Congestive Hepatopathy

 , , , , , and
, , , , , and
Abstract
:1. Introduction
2. Epidemiology
3. Pathophysiology
4. Clinical Presentation and Diagnosis
5. Biochemical Profile
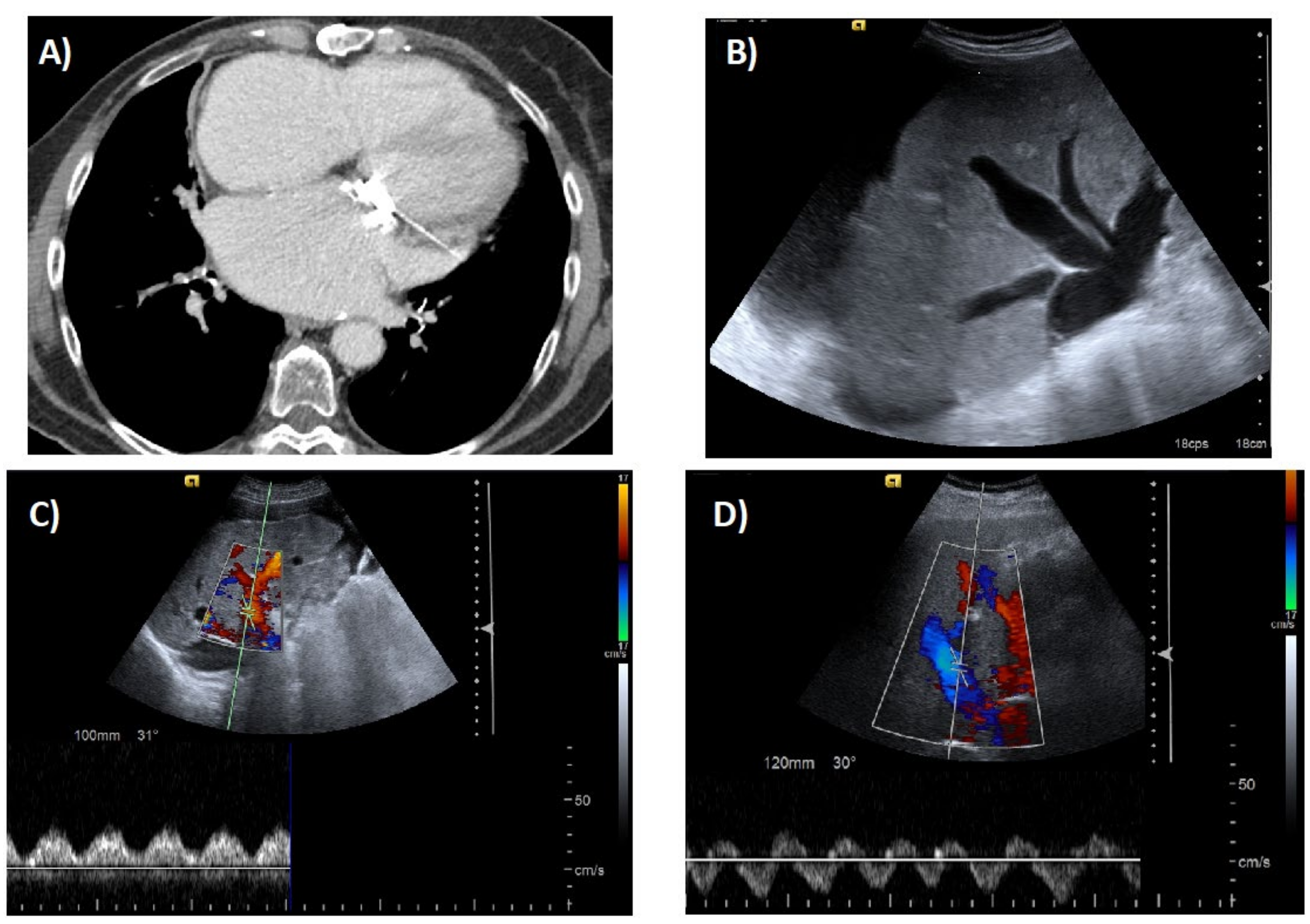
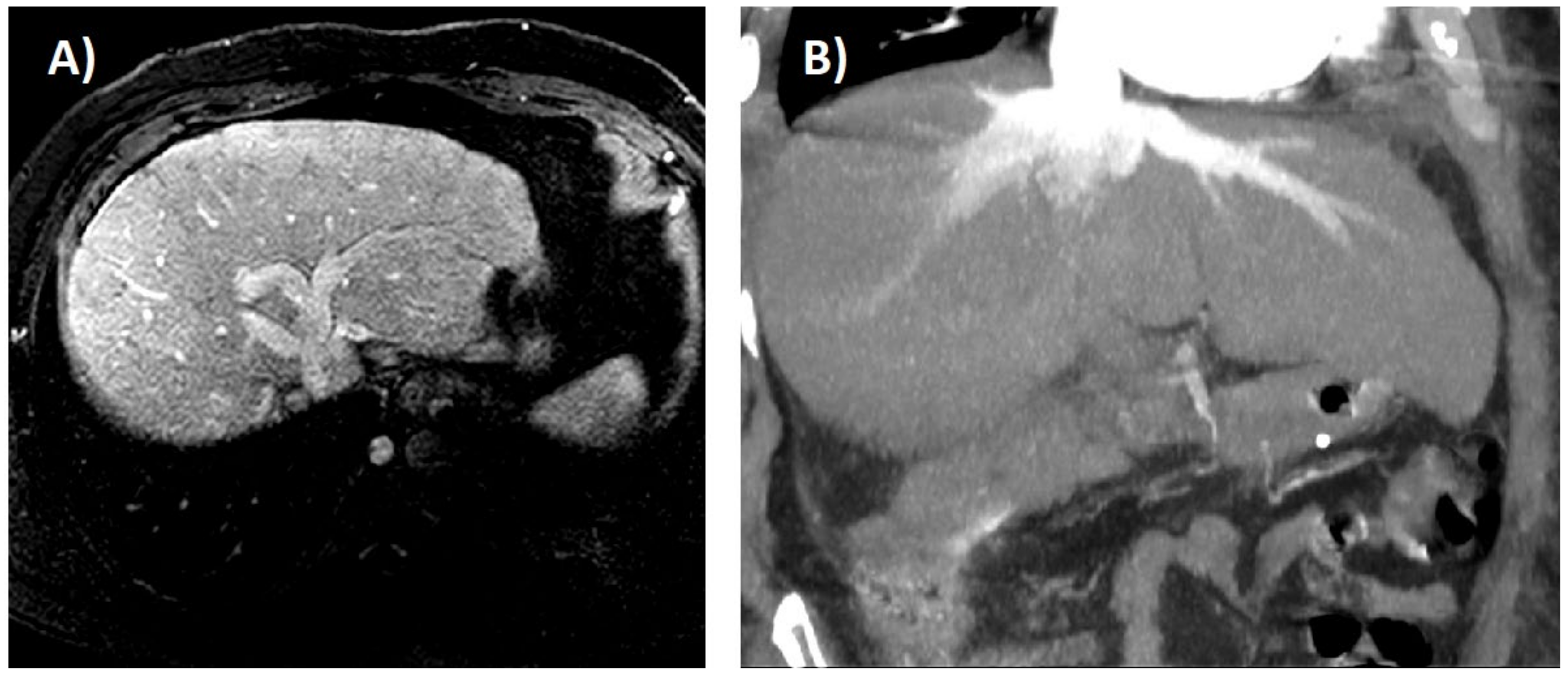
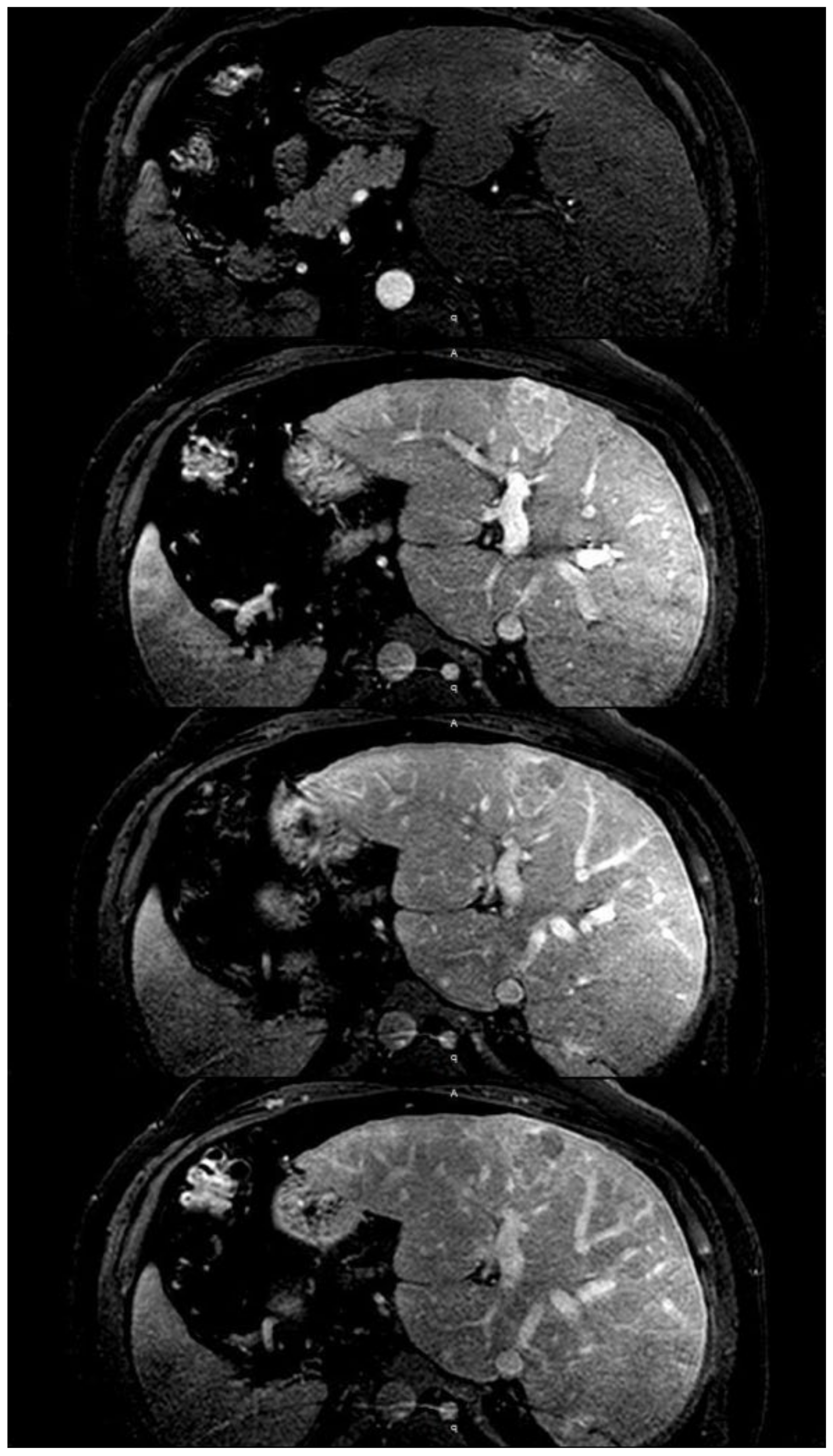
6. Imaging Tests
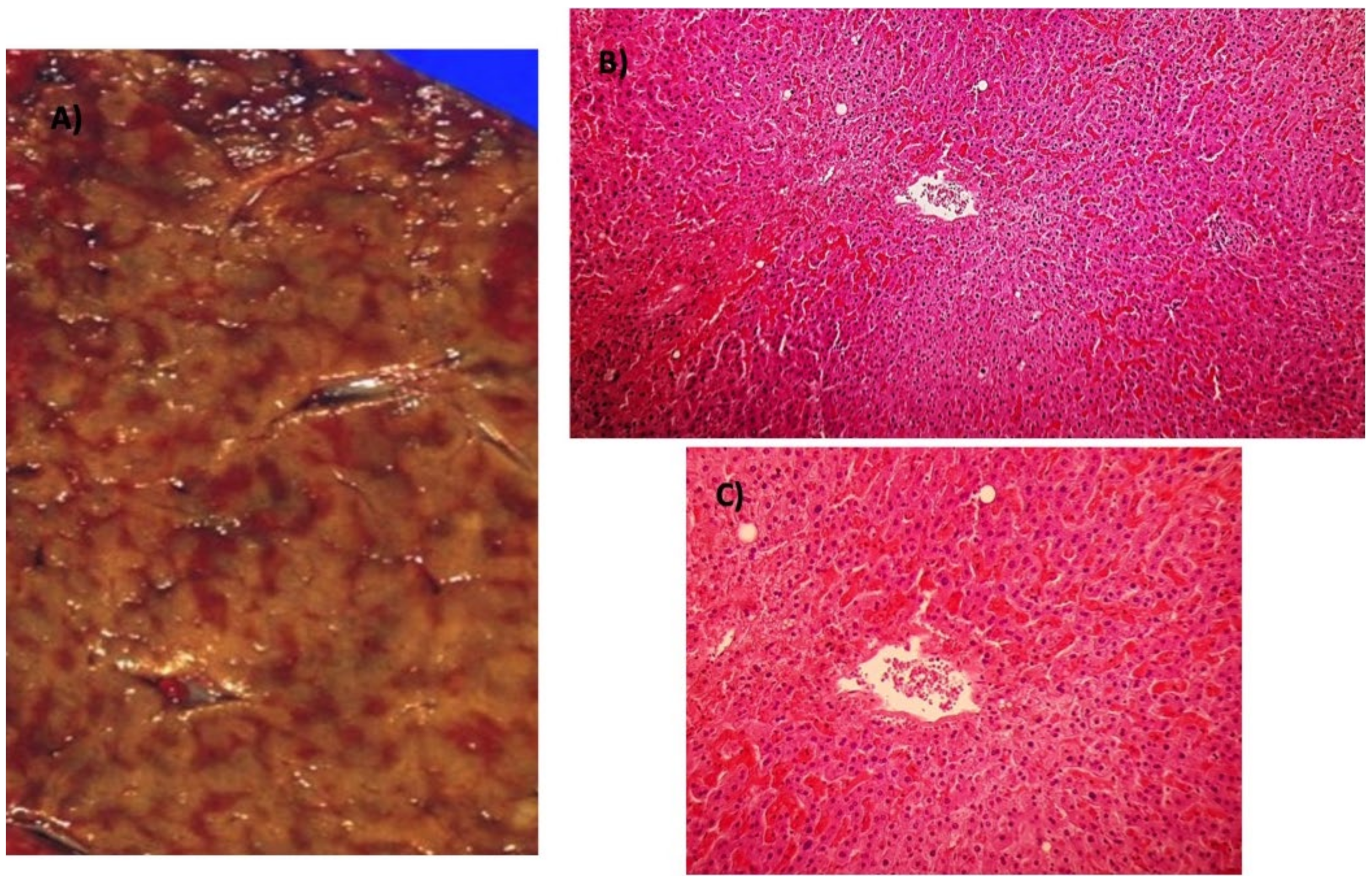
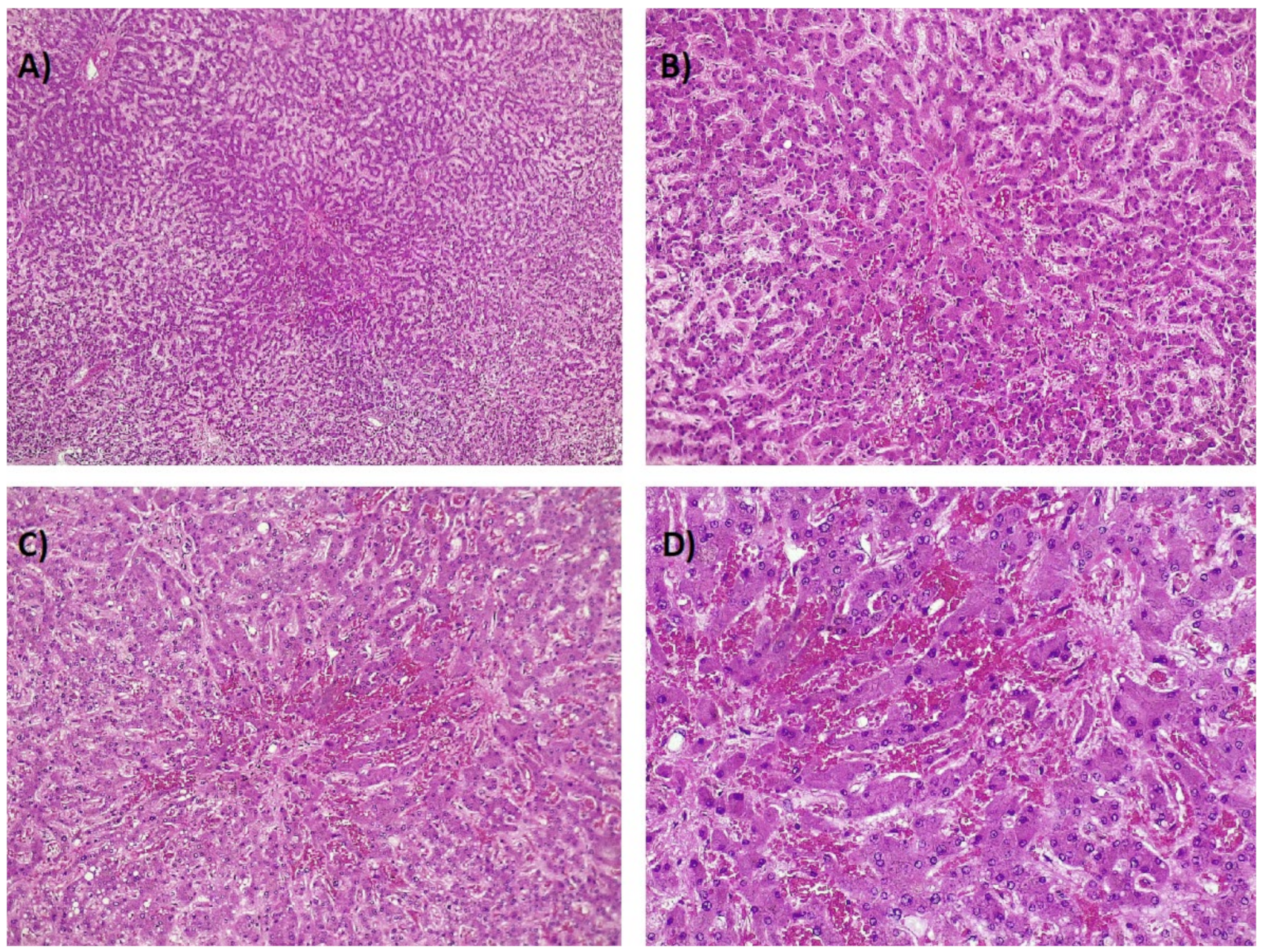
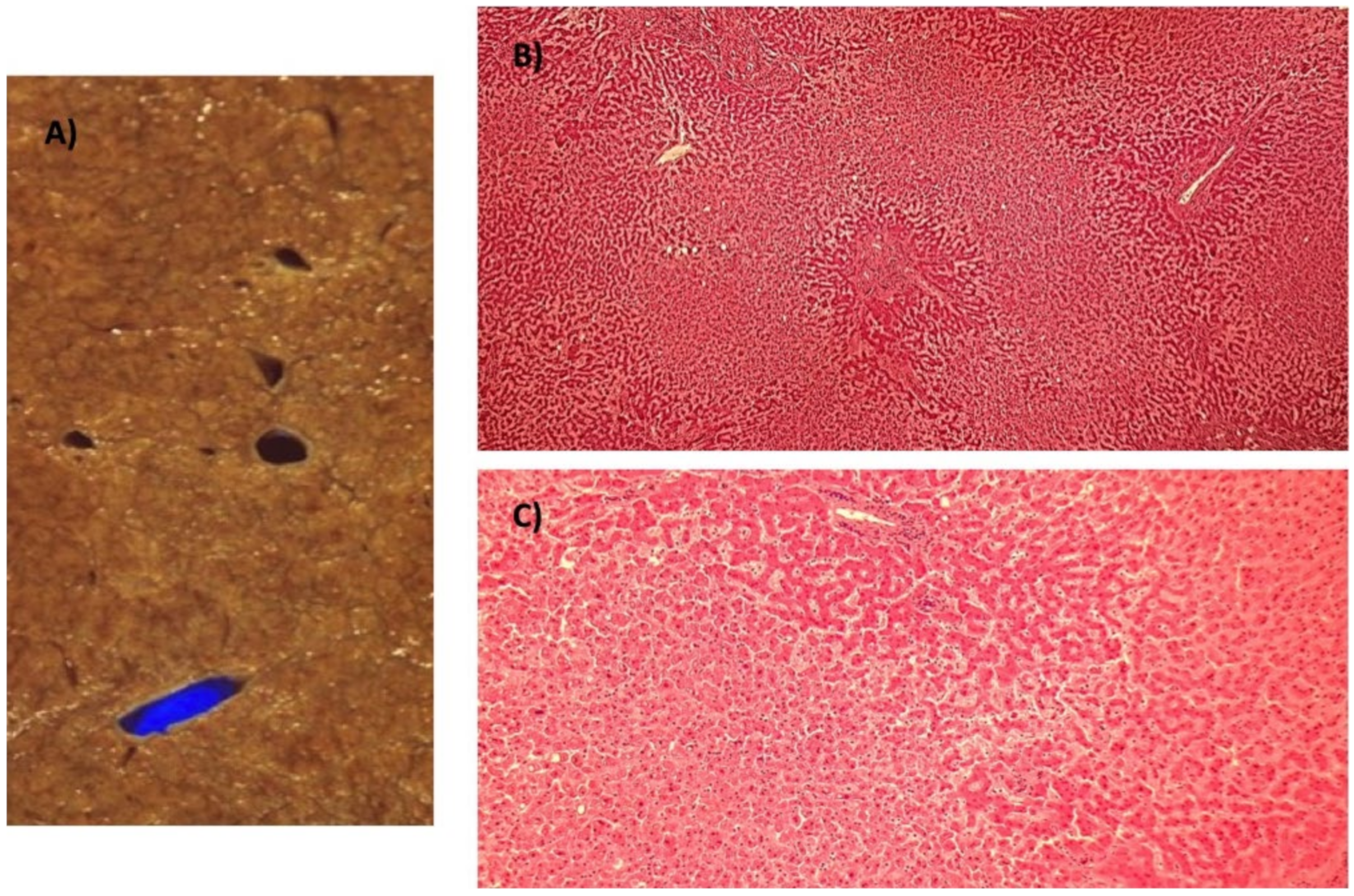
7. Histology
8. Non-Invasive Assessment of Liver Fibrosis
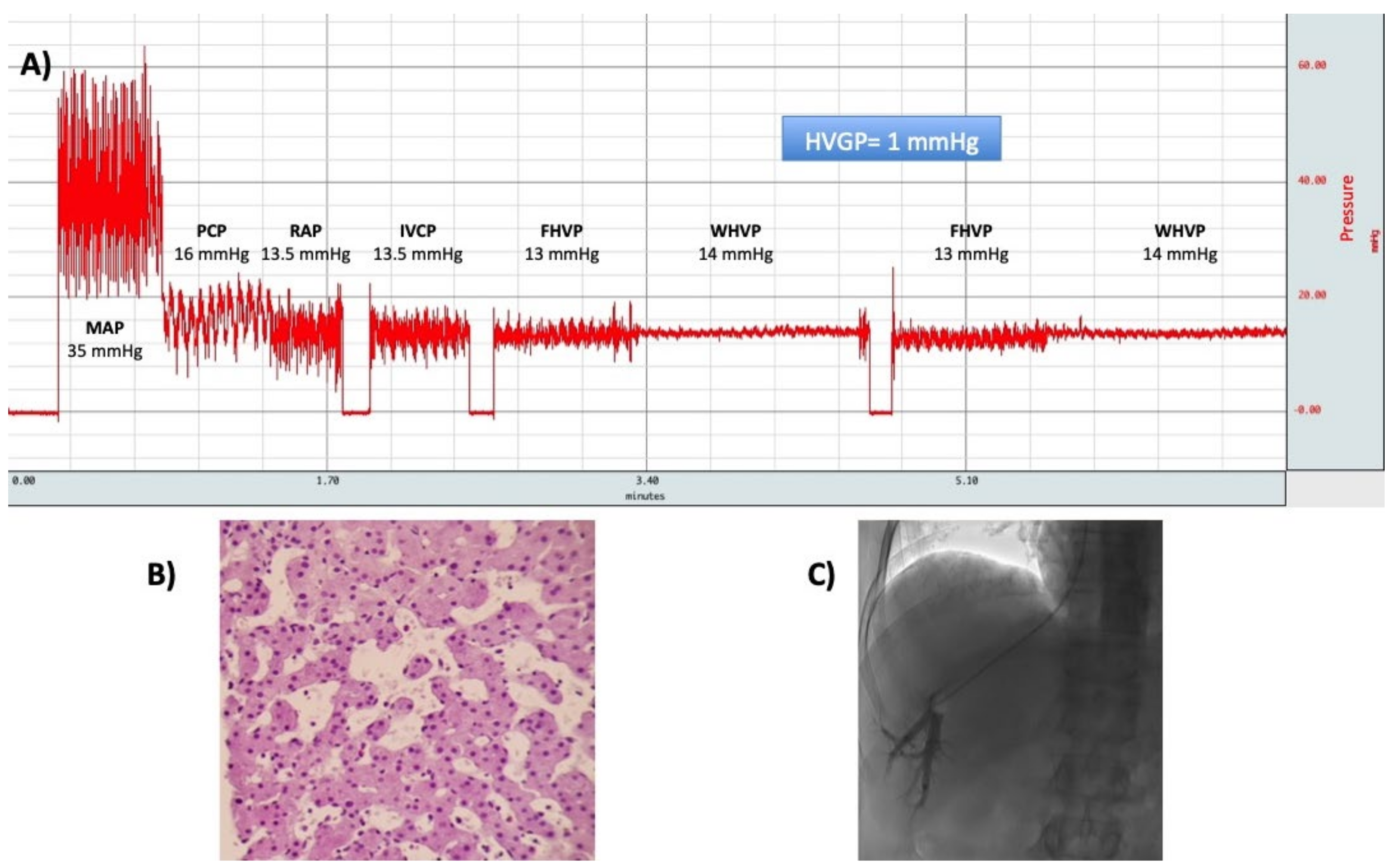
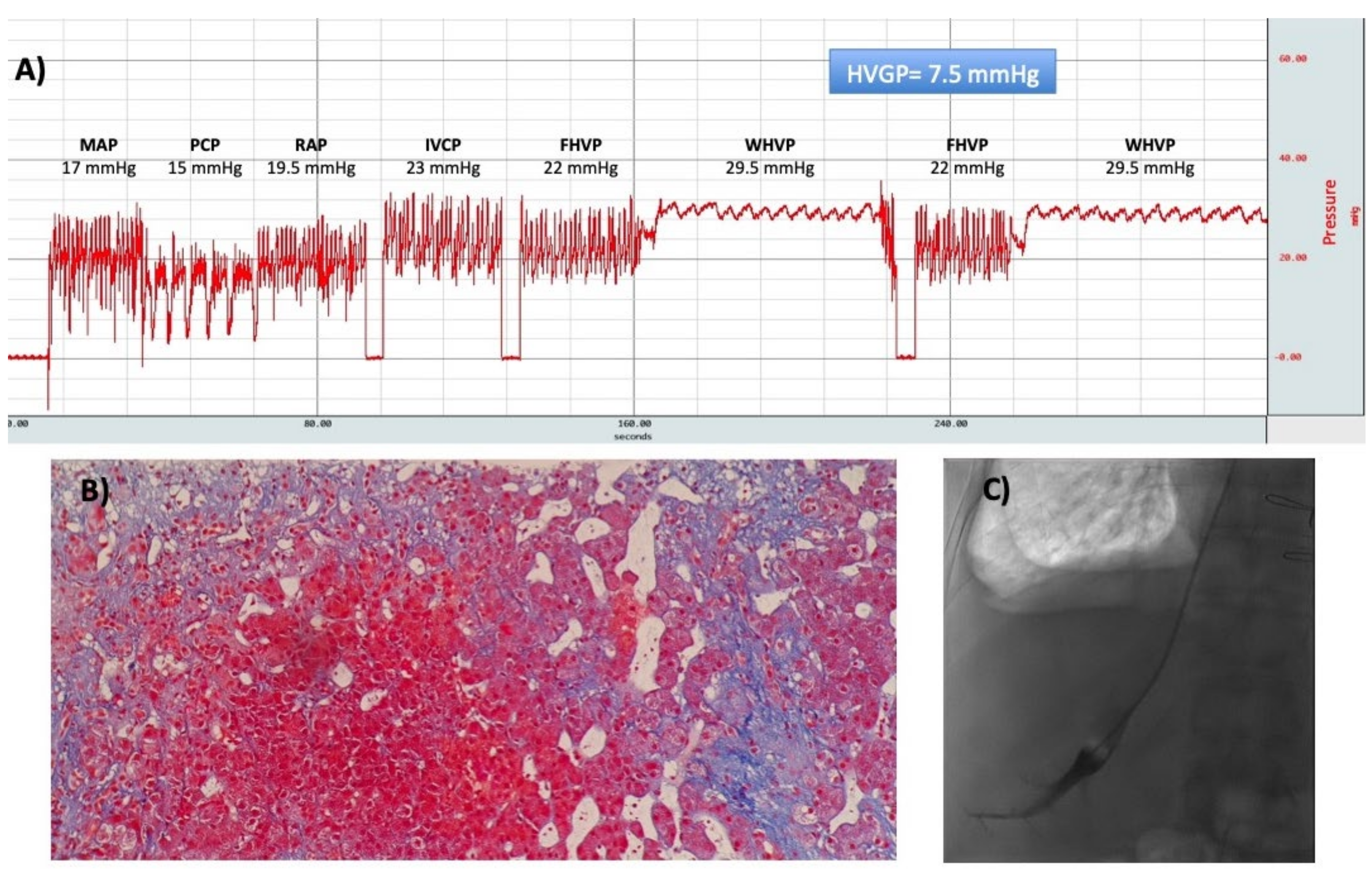
9. Hepatic Hemodynamic Study
10. Prognosis and Treatment
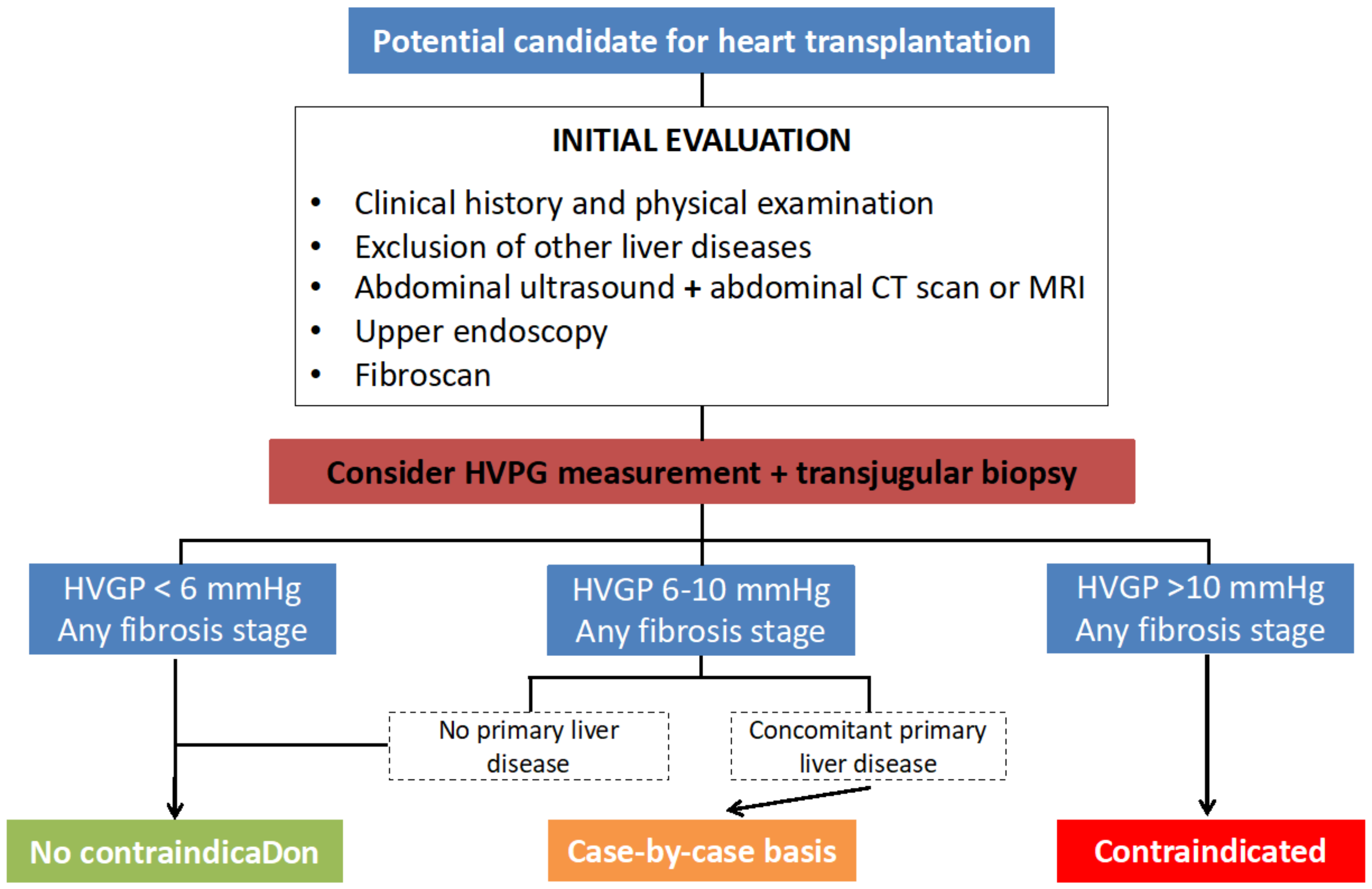
11. Determining Candidacy for Heart Transplantation
12. Conclusions
Funding
Conflicts of Interest
Abbreviations
| ACLI | acute cardiogenic liver injury |
| ALT | alanine aminotransferase |
| AST | aspartate aminotransferase |
| CH | congestive hepatopathy |
| CHLT | combined heart-liver transplantation |
| FAP | familial amyloid polyneuropathy |
| FHVP | free hepatic venous pressure |
| FNH | focal nodular hyperplasia |
| HF | heart failure |
| HT | heart transplantation |
| HVPG | hepatic venous pressure gradient |
| LDH | lactate dehydrogenase |
| MELD | Model for End-Stage Liver Disease |
| WHVP | wedged hepatic venous pressure |
References
- Ford, R.M.; Book, W.; Spivey, J.R. Liver disease related to the heart. Transpl. Rev. 2015, 29, 33–37. [Google Scholar] [CrossRef] [PubMed]
- Fouad, Y.M.; Yehia, R. Hepato-cardiac disorders. World J. Hepatol. 2014, 6, 41–54. [Google Scholar] [CrossRef] [PubMed]
- Cagli, K.; Basar, F.N.; Tok, D.; Turak, O.; Basar, O. How to interpret liver function tests in heart failure patients? Turk. J. Gastroenterol. Off. J. Turk. Soc. Gastroenterol. 2015, 26, 197–203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moller, S.; Bernardi, M. Interactions of the heart and the liver. Eur. Heart J. 2013, 34, 2804–2811. [Google Scholar] [CrossRef] [PubMed]
- Henrion, J. Hypoxic hepatitis. Liver Int. 2012, 32, 1039–1052. [Google Scholar] [CrossRef] [PubMed]
- Boland, E.; Willius, F. Changes in the liver produced by chronic passive congestion with special reference to the problem of cardiac cirrhosis. Arch. Intern. Med. 1938, 62, 723–739. [Google Scholar] [CrossRef]
- Sherlock, S. The liver in heart failure relation of anatomical, functional, and circulatory changes. Br. Heart J. 1951, 13, 273–293. [Google Scholar] [CrossRef] [Green Version]
- Samsky, M.D.; Patel, C.B.; DeWald, T.A.; Smith, A.D.; Felker, G.M.; Rogers, J.G.; Hernandez, A.F. Cardiohepatic interactions in heart failure: An overview and clinical implications. J. Am. Coll. Cardiol. 2013, 61, 2397–2405. [Google Scholar] [CrossRef] [Green Version]
- Kavoliuniene, A.; Vaitiekiene, A.; Cesnaite, G. Congestive hepatopathy and hypoxic hepatitis in heart failure: A cardiologist’s point of view. Int. J. Cardiol. 2013, 166, 554–558. [Google Scholar] [CrossRef]
- Hilscher, M.; Sanchez, W. Congestive hepatopathy. Clin. Liver Dis. 2016, 8, 68–71. [Google Scholar] [CrossRef] [Green Version]
- Myers, R.P.; Cerini, R.; Sayegh, R.; Moreau, R.; Degott, C.; Lebrec, D.; Lee, S.S. Cardiac hepatopathy: Clinical, hemodynamic, and histologic characteristics and correlations. Hepatology 2003, 37, 393–400. [Google Scholar] [CrossRef] [PubMed]
- Téllez, L.; de Santiago, E.R.; Albillos, A. Fontan-associated Liver Disease. Rev. Esp. Cardiol. 2018, 71, 192–202. [Google Scholar] [CrossRef] [PubMed]
- Emamaullee, J.; Zaidi, A.N.; Schiano, T.; Kahn, J.; Valentino, P.L.; Hofer, R.E.; Taner, T.; Wald, J.W.; Olthoff, K.M.; Bucuvalas, J.; et al. Fontan-Associated Liver Disease: Screening, Management, and Transplant Considerations. Circulation 2020, 142, 591–604. [Google Scholar] [CrossRef] [PubMed]
- Gordon-Walker, T.T.; Bove, K.; Veldtman, G. Fontan-associated liver disease: A review. J. Cardiol. 2019, 74, 223–232. [Google Scholar] [CrossRef] [PubMed]
- Lemmer, A.; Van Wagner, L.B.; Ganger, D. Assessment of Advanced Liver Fibrosis and the Risk for Hepatic Decompensation in Patients with Congestive Hepatopathy. Hepatology 2018, 68, 1633–1641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xanthopoulos, A.; Starling, R.C.; Kitai, T.; Triposkiadis, F. Heart Failure and Liver Disease: Cardiohepatic Interactions. JACC Heart Fail. 2019, 7, 87–97. [Google Scholar] [CrossRef]
