Alpha Ketoglutarate Exerts In Vitro Anti-Osteosarcoma Effects through Inhibition of Cell Proliferation, Induction of Apoptosis via the JNK and Caspase 9-Dependent Mechanism, and Suppression of TGF-β and VEGF Production and Metastatic Potential of Cells

 ,
,  ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
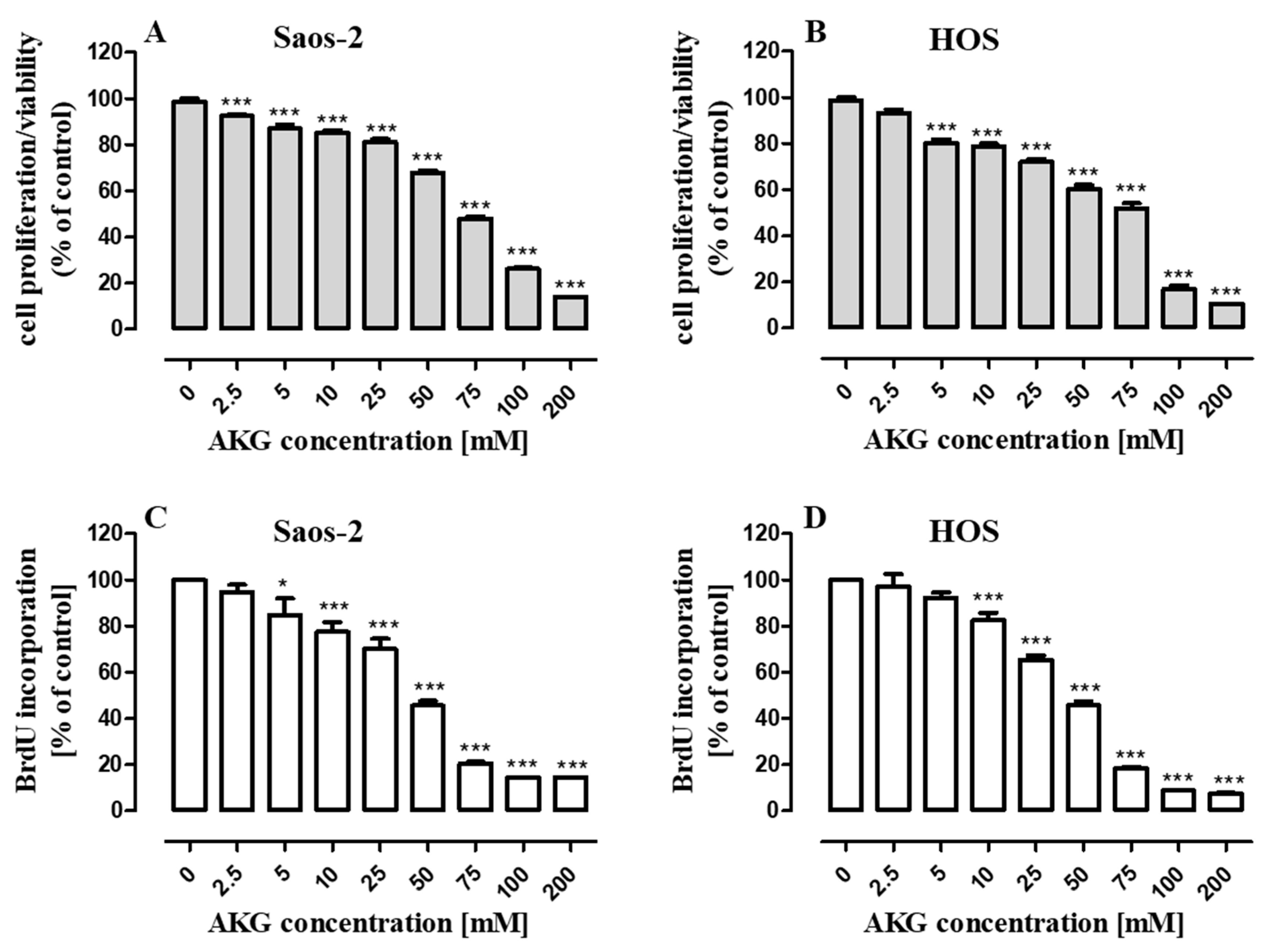
2.1. AKG Inhibits Proliferation of OS Cells
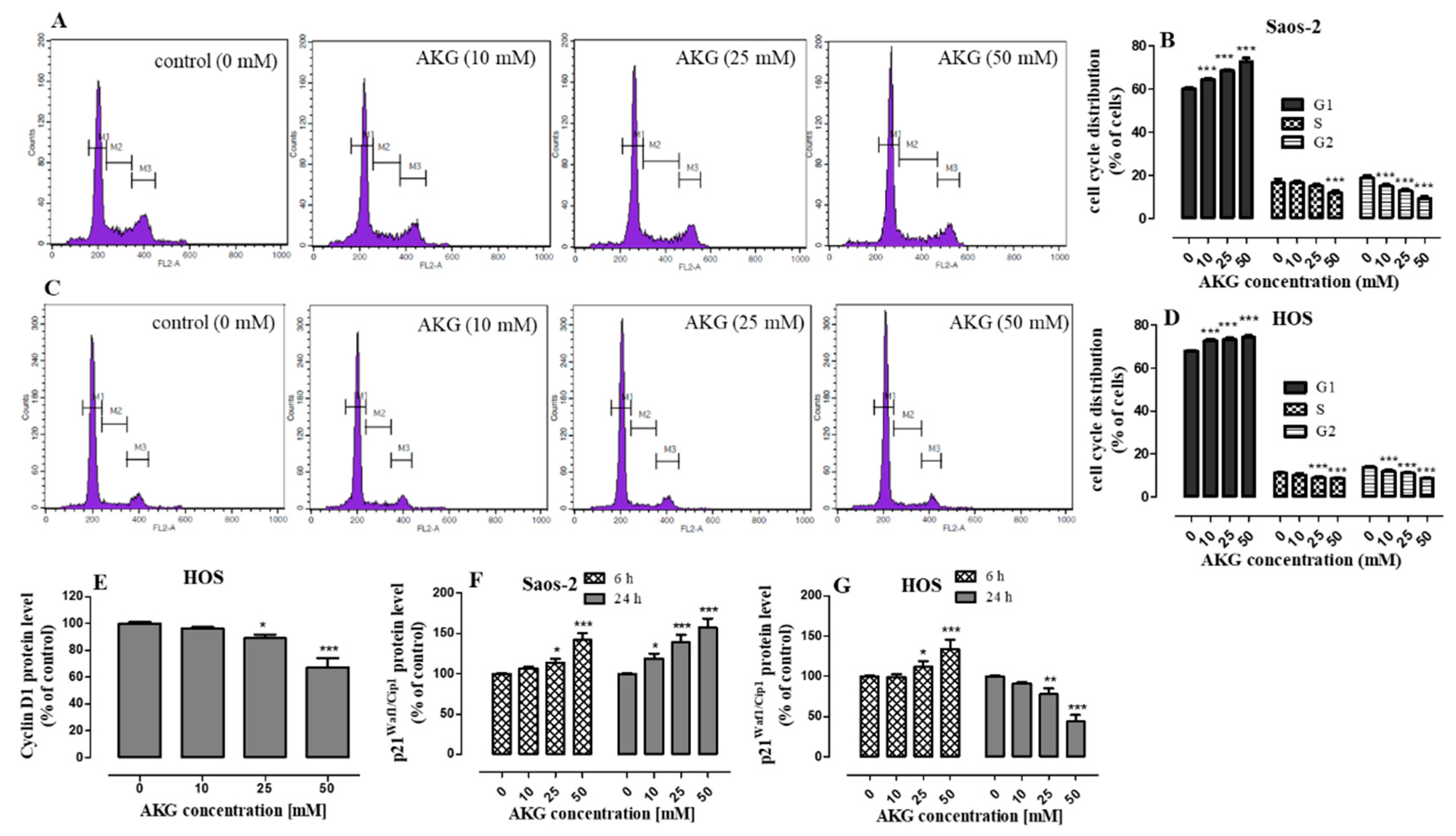
2.2. AKG Induces Cell Cycle Arrest in the G1 Phase in OS Cells through Modulation of the Expression of Cell Cycle-Associated Proteins
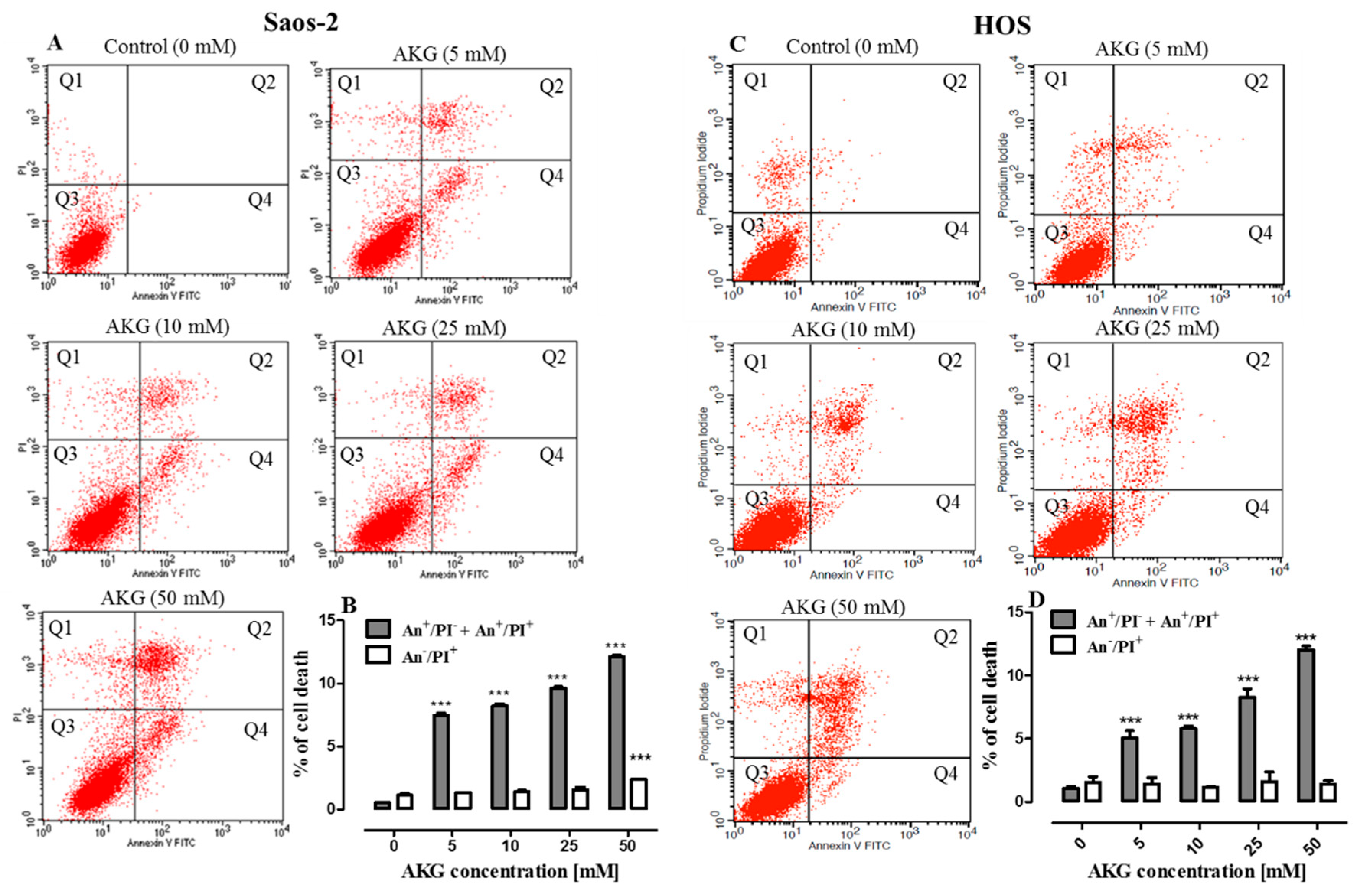
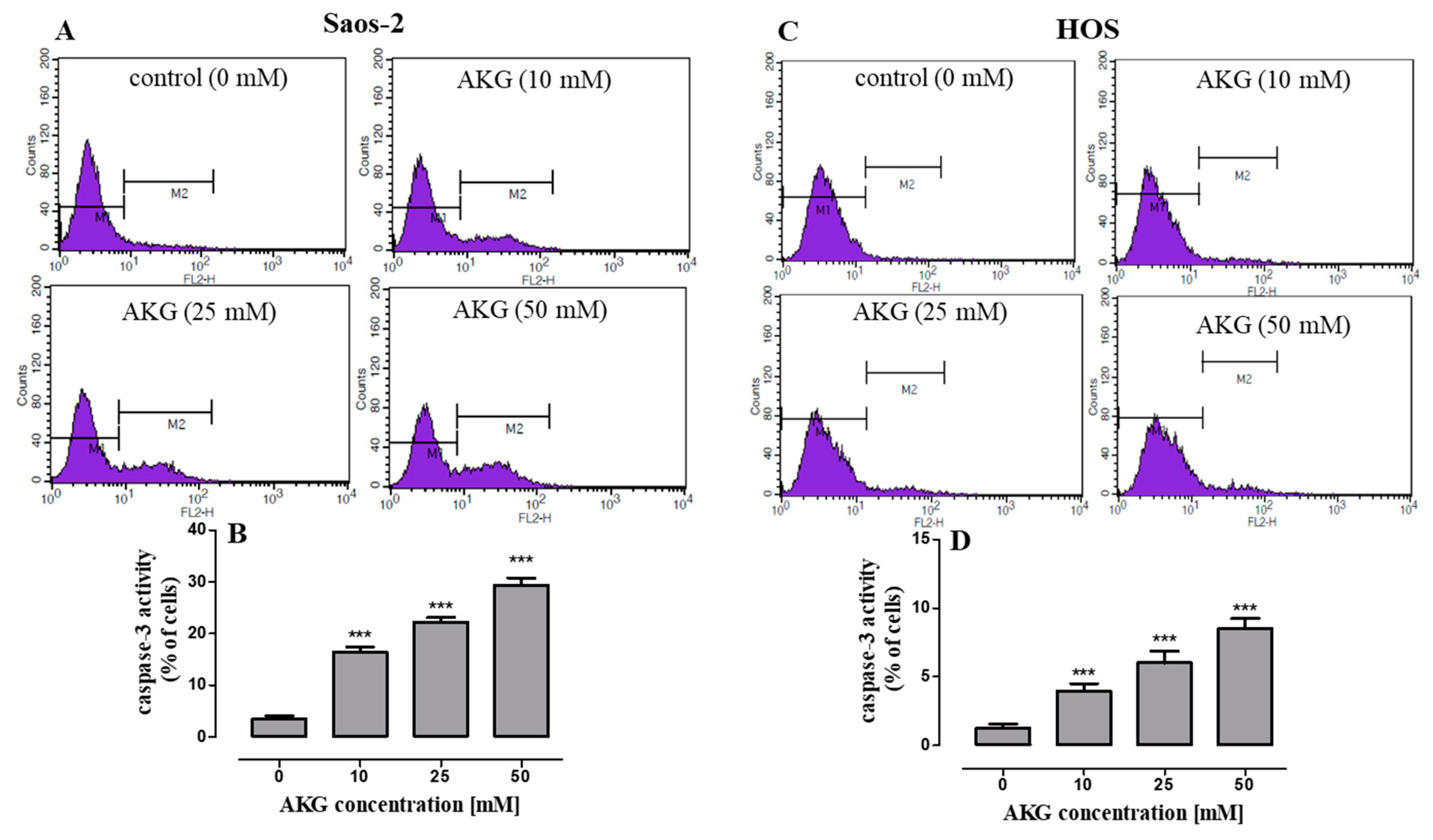
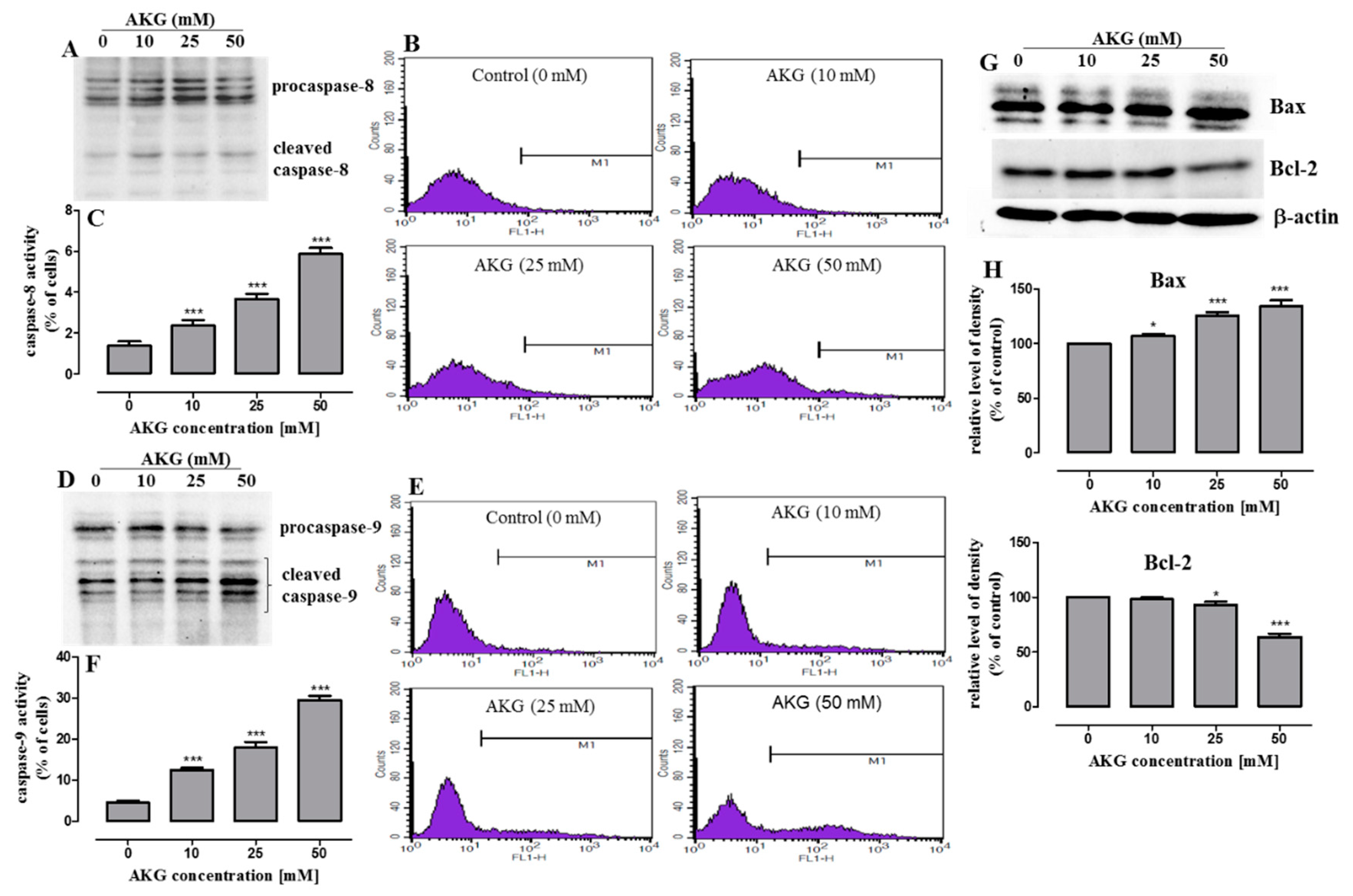
2.3. AKG Induces Cell Death in OS Cells through Apoptosis via an Intrinsic Caspase-Dependent Pathway
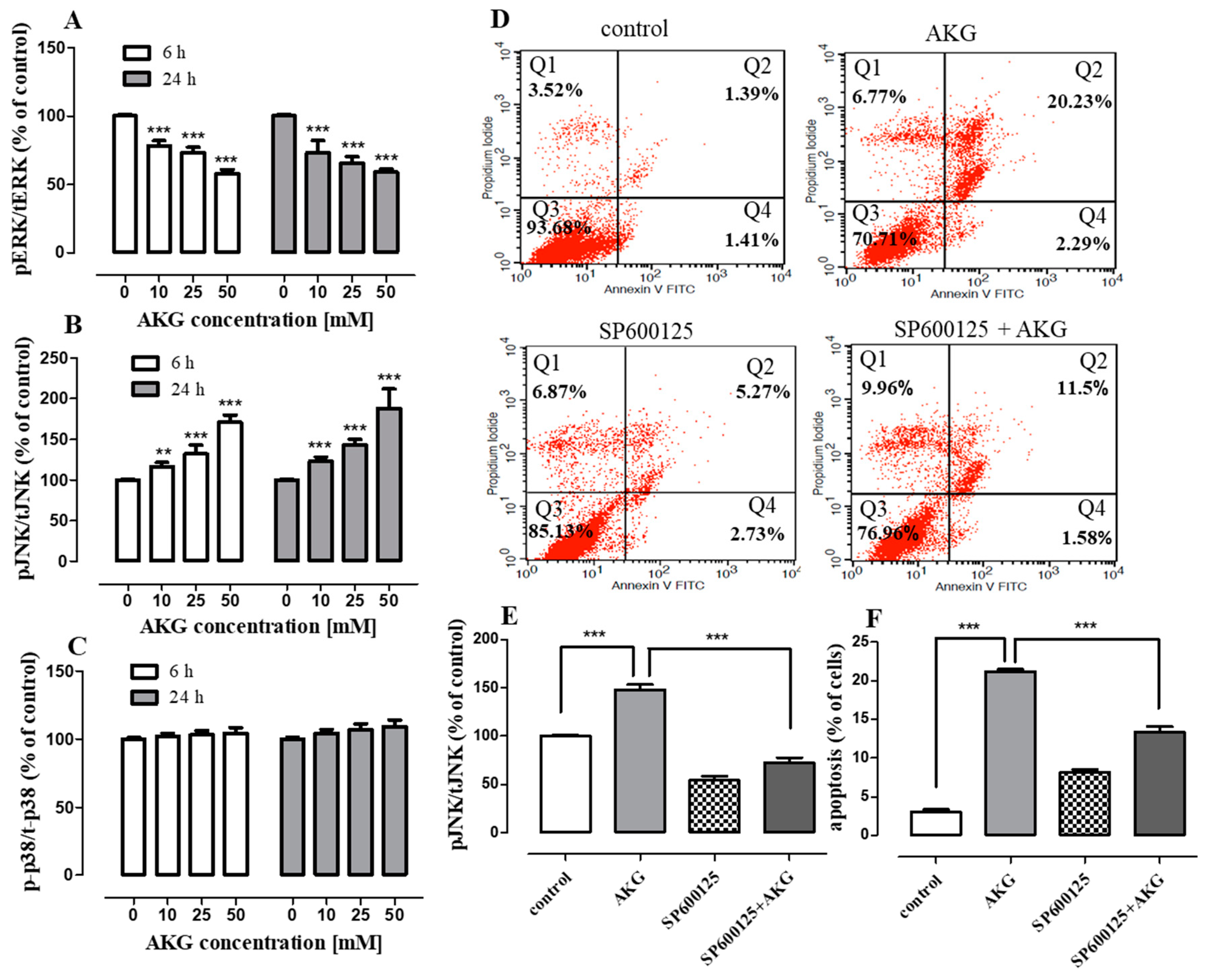
2.4. AKG Modulates the Phosphorylation of Mitogen-Activated Protein Kinases and Induces Apoptosis in OS Cells through a c-Jun N-Terminal Protein Kinase (JNK)-Dependent Mechanism
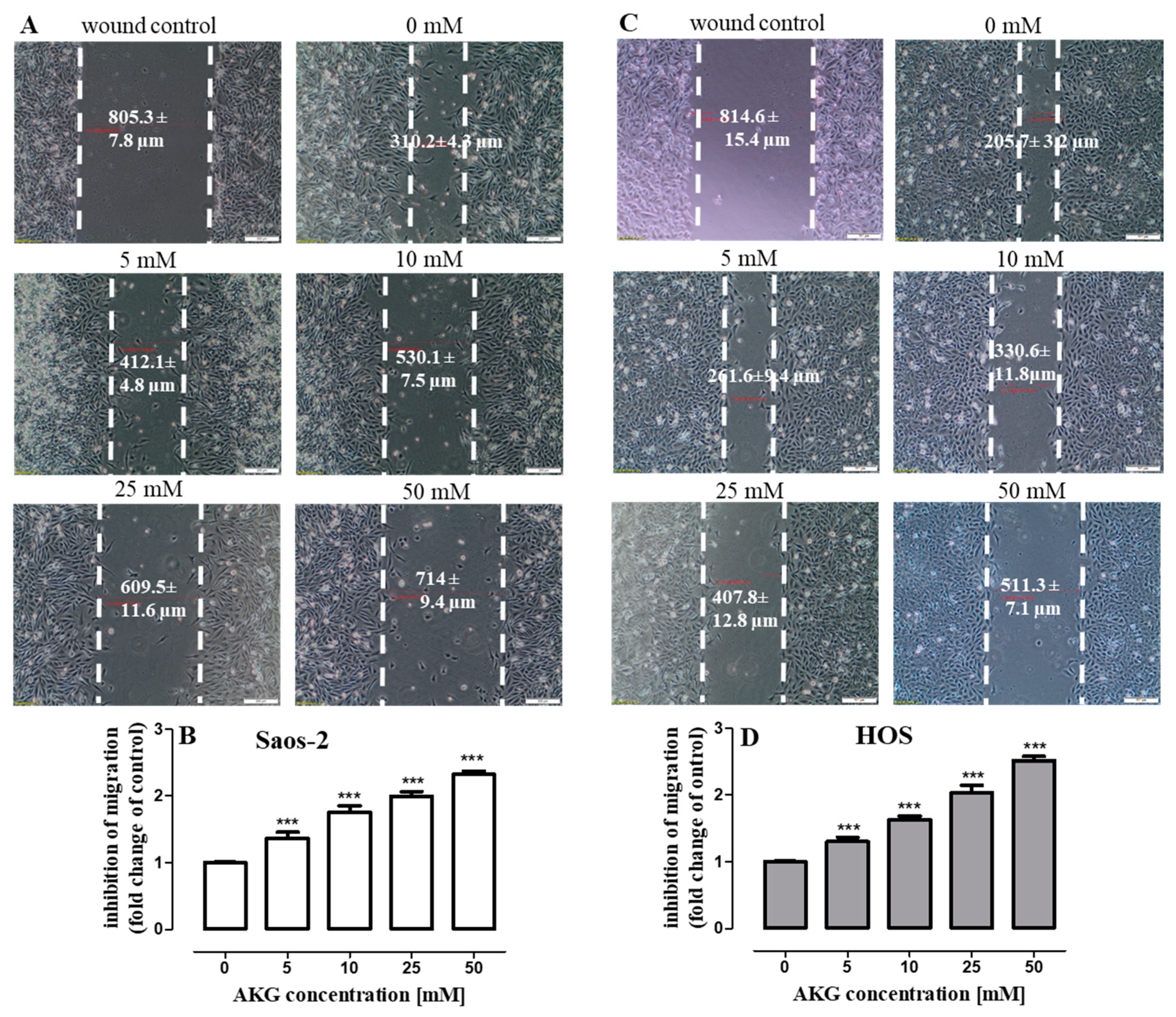
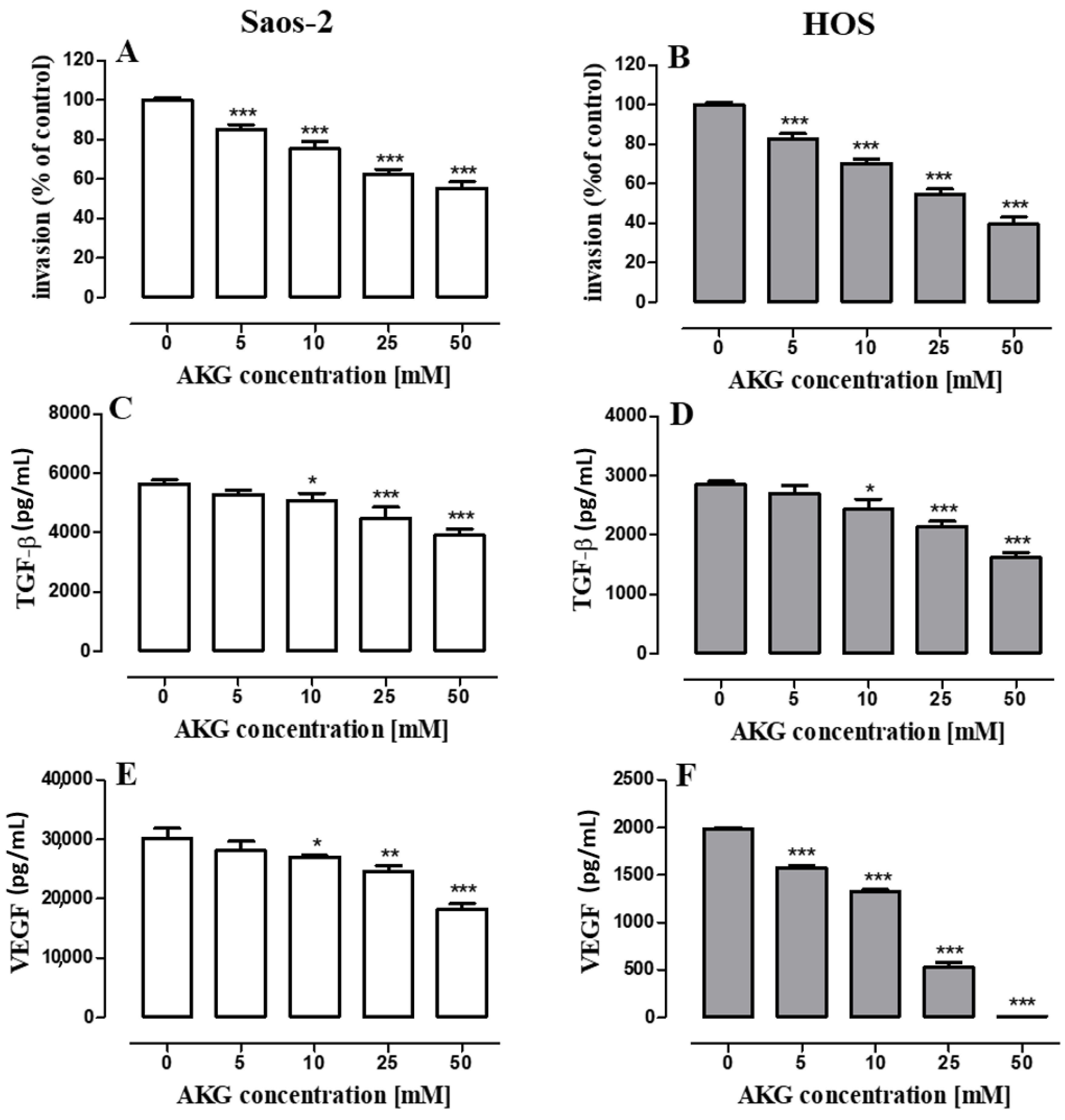
2.5. AKG Inhibits the Migration and Invasiveness of OS Cells and Decreases the Production of VEGF and TGF-β in These Cells
3. Discussion
4. Materials and Methods
4.1. Cell Culture and AKG
4.2. Cell Proliferation Assays
4.3. Cell Cycle Analysis
4.4. Flow Cytometry
4.5. Immunoblotting Analysis
4.6. ELISA Assays
4.7. PathScan ELISA Assays
4.8. Cell Migration Assay
4.9. Cell Invasion Assay
4.10. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Klein, M.J.; Siegal, G.P. Osteosarcoma. Am. J. Clin. Pathol. 2006, 125, 555–581. [Google Scholar] [CrossRef] [PubMed]
- Damron, T.A.; Ward, W.G.; Stewart, A. Osteosarcoma, chondrosarcoma, and Ewing’s sarcoma: National cancer data base report. Clin. Orthop. Relat. Res. 2007, 459, 40–47. [Google Scholar] [CrossRef] [PubMed]
- Mirabello, L.; Troisi, R.J.; Savage, S.A. International osteosarcoma incidence patterns in children and adolescents, middle ages and elderly persons. Int. J. Cancer 2009, 125, 229–234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Savage, S.A.; Mirabello, L. Using epidemiology and genomics to understand osteosarcoma etiology. Sarcoma 2011, 2011, 548151. [Google Scholar] [CrossRef] [Green Version]
- Gill, J.; Ahluwalia, M.K.; Geller, D.; Gorlick, R. New targets and approaches in osteosarcoma. Pharmacol. Ther. 2013, 137, 89–99. [Google Scholar] [CrossRef]
- Czarnecka, A.M.; Synoradzki, K.; Firlej, W.; Bartnik, E.; Sobczuk, P.; Fiedorowicz, M.; Grieb, P.; Rutkowski, P. Molecular biology of osteosarcoma. Cancers 2020, 12, 2130. [Google Scholar] [CrossRef]
