumor and Correlation with Sensitivity to Poly(ADP-Ribose) Polymerase Inhibitors. Clin. Cancer Res. 2010, 16, 2344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pilié, P.G.; Gay, C.M.; Byers, L.A.; Connor, M.J.; Yap, T.A. PARP Inhibitors: Extending Benefit Beyond BRCA—Mutant Cancers. Clin. Cancer Res. 2019, 25, 3759. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weigert, V.; Jost, T.; Hecht, M.; Knippertz, I.; Heinzerling, L.; Fietkau, R.; Distel, L.V. PARP inhibitors combined with ionizing radiation induce different effects in melanoma cells and healthy fibroblasts. BMC Cancer 2020, 20, 775. [Google Scholar] [CrossRef] [PubMed]
- O’Connor, M.J. Targeting the DNA Damage Response in Cancer. Mol. Cell 2015, 60, 547–560. [Google Scholar] [CrossRef] [Green Version]
- Konstantinopoulos, P.A.; Ceccaldi, R.; Shapiro, G.I.; Andrea, A.D. Homologous Recombination Deficiency: Exploiting the Fundamental Vulnerability of Ovarian Cancer. Cancer Discov. 2015, 5, 1137. [Google Scholar] [CrossRef] [Green Version]
- Hoppe, M.M.; Sundar, R.; Tan, D.S.P.; Jeyasekharan, A.D. Biomarkers for Homologous Recombination Deficiency in Cancer. J. Natl. Cancer Inst. 2018, 110, 704–713. [Google Scholar] [CrossRef] [Green Version]
- Lord, C.J.; Ashworth, A. BRCAness revisited. Nat. Rev. Cancer 2016, 16, 110–120. [Google Scholar] [CrossRef]
- Citrin, D.E. Recent Developments in Radiotherapy. New Engl. J. Med. 2017, 377, 1065–1075. [Google Scholar] [CrossRef]
- Burjakow, K.; Fietkau, R.; Putz, F.; Achterberg, N.; Lettmaier, S.; Knippen, S. Fractionated stereotactic radiation therapy for adrenal metastases: Contributing to local tumor control with low toxicity. Strahlenther. Onkol. 2019, 195, 236–245. [Google Scholar] [CrossRef]
- Weissmann, T.; Lettmaier, S.; Roesch, J.; Mengling, V.; Bert, C.; Iro, H.; Hornung, J.; Janka, R.; Semrau, S.; Fietkau, R.; et al. Paragangliomas of the Head and Neck: Local Control and Functional Outcome Following Fractionated Stereotactic Radiotherapy. Am. J. Clin. Oncol. 2019, 42, 818–823. [Google Scholar] [CrossRef]
- Baehr, A.; Trog, D.; Oertel, M.; Welsch, S.; Kröger, K.; Grauer, O.; Haverkamp, U.; Eich, H.T. Re-irradiation for recurrent glioblastoma multiforme: A critical comparison of different concepts. Strahlenther. Onkol. 2020, 196, 457–464. [Google Scholar] [CrossRef] [PubMed]
- Pawlik, T.M.; Keyomarsi, K. Role of cell cycle in mediating sensitivity to radiotherapy. Int. J. Radiat. Oncol. Biol. Phys. 2004, 59, 928–942. [Google Scholar] [CrossRef] [PubMed]
- Agami, R.; Bernards, R. Distinct Initiation and Maintenance Mechanisms Cooperate to Induce G1 Cell Cycle Arrest in Response to DNA Damage. Cell 2000, 102, 55–66. [Google Scholar] [CrossRef] [Green Version]
- Saqcena, M.; Menon, D.; Patel, D.; Mukhopadhyay, S.; Chow, V.; Foster, D.A. Amino acids and mTOR mediate distinct metabolic checkpoints in mammalian G1 cell cycle. PLoS ONE 2013, 8, e74157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allen, C.; Halbrook, J.; Nickoloff, J.A. Interactive Competition Between Homologous Recombination and Non-Homologous End Joining. Mol. Cancer Res. 2003, 1, 913. [Google Scholar] [PubMed]
- Tavecchio, M.; Munck, J.M.; Cano, C.; Newell, D.R.; Curtin, N.J. Further characterisation of the cellular activity of the DNA-PK inhibitor, NU7441, reveals potential cross-talk with homologous recombination. Cancer Chemother. Pharmacol. 2012, 69, 155–164. [Google Scholar] [CrossRef] [PubMed]
- Urushihara, Y.; Kobayashi, J.; Matsumoto, Y.; Komatsu, K.; Oda, S.; Mitani, H. DNA-PK inhibition causes a low level of H2AX phosphorylation and homologous recombination repair in Medaka (Oryzias latipes) cells. Biochem. Biophys. Res. Commun. 2012, 429, 131–136. [Google Scholar] [CrossRef]
- Ceccaldi, R.; Rondinelli, B.; D’Andrea, A.D. Repair Pathway Choices and Consequences at the Double-Strand Break. Trends Cell Biol. 2016, 26, 52–64. [Google Scholar] [CrossRef] [Green Version]
- Hill, R.; Lee, P.W. The DNA-dependent protein kinase (DNA-PK): More than just a case of making ends meet? Cell Cycle 2010, 9, 3460–3469. [Google Scholar] [CrossRef]
- Dong, J.; Zhang, T.; Ren, Y.; Wang, Z.; Ling, C.C.; He, F.; Li, G.C.; Wang, C.; Wen, B. Inhibiting DNA-PKcs in a non-homologous end-joining pathway in response to DNA double-strand breaks. Oncotarget 2017, 8, 22662–22673. [Google Scholar] [CrossRef] [Green Version]