- Kubo, S.H.; Walter, B.A.; John, D.H.; Clark, M.; Cody, R.J. Liver function abnormalities in chronic heart failure. Influence of systemic hemodynamics. Arch. Intern. Med. 1987, 147, 1227–1230. [Google Scholar] [CrossRef]
- Allen, L.A.; Felker, G.M.; Pocock, S.; McMurray, J.J.; Pfeffer, M.A.; Swedberg, K.; Wang, D.; Yusuf, S.; Michelson, E.L.; Granger, C.B. Liver function abnormalities and outcome in patients with chronic heart failure: Data from the Candesartan in Heart Failure: Assessment of Reduction in Mortality and Morbidity (CHARM) program. Eur. J. Heart Fail. 2009, 11, 170–177. [Google Scholar] [CrossRef]
- Gelow, J.M.; Desai, A.S.; Hochberg, C.P.; Glickman, J.N.; Givertz, M.M.; Fang, J.C. Clinical Predictors of Hepatic Fibrosis in Chronic Advanced Heart Failure. Circ. Heart Fail. 2010, 3, 59–64. [Google Scholar] [CrossRef] [Green Version]
- Van Deursen, V.M.; Damman, K.; Hillege, H.L.; van Beek, A.P.; van Veldhuisen, D.J.; Voors, A.A. Abnormal liver function in relation to hemodynamic profile in heart failure patients. J. Card. Fail. 2010, 16, 84–90. [Google Scholar] [CrossRef]
- Poelzl, G.; Ess, M.; Mussner-Seeber, C.; Pachinger, O.; Frick, M.; Ulmer, H. Liver dysfunction in chronic heart failure: Prevalence, characteristics and prognostic significance. Eur. J. Clin. Investig. 2012, 42, 153–163. [Google Scholar] [CrossRef]
- Megalla, S.; Holtzman, D.; Aronow, W.S.; Nazari, R.; Korenfeld, S.; Schwarcz, A.; Goldberg, Y.; Spevack, D.M. Predictors of cardiac hepatopathy in patients with right heart failure. Med. Sci. Monit. 2011, 17, Cr537–Cr541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zipprich, A.; Mehal, W.Z.; Ripoll, C.; Groszmann, R.J. A distinct nitric oxide and adenosine A1 receptor dependent hepatic artery vasodilatatory response in the CCl-cirrhotic liver. Liver Int. 2010, 30, 988–994. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lautt, W.W. Hepatic vasculature: A conceptual review. Gastroenterology 1977, 73, 1163–1169. [Google Scholar] [CrossRef]
- Jacobsen, K.R.; Ranek, L.; Tygstrup, N. Liver function and blood flow in normal man during infusion of vasopressin. Scand. J. Clin. Lab. Investig. 1969, 24, 279–284. [Google Scholar] [CrossRef] [PubMed]
- Simonetto, D.A.; Yang, H.Y.; Yin, M.; de Assuncao, T.M.; Kwon, J.H.; Hilscher, M.; Pan, S.; Yang, L.; Bi, Y.; Beyder, A.; et al. Chronic passive venous congestion drives hepatic fibrogenesis via sinusoidal thrombosis and mechanical forces. Hepatology 2015, 61, 648–659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marra, F.; DeFranco, R.; Grappone, C.; Milani, S.; Pinzani, M.; Pellegrini, G.; Laffi, G.; Gentilini, P. Expression of the thrombin receptor in human liver: Up-regulation during acute and chronic injury. Hepatology 1998, 27, 462–471. [Google Scholar] [CrossRef]
- Marra, F.; Grandaliano, G.; Valente, A.J.; Abboud, H.E. Thrombin stimulates proliferation of liver fat-storing cells and expression of monocyte chemotactic protein-1: Potential role in liver injury. Hepatology 1995, 22, 780–787. [Google Scholar]
- Wanless, I.R.; Liu, J.J.; Butany, J. Role of thrombosis in the pathogenesis of congestive hepatic fibrosis (cardiac cirrhosis). Hepatology 1995, 21, 1232–1237. [Google Scholar]
- Wanless, I.R. The Role of Vascular Injury and Congestion in the Pathogenesis of Cirrhosis: The Congestive Escalator and the Parenchymal Extinction Sequence. Curr. Hepatol. Rep. 2020, 19, 40–53. [Google Scholar] [CrossRef] [Green Version]
- Kiesewetter, C.H.; Sheron, N.; Vettukattill, J.J.; Hacking, N.; Stedman, B.; Millward-Sadler, H.; Haw, M.; Cope, R.; Salmon, A.P.; Sivaprakasam, M.C.; et al. Hepatic changes in the failing Fontan circulation. Heart 2007, 93, 579–584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kendall, T.J.; Stedman, B.; Hacking, N.; Haw, M.; Vettukattill, J.J.; Salmon, A.P.; Cope, R.; Sheron, N.; Millward-Sadler, H.; Veldtman, G.R.; et al. Hepatic fibrosis and cirrhosis in the Fontan circulation: A detailed morphological study. J. Clin. Pathol. 2008, 61, 504–508. [Google Scholar] [CrossRef] [PubMed]