- Akram, M. Citric Acid Cycle and Role of its Intermediates in Metabolism. Cell Biochem. Biophys. 2014, 68, 475–478. [Google Scholar] [CrossRef]
- Zdzisińska, B.; Żurek, A.; Kandefer-Szerszeń, M. Alpha-Ketoglutarate as a Molecule with Pleiotropic Activity: Well-Known and Novel Possibilities of Therapeutic Use. Arch. Immunol. Ther. Exp. (Warsz.) 2017, 65, 21–36. [Google Scholar] [CrossRef] [Green Version]
- Dalziel, K. Isocitrate dehydrogenase and related oxidative decarboxylases. FEBS Lett. 1980, 117, K45–K55. [Google Scholar] [CrossRef] [Green Version]
- Abla, H.; Sollazzo, M.; Gasparre, G.; Iommarini, L.; Porcelli, A.M. The multifaceted contribution of α-ketoglutarate to tumor progression: An opportunity to exploit? Semin. Cell Dev. Biol. 2020, 98, 26–33. [Google Scholar] [CrossRef]
- Tennant, D.A.; Frezza, C.; MacKenzie, E.D.; Nguyen, Q.D.; Zheng, L.; Selak, M.A.; Roberts, D.L.; Dive, C.; Watson, D.G.; Aboagye, E.O.; et al. Reactivating HIF prolyl hydroxylases under hypoxia results in metabolic catastrophe and cell death. Oncogene 2009, 28, 4009–4021. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- MacKenzie, E.D.; Selak, M.A.; Tennant, D.A.; Payne, L.J.; Crosby, S.; Frederiksen, C.M.; Watson, D.G.; Gottlieb, E. Cell-Permeating α-Ketoglutarate Derivatives Alleviate Pseudohypoxia in Succinate Dehydrogenase-Deficient Cells. Mol. Cell. Biol. 2007, 27, 3282–3289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blanquart, C.; Linot, C.; Cartron, P.-F.; Tomaselli, D.; Mai, A.; Bertrand, P. Epigenetic Metalloenzymes. Curr. Med. Chem. 2018, 26, 2748–2785. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Ibekwe, E.; Chornenkyy, Y. Metabolic alterations in cancer cells and the emerging role of oncometabolites as drivers of neoplastic change. Antioxidants 2018, 7, 16. [Google Scholar] [CrossRef] [Green Version]
- Xu, W.; Yang, H.; Liu, Y.; Yang, Y.; Wang, P.; Kim, S.H.; Ito, S.; Yang, C.; Wang, P.; Xiao, M.T.; et al. Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of α-ketoglutarate-dependent dioxygenases. Cancer Cell 2011, 19, 17–30. [Google Scholar] [CrossRef] [Green Version]
- Letouzé, E.; Martinelli, C.; Loriot, C.; Burnichon, N.; Abermil, N.; Ottolenghi, C.; Janin, M.; Menara, M.; Nguyen, A.T.; Benit, P.; et al. SDH Mutations Establish a Hypermethylator Phenotype in Paraganglioma. Cancer Cell 2013, 23, 739–752. [Google Scholar] [CrossRef] [Green Version]
- Atlante, S.; Visintin, A.; Marini, E.; Savoia, M.; Dianzani, C.; Giorgis, M.; Sürün, D.; Maione, F.; Schnütgen, F.; Farsetti, A.; et al. α-ketoglutarate dehydrogenase inhibition counteracts breast cancer-associated lung metastasis article. Cell Death Dis. 2018, 9, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Sun, X.; Zhu, M.J. Butyrate Inhibits Indices of Colorectal Carcinogenesis via Enhancing α-Ketoglutarate-Dependent DNA Demethylation of Mismatch Repair Genes. Mol. Nutr. Food Res. 2018, 62, e1700932. [Google Scholar] [CrossRef]