- Lapalombella, R. Two strikes against CLL. Blood 2016, 128, 470–471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Collis, S.J.; DeWeese, T.L.; Jeggo, P.A.; Parker, A.R. The life and death of DNA-PK. Oncogene 2005, 24, 949–961. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lips, J.; Kaina, B. DNA double-strand breaks trigger apoptosis in p53-deficient fibroblasts. Carcinogenesis 2001, 22, 579–585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Virág, L.; Robaszkiewicz, A.; Rodriguez-Vargas, J.M.; Oliver, F.J. Poly (ADP-ribose) signaling in cell death. Mol. Asp. Med. 2013, 34, 1153–1167. [Google Scholar] [CrossRef] [PubMed]
- Beebe, J.; Zhang, J.-T. CC-115, a dual mTOR/DNA-PK inhibitor in clinical trial, is a substrate of ABCG2, a risk factor for CC-115 resistance. J. Pharmacol. Exp. Ther. 2019. [Google Scholar] [CrossRef]
- Hosoi, H.; Dilling, M.B.; Shikata, T.; Liu, L.N.; Shu, L.; Ashmun, R.A.; Germain, G.S.; Abraham, R.T.; Houghton, P.J. Rapamycin Causes Poorly Reversible Inhibition of mTOR and Induces p53-independent Apoptosis in Human Rhabdomyosarcoma Cells. Cancer Res. 1999, 59, 886. [Google Scholar]
- Kim, L.C.; Cook, R.S.; Chen, J. mTORC1 and mTORC2 in cancer and the tumor microenvironment. Oncogene 2017, 36, 2191–2201. [Google Scholar] [CrossRef] [Green Version]
- Sini, P.; James, D.; Chresta, C.; Guichard, S. Simultaneous inhibition of mTORC1 and mTORC2 by mTOR kinase inhibitor AZD8055 induces autophagy and cell death in cancer cells. Autophagy 2010, 6, 553–554. [Google Scholar] [CrossRef] [Green Version]
- Walter, L.; Heinzerling, L. BRAF Inhibitors and Radiation Do Not Act Synergistically to Inhibit WT and V600E BRAF Human Melanoma. Anticancer. Res. 2018, 38, 1335–1341. [Google Scholar] [CrossRef]
- Hecht, M.; Harrer, T.; Körber, V.; Sarpong, E.O.; Moser, F.; Fiebig, N.; Schwegler, M.; Stürzl, M.; Fietkau, R.; Distel, L.V. Cytotoxic effect of Efavirenz in BxPC-3 pancreatic cancer cells is based on oxidative stress and is synergistic with ionizing radiation. Oncol. Lett. 2018, 15, 1728–1736. [Google Scholar] [CrossRef]
- Mannello, F.; Tonti, G.A. Concise review: No breakthroughs for human mesenchymal and embryonic stem cell culture: Conditioned medium, feeder layer, or feeder-free; medium with fetal calf serum, human serum, or enriched plasma; serum-free, serum replacement nonconditioned medium, or ad hoc formula? All that glitters is not gold! Stem Cells 2007, 25, 1603–1609. [Google Scholar] [CrossRef] [PubMed]
- Colzani, M.; Waridel, P.; Laurent, J.; Faes, E.; Ruegg, C.; Quadroni, M. Metabolic labeling and protein linearization technology allow the study of proteins secreted by cultured cells in serum-containing media. J. Proteome Res. 2009, 8, 4779–4788. [Google Scholar] [CrossRef] [PubMed]
- Buch, K.; Peters, T.; Nawroth, T.; Sanger, M.; Schmidberger, H.; Langguth, P. Determination of cell survival after irradiation via clonogenic assay versus multiple MTT Assay—A comparative study. Radiat. Oncol. 2012, 7, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bürkel, F.; Jost, T.; Hecht, M.; Heinzerling, L.; Fietkau, R.; Distel, L. Dual mTOR/DNA-PK Inhibitor CC-115 Induces Cell Death in Melanoma Cells and Has Radiosensitizing Potential. Int. J. Mol. Sci. 2020, 21, 9321. https://doi.org/10.3390/ijms21239321
Bürkel F, Jost T, Hecht M, Heinzerling L, Fietkau R, Distel L. Dual mTOR/DNA-PK Inhibitor CC-115 Induces Cell Death in Melanoma Cells and Has Radiosensitizing Potential. International Journal of Molecular Sciences. 2020; 21(23):9321. https://doi.org/10.3390/ijms21239321
Chicago/Turabian StyleBürkel, Felix, Tina Jost, Markus Hecht, Lucie Heinzerling, Rainer Fietkau, and Luitpold Distel. 2020. "Dual mTOR/DNA-PK Inhibitor CC-115 Induces Cell Death in Melanoma Cells and Has Radiosensitizing Potential" International Journal of Molecular Sciences 21, no. 23: 9321. https://doi.org/10.3390/ijms21239321
APA StyleBürkel, F., Jost, T., Hecht, M., Heinzerling, L., Fietkau, R., & Distel, L. (2020). Dual mTOR/DNA-PK Inhibitor CC-115 Induces Cell Death in Melanoma Cells and Has Radiosensitizing Potential. International Journal of Molecular Sciences, 21(23), 9321. https://doi.org/10.3390/ijms21239321