- Wu, F.M.; Kogon, B.; Earing, M.G.; Aboulhosn, J.A.; Broberg, C.S.; John, A.S.; Harmon, A.; Sainani, N.I.; Hill, A.J.; Odze, R.D.; et al. Liver health in adults with Fontan circulation: A multicenter cross-sectional study. J. Thorac. Cardiovasc. Surg. 2017, 153, 656–664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cerini, F.; Vilaseca, M.; Lafoz, E.; Garcia-Irigoyen, O.; Garcia-Caldero, H.; Tripathi, D.M.; Avila, M.; Reverter, J.C.; Bosch, J.; Gracia-Sancho, J.; et al. Enoxaparin reduces hepatic vascular resistance and portal pressure in cirrhotic rats. J. Hepatol. 2016, 64, 834–842. [Google Scholar] [CrossRef] [PubMed]
- Nakao, T.; Arii, S.; Kaido, T.; Mori, A.; Murata, T.; Matsumori, A.; Imamura, M. Heparin accelerates liver regeneration following portal branch ligation in normal and cirrhotic rats with increased plasma hepatocyte growth factor levels. J. Hepatol. 2002, 37, 87–92. [Google Scholar] [CrossRef] [Green Version]
- Abdel-Salam, O.M.; Baiuomy, A.R.; Ameen, A.; Hassan, N.S. A study of unfractionated and low molecular weight heparins in a model of cholestatic liver injury in the rat. Pharm. Res. 2005, 51, 59–67. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Zhang, J.; Huang, Q.; Zhu, H.; Zhang, X. Long-term administering low anticoagulant activity heparin can lessen rat hepatic fibrosis induced by either CCl(4) or porcine serum injection. Hepatol. Res. 2006, 36, 115–123. [Google Scholar] [CrossRef]
- Abe, W.; Ikejima, K.; Lang, T.; Okumura, K.; Enomoto, N.; Kitamura, T.; Takei, Y.; Sato, N. Low molecular weight heparin prevents hepatic fibrogenesis caused by carbon tetrachloride in the rat. J. Hepatol. 2007, 46, 286–294. [Google Scholar] [CrossRef]
- Assy, N.; Hussein, O.; Khalil, A.; Luder, A.; Szvalb, S.; Paizi, M.; Spira, G. The beneficial effect of aspirin and enoxaparin on fibrosis progression and regenerative activity in a rat model of cirrhosis. Dig. Dis. Sci. 2007, 52, 1187–1193. [Google Scholar] [CrossRef]
- Kukner, A.; Tore, F.; Firat, T.; Terzi, E.H.; Oner, H.; Balaban, Y.H.; Ozogul, C. The preventive effect of low molecular weight heparin on CCL(4)-induced necrosis and apoptosis in rat liver. Ann. Hepatol. 2010, 9, 445–454. [Google Scholar] [CrossRef]
- Lee, J.H.; Lee, H.; Joung, Y.K.; Jung, K.H.; Choi, J.H.; Lee, D.H.; Park, K.D.; Hong, S.S. The use of low molecular weight heparin-pluronic nanogels to impede liver fibrosis by inhibition the TGF-beta/Smad signaling pathway. Biomaterials 2011, 32, 1438–1445. [Google Scholar] [CrossRef] [PubMed]
- Shah, B.; Shah, G. Antifibrotic effect of heparin on liver fibrosis model in rats. World J. Gastrointest. Pharm. 2012, 3, 86–92. [Google Scholar] [CrossRef] [PubMed]
- Anstee, Q.M.; Goldin, R.D.; Wright, M.; Martinelli, A.; Cox, R.; Thursz, M.R. Coagulation status modulates murine hepatic fibrogenesis: Implications for the development of novel therapies. J. Thromb. Haemost. 2008, 6, 1336–1343. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.C.; Hsu, W.F.; Hsieh, Y.C.; Chan, C.C.; Yang, Y.Y.; Huang, Y.H.; Hou, M.C.; Lin, H.C. Dabigatran Reduces Liver Fibrosis in Thioacetamide-Injured Rats. Dig. Dis. Sci. 2019, 64, 102–112. [Google Scholar] [CrossRef]
- Vilaseca, M.; Garcia-Caldero, H.; Lafoz, E.; Garcia-Irigoyen, O.; Avila, M.A.; Reverter, J.C.; Bosch, J.; Hernandez-Gea, V.; Gracia-Sancho, J.; Garcia-Pagan, J.C. The anticoagulant rivaroxaban lowers portal hypertension in cirrhotic rats mainly by deactivating hepatic stellate cells. Hepatology 2017, 65, 2031–2044. [Google Scholar] [CrossRef] [Green Version]
- Villa, E.; Camma, C.; Marietta, M.; Luongo, M.; Critelli, R.; Colopi, S.; Tata, C.; Zecchini, R.; Gitto, S.; Petta, S.; et al. Enoxaparin prevents portal vein thrombosis and liver decompensation in patients with advanced cirrhosis. Gastroenterology 2012, 143, 1253–1260. [Google Scholar] [CrossRef] [Green Version]
- Fortea, J.I.; Zipprich, A.; Fernandez-Mena, C.; Puerto, M.; Bosoi, C.R.; Almagro, J.; Hollenbach, M.; Banares, J.; Rodriguez-Sanchez, B.; Cercenado, E.; et al. Enoxaparin does not ameliorate liver fibrosis or portal hypertension in rats with advanced cirrhosis. Liver Int. 2018, 38, 102–112. [Google Scholar] [CrossRef]