- Tseng, C.W.; Kuo, W.H.; Chan, S.H.; Chan, H.L.; Chang, K.J.; Wang, L.H. Transketolase Regulates the Metabolic Switch to Control Breast Cancer Cell Metastasis via the a-Ketoglutarate Signaling Pathway. Cancer Res. 2018, 78, 2799–2812. [Google Scholar] [CrossRef] [Green Version]
- Matsumoto, K.; Imagawa, S.; Obara, N.; Suzuki, N.; Takahashi, S.; Nagasawa, T.; Yamamoto, M. 2-Oxoglutarate downregulates expression of vascular endothelial growth factor and erythropoietin through decreasing hypoxia-inducible factor-1α and inhibits angiogenesis. J. Cell. Physiol. 2006, 209, 333–340. [Google Scholar] [CrossRef]
- Matsumoto, K.; Obara, N.; Ema, M.; Horie, M.; Naka, A.; Takahashi, S.; Imagawa, S. Antitumor effects of 2-oxoglutarate through inhibition of angiogenesis in a murine tumor model. Cancer Sci. 2009, 100, 1639–1647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rzeski, W.; Walczak, K.; Juszczak, M.; Langner, E.; PoŻarowski, P.; Kandefer-Szerszeń, M.; Pierzynowski, S.G. Alpha-ketoglutarate (AKG) inhibits proliferation of colon adenocarcinoma cells in normoxic conditions. Scand. J. Gastroenterol. 2012, 47, 565–571. [Google Scholar] [CrossRef] [PubMed]
- Tennant, D.A.; Gottlieb, E. HIF prolyl hydroxylase-3 mediates alpha-ketoglutarate-induced apoptosis and tumor suppression. J. Mol. Med. 2010, 88, 839–849. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Liu, Y.; Qin, C.; Pan, Z.; Luo, J.; Yu, A.; Cheng, Z. Up-regulated isocitrate dehydrogenase 1 suppresses proliferation, migration and invasion in osteosarcoma: In vitro and in vivo. Cancer Lett. 2014, 346, 114–121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morris, J.P.; Yashinskie, J.J.; Koche, R.; Chandwani, R.; Tian, S.; Chen, C.C.; Baslan, T.; Marinkovic, Z.S.; Sánchez-Rivera, F.J.; Leach, S.D.; et al. α-Ketoglutarate links p53 to cell fate during tumour suppression. Nature 2019, 573, 595–599. [Google Scholar] [CrossRef]
- Morrow, J.J.; Khanna, C. Osteosarcoma genetics and epigenetics: Emerging biology and candidate therapies. Crit. Rev. Oncog. 2015, 20, 173–197. [Google Scholar] [CrossRef] [Green Version]
- Amary, M.F.; Bacsi, K.; Maggiani, F.; Damato, S.; Halai, D.; Berisha, F.; Pollock, R.; O’Donnell, P.; Grigoriadis, A.; Diss, T.; et al. IDH1 and IDH2 mutations are frequent events in central chondrosarcoma and central and periosteal chondromas but not in other mesenchymal tumours. J. Pathol. 2011, 224, 334–343. [Google Scholar] [CrossRef]