- El Hadi, H.; Di Vincenzo, A.; Vettor, R.; Rossato, M. Relationship between Heart Disease and Liver Disease: A Two-Way Street. Cells 2020, 9, 567. [Google Scholar] [CrossRef] [Green Version]
- Kato, T.; Niizuma, S.; Inuzuka, Y.; Kawashima, T.; Okuda, J.; Kawamoto, A.; Tamaki, Y.; Iwanaga, Y.; Soga, T.; Kita, T.; et al. Analysis of liver metabolism in a rat model of heart failure. Int. J. Cardiol. 2012, 161, 130–136. [Google Scholar] [CrossRef]
- Grueter, C.E.; van Rooij, E.; Johnson, B.A.; DeLeon, S.M.; Sutherland, L.B.; Qi, X.; Gautron, L.; Elmquist, J.K.; Bassel-Duby, R.; Olson, E.N. A cardiac microRNA governs systemic energy homeostasis by regulation of MED13. Cell 2012, 149, 671–683. [Google Scholar] [CrossRef] [Green Version]
- Nakamura, M.; Sadoshima, J. Heart over mind: Metabolic control of white adipose tissue and liver. EMBO Mol. Med. 2014, 6, 1521–1524. [Google Scholar] [CrossRef] [PubMed]
- Baskin, K.K.; Grueter, C.E.; Kusminski, C.M.; Holland, W.L.; Bookout, A.L.; Satapati, S.; Kong, Y.M.; Burgess, S.C.; Malloy, C.R.; Scherer, P.E.; et al. MED13-dependent signaling from the heart confers leanness by enhancing metabolism in adipose tissue and liver. EMBO Mol. Med. 2014, 6, 1610–1621. [Google Scholar] [CrossRef]
- Waseem, N.; Chen, P.H. Hypoxic Hepatitis: A Review and Clinical Update. J. Clin. Transl. Hepatol. 2016, 4, 263–268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fuhrmann, V.; Kneidinger, N.; Herkner, H.; Heinz, G.; Nikfardjam, M.; Bojic, A.; Schellongowski, P.; Angermayr, B.; Kitzberger, R.; Warszawska, J.; et al. Hypoxic hepatitis: Underlying conditions and risk factors for mortality in critically ill patients. Intensive Care Med. 2009, 35, 1397–1405. [Google Scholar] [CrossRef] [PubMed]
- Killip, T.; Payne, M.A. High serum transaminase activity in heart disease. Circulatory failure and hepatic necrosis. Circulation 1960, 21, 646–660. [Google Scholar] [CrossRef] [Green Version]
- Richman, S.M.; Delman, A.J.; Grob, D. Alterations in indices of liver function in congestive heart failure with particular reference to serum enzymes. Am. J. Med. 1961, 30, 211–225. [Google Scholar] [CrossRef]
- Seeto, R.K.; Fenn, B.; Rockey, D.C. Ischemic hepatitis: Clinical presentation and pathogenesis. Am. J. Med. 2000, 109, 109–113. [Google Scholar] [CrossRef]
- Henrion, J.; Schapira, M.; Luwaert, R.; Colin, L.; Delannoy, A.; Heller, F.R. Hypoxic hepatitis: Clinical and hemodynamic study in 142 consecutive cases. Medicine 2003, 82, 392–406. [Google Scholar] [CrossRef]
- Tapper, E.B.; Sengupta, N.; Bonder, A. The Incidence and Outcomes of Ischemic Hepatitis: A Systematic Review with Meta-analysis. Am. J. Med. 2015, 128, 1314–1321. [Google Scholar] [CrossRef]
- Henrion, J.; Descamps, O.; Luwaert, R.; Schapira, M.; Parfonry, A.; Heller, F. Hypoxic hepatitis in patients with cardiac failure: Incidence in a coronary care unit and measurement of hepatic blood flow. J. Hepatol. 1994, 21, 696–703. [Google Scholar] [CrossRef]
- Birrer, R.; Takuda, Y.; Takara, T. Hypoxic hepatopathy: Pathophysiology and prognosis. Intern. Med. 2007, 46, 1063–1070. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farias, A.Q.; Silvestre, O.M.; Garcia-Tsao, G.; da Seguro, L.F.C.; de Mazo, D.F.C.; Bacal, F.; Andrade, J.L.; Goncalves, L.L.; Strunz, C.; Ramos, D.S.; et al. Serum B-type natriuretic peptide in the initial workup of patients with new onset ascites: A diagnostic accuracy study. Hepatology 2014, 59, 1043–1051. [Google Scholar] [CrossRef] [PubMed]
- Runyon, B.A. Cardiac ascites: A characterization. J. Clin. Gastroenterol. 1988, 10, 410–412. [Google Scholar] [CrossRef] [PubMed]
- McConnell, M.; Iwakiri, Y. Biology of portal hypertension. Hepatol. Int. 2018, 12, 11–23. [Google Scholar] [CrossRef]
- Sheer, T.A.; Joo, E.; Runyon, B.A. Usefulness of serum N-terminal-ProBNP in distinguishing ascites due to cirrhosis from ascites due to heart failure. J. Clin. Gastroenterol. 2010, 44, e23–e26. [Google Scholar] [CrossRef]
- Lau, G.T.; Tan, H.C.; Kritharides, L. Type of liver dysfunction in heart failure and its relation to the severity of tricuspid regurgitation. Am. J. Cardiol. 2002, 90, 1405–1409. [Google Scholar] [CrossRef]
- De Gonzalez, A.K.K.; Lefkowitch, J.H. Heart Disease and the Liver: Pathologic Evaluation. Gastroenterol. Clin. N. Am. 2017, 46, 421–435. [Google Scholar] [CrossRef]
- Aboelsoud, M.M.; Javaid, A.I.; Al-Qadi, M.O.; Lewis, J.H. Hypoxic hepatitis—Its biochemical profile, causes and risk factors of mortality in critically-ill patients: A cohort study of 565 patients. J. Crit. Care 2017, 41, 9–15. [Google Scholar] [CrossRef]
- Raurich, J.M.; Llompart-Pou, J.A.; Ferreruela, M.; Colomar, A.; Molina, M.; Royo, C.; Ayestaran, I.; Ibanez, J. Hypoxic hepatitis in critically ill patients: Incidence, etiology and risk factors for mortality. J. Anesth. 2011, 25, 50–56. [Google Scholar] [CrossRef]
- Cassidy, W.M.; Reynolds, T.B. Serum lactic dehydrogenase in the differential diagnosis of acute hepatocellular injury. J. Clin. Gastroenterol. 1994, 19, 118–121. [Google Scholar] [CrossRef]
- Woreta, T.A.; Alqahtani, S.A. Evaluation of abnormal liver tests. Med. Clin. N. Am. 2014, 98, 1–16. [Google Scholar] [CrossRef]
- Green, R.M.; Flamm, S. AGA technical review on the evaluation of liver chemistry tests. Gastroenterology 2002, 123, 1367–1384. [Google Scholar] [CrossRef] [Green Version]
- Morales, A.; Hirsch, M.; Schneider, D.; González, D. Congestive hepatopathy: The role of the radiologist in the diagnosis. Diagn. Interv. Radiol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Wells, M.L.; Venkatesh, S.K. Congestive hepatopathy. Abdom. Radiol. 2018, 43, 2037–2051. [Google Scholar] [CrossRef] [PubMed]
- Rychik, J.; Atz, A.M.; Celermajer, D.S.; Deal, B.J.; Gatzoulis, M.A.; Gewillig, M.H.; Hsia, T.Y.; Hsu, D.T.; Kovacs, A.H.; McCrindle, B.W.; et al. Evaluation and Management of the Child and Adult with Fontan Circulation: A Scientific Statement From the American Heart Association. Circulation 2019. [Google Scholar] [CrossRef] [PubMed]
- Dhall, D.; Kim, S.A.; Mc Phaul, C.; Kransdorf, E.P.; Kobashigawa, J.A.; Sundaram, V.; Mirocha, J.; Guindi, M. Heterogeneity of Fibrosis in Liver Biopsies of Patients with Heart Failure Undergoing Heart Transplant Evaluation. Am. J. Surg. Pathol. 2018, 42, 1617–1624. [Google Scholar] [CrossRef] [PubMed]
- Dai, D.F.; Swanson, P.E.; Krieger, E.V.; Liou, I.W.; Carithers, R.L.; Yeh, M.M. Congestive hepatic fibrosis score: A novel histologic assessment of clinical severity. Mod. Pathol. 2014, 27, 1552–1558. [Google Scholar] [CrossRef] [Green Version]
- Bosch, D.E.; Koro, K.; Richards, E.; Hoch, B.L.; Jalikis, F.; Koch, L.K.; Swanson, P.E.; Truong, C.D.; Liou, I.; Yu, L.; et al. Validation of a Congestive Hepatic Fibrosis Scoring System. Am. J. Surg. Pathol. 2019, 43, 766–772. [Google Scholar] [CrossRef]
- Surrey, L.F.; Russo, P.; Rychik, J.; Goldberg, D.J.; Dodds, K.; O’Byrne, M.L.; Glatz, A.C.; Rand, E.B.; Lin, H.C. Prevalence and characterization of fibrosis in surveillance liver biopsies of patients with Fontan circulation. Hum. Pathol. 2016, 57, 106–115. [Google Scholar] [CrossRef]
- Bayraktar, U.-D.; Seren, S.; Bayraktar, Y. Hepatic venous outflow obstruction: Three similar syndromes. World J. Gastroenterol. 2007, 13, 1912–1927. [Google Scholar] [CrossRef] [Green Version]
- Louie, C.Y.; Pham, M.X.; Daugherty, T.J.; Kambham, N.; Higgins, J.P. The liver in heart failure: A biopsy and explant series of the histopathologic and laboratory findings with a particular focus on pre-cardiac transplant evaluation. Mod. Pathol. 2015, 28, 932–943. [Google Scholar] [CrossRef] [PubMed]
- Mallory, T.B.; Sullivan, E.R.; Burnett, C.H.; Simeone, F.A.; Shapiro, S.L.; Beecher, H.K., VII. The general pathology of traumatic shock. Surgery 1950, 27, 629–644. [Google Scholar] [CrossRef]
- Brunson, J.G.; Eckman, P.L.; Campbell, J.B. Increasing prevalence of unexplained liver necrosis. N. Engl. J. Med. 1957, 257, 52–56. [Google Scholar] [CrossRef] [PubMed]
- De la Monte, S.M.; Arcidi, J.M.; Moore, G.W.; Hutchins, G.M. Midzonal necrosis as a pattern of hepatocellular injury after shock. Gastroenterology 1984, 86, 627–631. [Google Scholar] [CrossRef]