- Anderson, N.M.; Mucka, P.; Kern, J.G.; Feng, H. The emerging role and targetability of the TCA cycle in cancer metabolism. Protein Cell 2018, 9, 216–237. [Google Scholar] [CrossRef]
- Liu, X.; Kato, Y.; Kaneko, M.K.; Sugawara, M.; Ogasawara, S.; Tsujimoto, Y.; Naganuma, Y.; Yamakawa, M.; Tsuchiya, T.; Takagi, M. Isocitrate dehydrogenase 2 mutation is a frequent event in osteosarcoma detected by a multi-specific monoclonal antibody MsMab-1. Cancer Med. 2013, 2, 803–814. [Google Scholar] [CrossRef]
- Hu, X.; Yu, A.-X.; Qi, B.-W.; Fu, T.; Wu, G.; Zhou, M.; Luo, J.; Xu, J.-H. The expression and significance of IDH1 and p53 in osteosarcoma. J. Exp. Clin. Cancer Res. 2010, 29, 43. [Google Scholar] [CrossRef] [Green Version]
- Yi, W.R.; Li, Z.H.; Qi, B.W.; Hu, X.; Yu, A.X. Downregulation of IDH2 exacerbates the malignant progression of osteosarcoma cells via increased NF-?B and MMP-9 activation. Oncol. Rep. 2016, 35, 2277–2285. [Google Scholar] [CrossRef] [PubMed]
- Ewen, M.E.; Sluss, H.K.; Sherr, C.J.; Matsushime, H.; Kato, J.Y.; Livingston, D.M. Functional interactions of the retinoblastoma protein with mammalian D-type cyclins. Cell 1993, 73, 487–497. [Google Scholar] [CrossRef]
- Dhanasekaran, D.N.; Premkumar Reddy, E. JNK-signaling: A multiplexing hub in programmed cell death. Genes Cancer 2017, 8, 682–694. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, X.; Zhao, J.; Bai, J.; Shen, H.; Zhang, B.; Deng, L.; Sun, C.; Liu, Y.; Zhang, J.; Zheng, J. Risk and clinicopathological features of osteosarcoma metastasis to the lung: A population-based study. J. Bone Oncol. 2019, 16, 100230. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Yang, J.; Zhao, N.; Wang, C.; Kamar, S.; Zhou, Y.; He, Z.; Yang, J.; Sun, B.; Shi, X.; et al. Progress in the chemotherapeutic treatment of osteosarcoma. Oncol. Lett. 2018, 16, 6228–6237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Żurek, A.; Mizerska-Kowalska, M.; Sławińska-Brych, A.; Kaławaj, K.; Bojarska-Junak, A.; Kandefer-Szerszeń, M.; Zdzisińska, B. Alpha ketoglutarate exerts a pro-osteogenic effect in osteoblast cell lines through activation of JNK and mTOR/S6K1/S6 signaling pathways. Toxicol. Appl. Pharmacol. 2019, 374, 53–64. [Google Scholar] [CrossRef]
- Qie, S.; Diehl, J.A. Cyclin D1, cancer progression, and opportunities in cancer treatment. J. Mol. Med. 2016, 94, 1313–1326. [Google Scholar] [CrossRef] [Green Version]
- Si, X.H.; Liu, Z. Expression of cyclin D1 and CDK4 in osteosarcoma of the jaws. Chin. J. Cancer Res. 2001, 13, 140–143. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Shen, J.K.; Yu, Z.; Hornicek, F.J.; Kan, Q.; Duan, Z. Expression and therapeutic implications of cyclin-dependent kinase 4 (CDK4) in osteosarcoma. Biochim. Biophys. Acta-Mol. Basis Dis. 2018, 1864, 1573–1582. [Google Scholar] [CrossRef]