- European Association for Study of the Liver; Asociacion Latinoamericana para el Estudio del Higado. EASL-ALEH Clinical Practice Guidelines: Non-invasive tests for evaluation of liver disease severity and prognosis. J. Hepatol. 2015, 63, 237–264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lemmer, A.; VanWagner, L.; Ganger, D. Congestive hepatopathy: Differentiating congestion from fibrosis. Clin. Liver Dis. 2017, 10, 139–143. [Google Scholar] [CrossRef]
- Diamond, T.; Ovchinsky, N. Fontan-associated liver disease: Monitoring progression of liver fibrosis. Clin. Liver Dis. 2018, 11, 1–5. [Google Scholar] [CrossRef] [Green Version]
- Evans, W.N.; Acherman, R.J.; Ciccolo, M.L.; Carrillo, S.A.; Galindo, A.; Rothman, A.; Winn, B.J.; Yumiaco, N.S.; Restrepo, H. MELD-XI Scores Correlate with Post-Fontan Hepatic Biopsy Fibrosis Scores. Pediatr. Cardiol. 2016, 37, 1274–1277. [Google Scholar] [CrossRef]
- Friedrich-Rust, M.; Koch, C.; Rentzsch, A.; Sarrazin, C.; Schwarz, P.; Herrmann, E.; Lindinger, A.; Sarrazin, U.; Poynard, T.; Schafers, H.J.; et al. Noninvasive assessment of liver fibrosis in patients with Fontan circulation using transient elastography and biochemical fibrosis markers. J. Thorac. Cardiovasc. Surg. 2008, 135, 560–567. [Google Scholar] [CrossRef] [Green Version]
- Singh, S.; Muir, A.J.; Dieterich, D.T.; Falck-Ytter, Y.T. American Gastroenterological Association Institute Technical Review on the Role of Elastography in Chronic Liver Diseases. Gastroenterology 2017, 152, 1544–1577. [Google Scholar] [CrossRef] [Green Version]
- Berzigotti, A. Non-invasive evaluation of portal hypertension using ultrasound elastography. J. Hepatol. 2017, 67, 399–411. [Google Scholar] [CrossRef] [PubMed]
- Silva-Sepulveda, J.A.; Fonseca, Y.; Vodkin, I.; Vaughn, G.; Newbury, R.; Vavinskaya, V.; Dwek, J.; Perry, J.C.; Reshamwala, P.; Baehling, C.; et al. Evaluation of Fontan liver disease: Correlation of transjugular liver biopsy with magnetic resonance and hemodynamics. Congenit. Heart Dis. 2019, 14, 600–608. [Google Scholar] [CrossRef] [PubMed]
- Sugimoto, M.; Oka, H.; Kajihama, A.; Nakau, K.; Kuwata, S.; Kurishima, C.; Azuma, H. Non-invasive assessment of liver fibrosis by magnetic resonance elastography in patients with congenital heart disease undergoing the Fontan procedure and intracardiac repair. J. Cardiol. 2016, 68, 202–208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, G.; Li, F.; Mao, Y. Portal pressure monitoring-state-of-the-art and future perspective. Ann. Transl. Med. 2019, 7, 583. [Google Scholar] [CrossRef]
- Reiberger, T.; Schwabl, P.; Trauner, M.; Peck-Radosavljevic, M.; Mandorfer, M. Measurement of the Hepatic Venous Pressure Gradient and Transjugular Liver Biopsy. J. Vis. Exp. 2020. [Google Scholar] [CrossRef]
- Bosch, J.; Abraldes, J.G.; Berzigotti, A.; Garcia-Pagan, J.C. The clinical use of HVPG measurements in chronic liver disease. Nat. Rev. Gastroenterol. Hepatol. 2009, 6, 573–582. [Google Scholar] [CrossRef]
- Burroughs, A.K.; Thalheimer, U. Hepatic venous pressure gradient in 2010: Optimal measurement is key. Hepatology 2010, 51, 1894–1896. [Google Scholar] [CrossRef]
- Abraldes, J.G.; Sarlieve, P.; Tandon, P. Measurement of portal pressure. Clin. Liver Dis. 2014, 18, 779–792. [Google Scholar] [CrossRef]
- Dohan, A.; Guerrache, Y.; Boudiaf, M.; Gavini, J.P.; Kaci, R.; Soyer, P. Transjugular liver biopsy: Indications, technique and results. Diagn. Interv. Imaging 2014, 95, 11–15. [Google Scholar] [CrossRef] [Green Version]
- Ruttmann, E.; Brant, L.J.; Concin, H.; Diem, G.; Rapp, K.; Ulmer, H. Gamma-glutamyltransferase as a risk factor for cardiovascular disease mortality: An epidemiological investigation in a cohort of 163, 944 Austrian adults. Circulation 2005, 112, 2130–2137. [Google Scholar] [CrossRef] [Green Version]