- Abbas, T.; Dutta, A. P21 in cancer: Intricate networks and multiple activities. Nat. Rev. Cancer 2009, 9, 400–414. [Google Scholar] [CrossRef]
- He, Y.; Yu, B. MicroRNA-93 promotes cell proliferation by directly targeting P21 in osteosarcoma cells. Exp. Ther. Med. 2017, 13, 2003–2011. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, X.; Deng, M.; Ma, L.; Zhou, J.; Xiao, Y.; Zhou, X.; Zhang, C.; Wu, M. Inhibitory effects of forkhead box L1 gene on osteosarcoma growth through the induction of cell cycle arrest and apoptosis. Oncol. Rep. 2015, 34, 265–271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.M.; Zhang, J.; Xia, Y.M.; Wang, X.X.; Li, J. The natural sweetener metabolite steviol inhibits the proliferation of human osteosarcoma U2OS cell line. Oncol. Lett. 2018, 15, 5250–5256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Yang, Z.; Li, Y.; Xia, J.; Li, D.; Li, H.; Ren, M.; Liao, Y.; Yu, S.; Chen, Y.; et al. Cell apoptosis, autophagy and necroptosis in osteosarcoma treatment. Oncotarget 2016, 7, 44763–44778. [Google Scholar] [CrossRef] [Green Version]
- Redza-Dutordoir, M.; Averill-Bates, D.A. Activation of apoptosis signalling pathways by reactive oxygen species. Biochim. Biophys. Acta-Mol. Cell Res. 2016, 1863, 2977–2992. [Google Scholar] [CrossRef]
- Boutros, T.; Chevet, E.; Metrakos, P. Mitogen-Activated Protein (MAP) kinase/MAP kinase phosphatase regulation: Roles in cell growth, death, and cancer. Pharmacol. Rev. 2008, 60, 261–310. [Google Scholar] [CrossRef]
- Guo, Y.; Pan, W.; Liu, S.; Shen, Z.; Xu, Y.; Hu, L. ERK/MAPK signalling pathway and tumorigenesis (Review). Exp. Ther. Med. 2020, 19, 1997–2007. [Google Scholar] [CrossRef] [Green Version]
- Wu, Q.; Wu, W.; Fu, B.; Shi, L.; Wang, X.; Kuca, K. JNK signaling in cancer cell survival. Med. Res. Rev. 2019, 39, 2082–2104. [Google Scholar] [CrossRef]
- Zhang, Y.; Chen, P.; Hong, H.; Wang, L.; Zhou, Y.; Lang, Y. JNK pathway mediates curcumin-induced apoptosis and autophagy in osteosarcoma MG63 cells. Exp. Ther. Med. 2017, 14, 593–599. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Li, H.; Chen, S.; Wang, Z.; Yao, Y.; Chen, T.; Ye, Z.; Lin, P. Andrographolide induces apoptosis in human osteosarcoma cells via the ROS/JNK pathway. Int. J. Oncol. 2020, 56, 1417–1428. [Google Scholar] [CrossRef] [Green Version]