- Assenza, G.E.; Graham, D.A.; Landzberg, M.J.; Valente, A.M.; Singh, M.N.; Bashir, A.; Fernandes, S.; Mortele, K.J.; Ukomadu, C.; Volpe, M.; et al. MELD-XI score and cardiac mortality or transplantation in patients after Fontan surgery. Heart 2013, 99, 491–496. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.S.; Kato, T.S.; Farr, M.; Wu, C.; Givens, R.C.; Collado, E.; Mancini, D.M.; Schulze, P.C. Hepatic dysfunction in ambulatory patients with heart failure: Application of the MELD scoring system for outcome prediction. J. Am. Coll. Cardiol. 2013, 61, 2253–2261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deo, S.V.; Al-Kindi, S.G.; Altarabsheh, S.E.; Hang, D.; Kumar, S.; Ginwalla, M.B.; ElAmm, C.A.; Sareyyupoglu, B.; Medalion, B.; Oliveira, G.H.; et al. Model for end-stage liver disease excluding international normalized ratio (MELD-XI) score predicts heart transplant outcomes: Evidence from the registry of the United Network for Organ Sharing. J. Heart Lung Transpl. 2016, 35, 222–227. [Google Scholar] [CrossRef] [PubMed]
- Ponikowski, P.; Voors, A.A.; Anker, S.D.; Bueno, H.; Cleland, J.G.; Coats, A.J.; Falk, V.; Gonzalez-Juanatey, J.R.; Harjola, V.P.; Jankowska, E.A.; et al. 2016 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure: The Task Force for the diagnosis and treatment of acute and chronic heart failure of the European Society of Cardiology (ESC). Developed with the special contribution of the Heart Failure Association (HFA) of the ESC. Eur. J. Heart Fail. 2016, 18, 891–975. [Google Scholar] [CrossRef] [PubMed]
- Yancy, C.W.; Jessup, M.; Bozkurt, B.; Butler, J.; Casey, D.E., Jr.; Drazner, M.H.; Fonarow, G.C.; Geraci, S.A.; Horwich, T.; Januzzi, J.L.; et al. 2013 ACCF/AHA guideline for the management of heart failure: A report of the American College of Cardiology Foundation/American Heart Association Task Force on practice guidelines. Circulation 2013, 128, e240–e327. [Google Scholar] [CrossRef] [PubMed]
- Taylor, R.M.; Tujios, S.; Jinjuvadia, K.; Davern, T.; Shaikh, O.S.; Han, S.; Chung, R.T.; Lee, W.M.; Fontana, R.J. Short and long-term outcomes in patients with acute liver failure due to ischemic hepatitis. Dig. Dis. Sci. 2012, 57, 777–785. [Google Scholar] [CrossRef]
- Bizouarn, P.; Ausseur, A.; Desseigne, P.; Le Teurnier, Y.; Nougarede, B.; Train, M.; Michaud, J.L. Early and late outcome after elective cardiac surgery in patients with cirrhosis. Ann. Thorac. Surg. 1999, 67, 1334–1338. [Google Scholar] [CrossRef]
- Shaheen, A.A.; Kaplan, G.G.; Hubbard, J.N.; Myers, R.P. Morbidity and mortality following coronary artery bypass graft surgery in patients with cirrhosis: A population-based study. Liver Int. 2009, 29, 1141–1151. [Google Scholar] [CrossRef]
- Wallwork, K.; Ali, J.M.; Abu-Omar, Y.; De Silva, R. Does liver cirrhosis lead to inferior outcomes following cardiac surgery? Interact. Cardiovasc. Thorac. Surg. 2018, 28, 102–107. [Google Scholar] [CrossRef]
- Northup, P.G.; Friedman, L.S.; Kamath, P.S. AGA Clinical Practice Update on Surgical Risk Assessment and Perioperative Management in Cirrhosis: Expert Review. Clin. Gastroenterol. Hepatol. 2019, 17, 595–606. [Google Scholar] [CrossRef] [Green Version]
- Hsu, R.B.; Chang, C.I.; Lin, F.Y.; Chou, N.K.; Chi, N.H.; Wang, S.S.; Chu, S.H. Heart transplantation in patients with liver cirrhosis. Eur. J. Cardiothorac. Surg. 2008, 34, 307–312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lebray, P.; Varnous, S. Combined heart and liver transplantation: State of knowledge and outlooks. Clin. Res. Hepatol. Gastroenterol. 2019, 43, 123–130. [Google Scholar] [CrossRef] [PubMed]
- Ngu, N.L.Y.; Majeed, A.; Roberts, S.K.; Bergin, P.; Kemp, W. Outcomes of patients with cardiac cirrhosis undergoing heart transplantation. Clin. Transpl. 2020, 34, e13898. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Tsao, G.; Friedman, S.; Iredale, J.; Pinzani, M. Now there are many (stages) where before there was one: In search of a pathophysiological classification of cirrhosis. Hepatology 2010, 51, 1445–1449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia-Tsao, G. Regression of HCV cirrhosis: Time will tell. Hepatology 2018, 67, 1651–1653. [Google Scholar] [CrossRef] [Green Version]
- Berzigotti, A.; Reig, M.; Abraldes, J.G.; Bosch, J.; Bruix, J. Portal hypertension and the outcome of surgery for hepatocellular carcinoma in compensated cirrhosis: A systematic review and meta-analysis. Hepatology 2015, 61, 526–536. [Google Scholar] [CrossRef]
- Reverter, E.; Cirera, I.; Albillos, A.; Debernardi-Venon, W.; Abraldes, J.G.; Llop, E.; Flores, A.; Martínez-Palli, G.; Blasi, A.; Martínez, J.; et al. The prognostic role of hepatic venous pressure gradient in cirrhotic patients undergoing elective extrahepatic surgery. J. Hepatol. 2019, 71, 942–950. [Google Scholar] [CrossRef]
- Gong, T.; Hall, S. Considerations and experience driving expansion of combined heart-liver transplantation. Curr. Opin. Organ. Transpl. 2020, 25, 496–500. [Google Scholar] [CrossRef]
- Chih, S.; McDonald, M.; Dipchand, A.; Kim, D.; Ducharme, A.; Kaan, A.; Abbey, S.; Toma, M.; Anderson, K.; Davey, R.; et al. Canadian Cardiovascular Society/Canadian Cardiac Transplant Network Position Statement on Heart Transplantation: Patient Eligibility, Selection, and Post-Transplantation Care. Can. J. Cardiol. 2020, 36, 335–356. [Google Scholar] [CrossRef] [Green Version]
- Zhao, K.; McLean, R.C.; Hoteit, M.A.; Olthoff, K.M. Combined Heart and Liver Transplant: Indication, Patient Selection, and Allocation Policy. Clin. Liver Dis. 2019, 13, 170–175. [Google Scholar] [CrossRef]
- Cotter, T.G.; Wang, J.; Peeraphatdit, T.; Sandıkçı, B.; Ayoub, F.; Kim, G.; Te, H.; Jeevanandam, V.; di Sabato, D.; Charlton, M. Simultaneous Heart-Liver Transplantation for Congenital Heart Disease in the USA: Rapidly Increasing with Acceptable Outcomes. Hepatology 2020. [Google Scholar] [CrossRef] [PubMed]
- Rizvi, S.S.A.; Challapalli, J.; Maynes, E.J.; Weber, M.P.; Choi, J.H.; O’Malley, T.J.; Entwistle, J.W.; Morris, R.J.; Samuels, L.E.; Massey, H.T.; et al. Indications and outcomes of combined heart-liver transplant: A systematic review and met-analysis. Transpl. Rev. 2020, 34, 100517. [Google Scholar] [CrossRef] [PubMed]
- Khush, K.K.; Cherikh, W.S.; Chambers, D.C.; Goldfarb, S.; Hayes, D., Jr.; Kucheryavaya, A.Y.; Levvey, B.J.; Meiser, B.; Rossano, J.W.; Stehlik, J. The International Thoracic Organ Transplant Registry of the International Society for Heart and Lung Transplantation: Thirty-fifth Adult Heart Transplantation Report-2018; Focus Theme: Multiorgan Transplantation. J. Heart Lung Transpl. 2018, 37, 1155–1168. [Google Scholar] [CrossRef] [PubMed]
- Bryant, R.; Rizwan, R.; Zafar, F.; Shah, S.A.; Chin, C.; Tweddell, J.S.; Morales, D.L. Contemporary Outcomes of Combined Heart-Liver Transplant in Patients with Congenital Heart Disease. Transplantation 2018, 102, e67–e73. [Google Scholar] [CrossRef] [PubMed]
- Chou, A.S.; Habertheuer, A.; Chin, A.L.; Sultan, I.; Vallabhajosyula, P. Heart-Kidney and Heart-Liver Transplantation Provide Immunoprotection to the Cardiac Allograft. Ann. Thorac. Surg. 2019, 108, 458–466. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Basic * | In-Depth * | Investigational * | |
|---|---|---|---|
| Childhood (every 3–4 years) |
|
|
|
| Adolescence (every 1–3 years) |
|
|
|
| Adulthood (every 1–2 years) |
|
|
|
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fortea, J.I.; Puente, Á.; Cuadrado, A.; Huelin, P.; Pellón, R.; González Sánchez, F.J.; Mayorga, M.; Cagigal, M.L.; García Carrera, I.; Cobreros, M.; et al. Congestive Hepatopathy. Int. J. Mol. Sci. 2020, 21, 9420. https://doi.org/10.3390/ijms21249420
Fortea JI, Puente Á, Cuadrado A, Huelin P, Pellón R, González Sánchez FJ, Mayorga M, Cagigal ML, García Carrera I, Cobreros M, et al. Congestive Hepatopathy. International Journal of Molecular Sciences. 2020; 21(24):9420. https://doi.org/10.3390/ijms21249420
Chicago/Turabian StyleFortea, José Ignacio, Ángela Puente, Antonio Cuadrado, Patricia Huelin, Raúl Pellón, Francisco José González Sánchez, Marta Mayorga, María Luisa Cagigal, Inés García Carrera, Marina Cobreros, and et al. 2020. "Congestive Hepatopathy" International Journal of Molecular Sciences 21, no. 24: 9420. https://doi.org/10.3390/ijms21249420
APA StyleFortea, J. I., Puente, Á., Cuadrado, A., Huelin, P., Pellón, R., González Sánchez, F. J., Mayorga, M., Cagigal, M. L., García Carrera, I., Cobreros, M., Crespo, J., & Fábrega, E. (2020). Congestive Hepatopathy. International Journal of Molecular Sciences, 21(24), 9420. https://doi.org/10.3390/ijms21249420