- Tsuruta, F.; Sunayama, J.; Mori, Y.; Hattori, S.; Shimizu, S.; Tsujimoto, Y.; Yoshioka, K.; Masuyama, N.; Gotoh, Y. JNK promotes Bax translocation to mitochondria through phosphorylation of 14-3-3 proteins. EMBO J. 2004, 23, 1889–1899. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papadakis, E.S.; Finegan, K.G.; Wang, X.; Robinson, A.C.; Guo, C.; Kayahara, M.; Tournier, C. The regulation of Bax by c-Jun N-terminal protein kinase (JNK) is a prerequisite to the mitochondrial-induced apoptotic pathway. FEBS Lett. 2006, 580, 1320–1326. [Google Scholar] [CrossRef] [PubMed]
- Chandhanayingyong, C.; Kim, Y.; Staples, J.R.; Hahn, C.; Lee, F.Y. MAPK/ERK signaling in osteosarcomas, Ewing sarcomas and chondrosarcomas: Therapeutic implications and future directions. Sarcoma 2012, 2012, 404810. [Google Scholar] [CrossRef] [PubMed]
- Noh, K.; Kim, K.O.; Patel, N.R.; Staples, J.R.; Minematsu, H.; Nair, K.; Lee, F.Y.I. Targeting inflammatory kinase as an adjuvant treatment for osteosarcomas. J. Bone Jt. Surg.-Ser. A 2011, 93, 723–732. [Google Scholar] [CrossRef]
- Sasaki, K.; Hitora, T.; Nakamura, O.; Kono, R.; Yamamoto, T. The role of MAPK pathway in bone and soft tissue tumors. Anticancer Res. 2011, 31, 549–553. [Google Scholar]
- Yu, Y.; Luk, F.; Yang, J.-L.; Walsh, W.R. Ras/Raf/MEK/ERK pathway is associated with lung metastasis of osteosarcoma in an orthotopic mouse model. Anticancer Res. 2011, 31, 1147–1152. [Google Scholar]
- Salas, S.; Jiguet-Jiglaire, C.; Campion, L.; Bartoli, C.; Frassineti, F.; Deville, J.L.; Maues De Paula, A.; Forest, F.; Jézéquel, P.; Gentet, J.C.; et al. Correlation between ERK1 and STAT3 expression and chemoresistance in patients with conventional osteosarcoma. BMC Cancer 2014, 14, 606. [Google Scholar] [CrossRef] [Green Version]
- Pan, P.-J.; Liu, Y.-C.; Hsu, F.-T. Protein Kinase B and Extracellular Signal-Regulated Kinase Inactivation is Associated with Regorafenib-Induced Inhibition of Osteosarcoma Progression In Vitro and In Vivo. J. Clin. Med. 2019, 8, 900. [Google Scholar] [CrossRef] [Green Version]
- Verrecchia, F.; Rédini, F. Transforming growth factor-β signaling plays a pivotal role in the interplay between osteosarcoma cells and their microenvironment. Front. Oncol. 2018, 8, 133. [Google Scholar] [CrossRef]
- Lamora, A.; Talbot, J.; Mullard, M.; Brounais-Le Royer, B.; Redini, F.; Verrecchia, F. TGF-β Signaling in Bone Remodeling and Osteosarcoma Progression. J. Clin. Med. 2016, 5, 96. [Google Scholar] [CrossRef]
- Sung, J.Y.; Park, S.Y.; Kim, J.H.; Kang, H.G.; Yoon, J.H.; Na, Y.S.; Kim, Y.N.; Park, B.K. Interferon consensus sequence-binding protein (ICSBP) promotes epithelial-to-mesenchymal transition (EMT)-like phenomena, cell-motility, and invasion via TGF-β signaling in U2OS cells. Cell Death Dis. 2014, 5, e1224. [Google Scholar] [CrossRef]
- Lee, S.H.; Jeong, D.; Han, Y.S.; Baek, M.J. Pivotal role of vascular endothelial growth factor pathway in tumor angiogenesis. Ann. Surg. Treat. Res. 2015, 89, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Ohba, T.; Cates, J.M.M.; Cole, H.A.; Slosky, D.A.; Haro, H.; Ando, T.; Schwartz, H.S.; Schoenecker, J.G. Autocrine VEGF/VEGFR1 signaling in a subpopulation of cells associates with aggressive osteosarcoma. Mol. Cancer Res. 2014, 12, 1100–1111. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Yang, D.; Sun, Y.; Sun, B.; Wang, G.; Trent, J.C.; Araujo, D.M.; Chen, K.; Zhang, W. Genetic amplification of the vascular endothelial growth factor (VEGF) pathway genes, including VEGFA, in human osteosarcoma. Cancer 2011, 117, 4925–4938. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, D.; Zhang, Y.J.; Zhu, K.W.; Wang, W.C. A systematic review of vascular endothelial growth factor expression as a biomarker of prognosis in patients with osteosarcoma. Tumor Biol. 2013, 34, 1895–1899. [Google Scholar] [CrossRef] [PubMed]
- Kaya, M.; Wada, T.; Akatsuka, T.; Kawaguchi, S.; Nagoya, S.; Shindoh, M.; Higashino, F.; Mezawa, F.; Okada, F.; Ishii, S. Vascular endothelial growth factor expression in untreated osteosarcoma is predictive of pulmonary metastasis and poor prognosis. Clin. Cancer Res. 2000, 6, 572–577. [Google Scholar]
- Bajpai, J.; Sharma, M.; Sreenivas, V.; Kumar, R.; Gamnagatti, S.; Khan, S.A.; Rastogi, S.; Malhotra, A.; Bakhshi, S. VEGF expression as a prognostic marker in osteosarcoma. Pediatr. Blood Cancer 2009, 53, 1035–1039. [Google Scholar] [CrossRef] [PubMed]
- Peng, N.; Gao, S.; Guo, X.; Wang, G.; Cheng, C.; Li, M.; Liu, K. Silencing of VEGF inhibits human osteosarcoma angiogenesis and promotes cell apoptosis via VEGF/PI3K/AKT signaling pathway. Am. J. Transl. Res. 2016, 8, 1005–1015. [Google Scholar]
- Wunder, J.S.; Gokgoz, N.; Parkes, R.; Bull, S.B.; Eskandarian, S.; Davis, A.M.; Beauchamp, C.P.; Conrad, E.U.; Grimer, R.J.; Healey, J.H.; et al. TP53 mutations and outcome in osteosarcoma: A prospective, multicenter study. J. Clin. Oncol. 2005, 23, 1483–1490. [Google Scholar] [CrossRef]
- Chen, X.; Bahrami, A.; Pappo, A.; Easton, J.; Dalton, J.; Hedlund, E.; Ellison, D.; Shurtleff, S.; Wu, G.; Wei, L.; et al. Recurrent somatic structural variations contribute to tumorigenesis in pediatric osteosarcoma. Cell Rep. 2014, 7, 104–112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Slawinska-Brych, A.; Zdzisinska, B.; Mizerska-Dudka, M.; Kandefer-Szerszen, M. Induction of apoptosis in multiple myeloma cells by a statin-thalidomide combination can be enhanced by p38 MAPK inhibition. Leuk. Res. 2013, 37, 586–594. [Google Scholar] [CrossRef] [PubMed]
- Sławińska-Brych, A.; Król, S.K.; Dmoszyńska-Graniczka, M.; Zdzisińska, B.; Stepulak, A.; Gagoś, M. Xanthohumol inhibits cell cycle progression and proliferation of larynx cancer cells in vitro. Chem. Biol. Interact. 2015, 240, 110–118. [Google Scholar] [CrossRef] [PubMed]
- Bartnik, M.; Sławińska-Brych, A.; Żurek, A.; Kandefer-Szerszeń, M.; Zdzisińska, B. 8-methoxypsoralen reduces AKT phosphorylation, induces intrinsic and extrinsic apoptotic pathways, and suppresses cell growth of SK-N-AS neuroblastoma and SW620 metastatic colon cancer cells. J. Ethnopharmacol. 2017, 207, 19–29. [Google Scholar] [CrossRef] [PubMed]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kaławaj, K.; Sławińska-Brych, A.; Mizerska-Kowalska, M.; Żurek, A.; Bojarska-Junak, A.; Kandefer-Szerszeń, M.; Zdzisińska, B. Alpha Ketoglutarate Exerts In Vitro Anti-Osteosarcoma Effects through Inhibition of Cell Proliferation, Induction of Apoptosis via the JNK and Caspase 9-Dependent Mechanism, and Suppression of TGF-β and VEGF Production and Metastatic Potential of Cells. Int. J. Mol. Sci. 2020, 21, 9406. https://doi.org/10.3390/ijms21249406
Kaławaj K, Sławińska-Brych A, Mizerska-Kowalska M, Żurek A, Bojarska-Junak A, Kandefer-Szerszeń M, Zdzisińska B. Alpha Ketoglutarate Exerts In Vitro Anti-Osteosarcoma Effects through Inhibition of Cell Proliferation, Induction of Apoptosis via the JNK and Caspase 9-Dependent Mechanism, and Suppression of TGF-β and VEGF Production and Metastatic Potential of Cells. International Journal of Molecular Sciences. 2020; 21(24):9406. https://doi.org/10.3390/ijms21249406
Chicago/Turabian StyleKaławaj, Katarzyna, Adrianna Sławińska-Brych, Magdalena Mizerska-Kowalska, Aleksandra Żurek, Agnieszka Bojarska-Junak, Martyna Kandefer-Szerszeń, and Barbara Zdzisińska. 2020. "Alpha Ketoglutarate Exerts In Vitro Anti-Osteosarcoma Effects through Inhibition of Cell Proliferation, Induction of Apoptosis via the JNK and Caspase 9-Dependent Mechanism, and Suppression of TGF-β and VEGF Production and Metastatic Potential of Cells" International Journal of Molecular Sciences 21, no. 24: 9406. https://doi.org/10.3390/ijms21249406
APA StyleKaławaj, K., Sławińska-Brych, A., Mizerska-Kowalska, M., Żurek, A., Bojarska-Junak, A., Kandefer-Szerszeń, M., & Zdzisińska, B. (2020). Alpha Ketoglutarate Exerts In Vitro Anti-Osteosarcoma Effects through Inhibition of Cell Proliferation, Induction of Apoptosis via the JNK and Caspase 9-Dependent Mechanism, and Suppression of TGF-β and VEGF Production and Metastatic Potential of Cells. International Journal of Molecular Sciences, 21(24), 9406. https://doi.org/10.3390/ijms21249406