TAK1 Mediates ROS Generation Triggered by the Specific Cephalosporins through Noncanonical Mechanisms


 , and
, and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
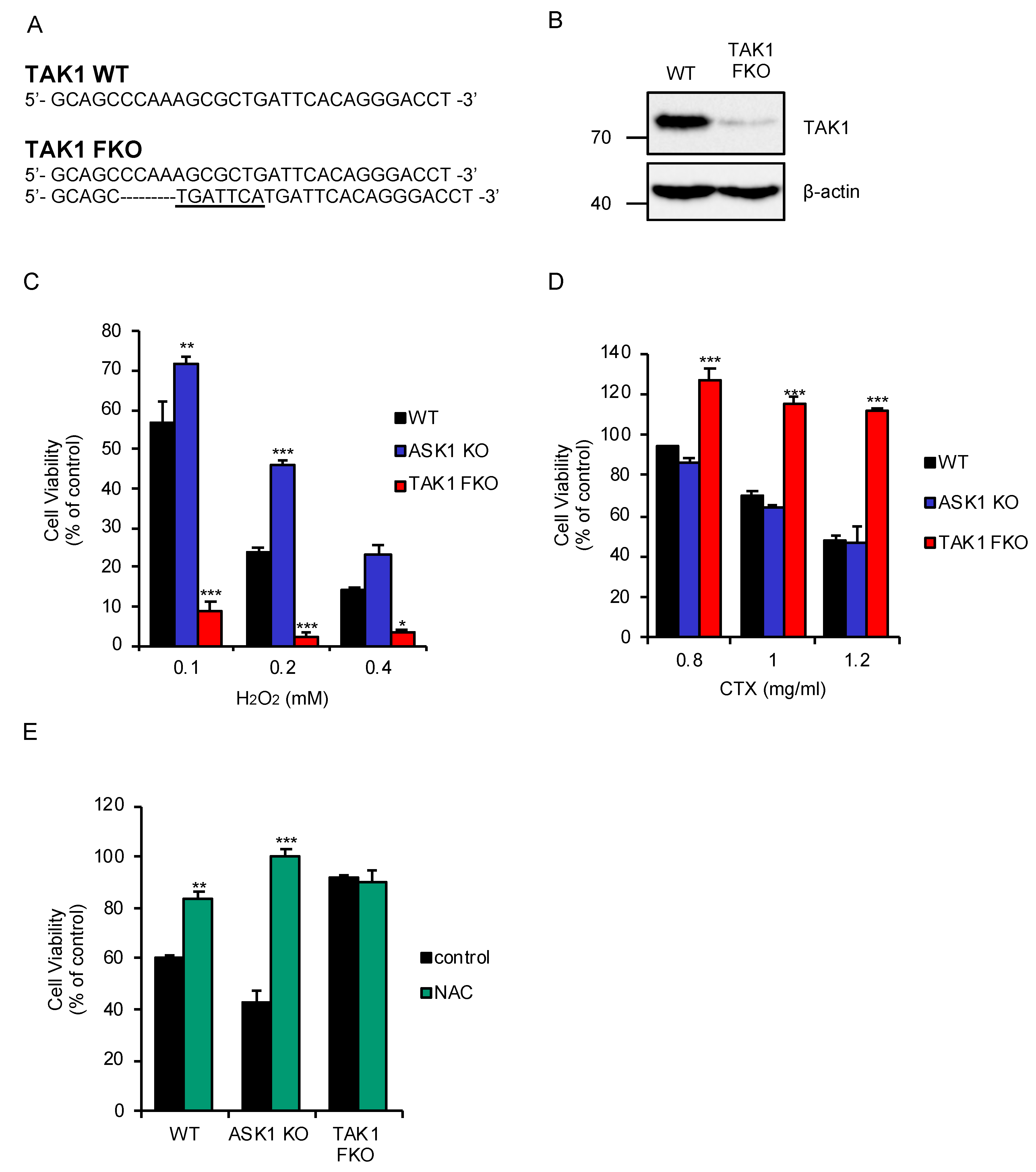
2.1. TAK1 Is Required for Oxidative Stress-Induced Cell Death Stimulated by CTX
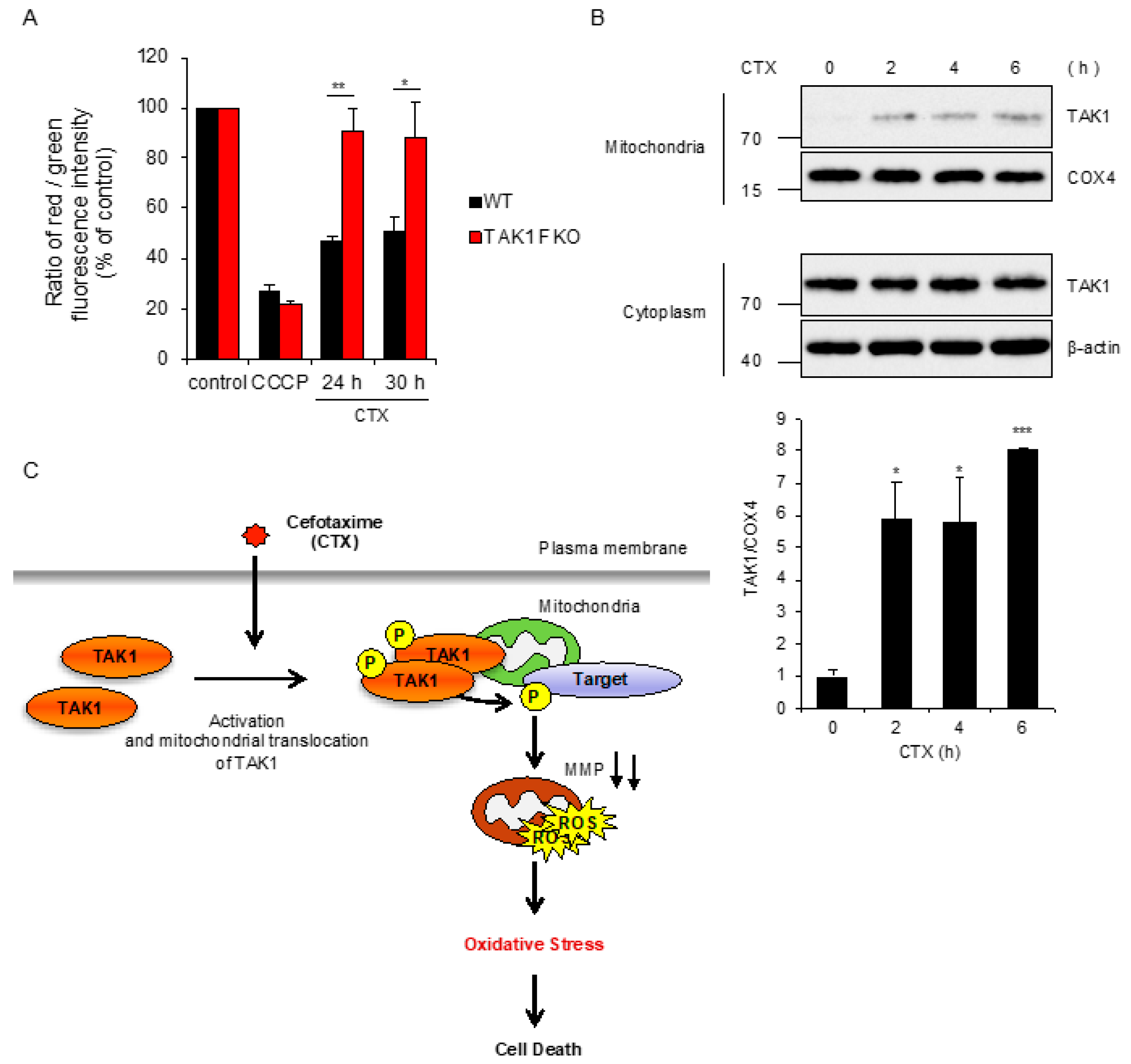
2.2. TAK1 Is Required for CTX-Induced ROS Generation
2.3. Activation of the MAPK and NF-κB Pathways Are not Required for CTX-Induced ROS Generation
2.4. CTX-Driven Mitochondrial Damage Is Mitigated by Loss of TAK1
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Reagents
4.2. Colorimetric Cell Viability Assay
4.3. Immunoblot Analysis
4.4. Generation of Knockout Cell Lines
4.5. Bioimaging and Quantification of ROS
4.6. Isolation of Mitochondrial Fraction
4.7. Mitochondrial Membrane Potential Assay
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hirata, Y.; Takahashi, M.; Morishita, T.; Noguchi, T.; Matsuzawa, A. Post-Translational Modifications of the TAK1-TAB Complex. Int. J. Mol. Sci. 2017, 18, 205. [Google Scholar] [CrossRef]
- Dai, L.; Thu, C.A.; Liu, X.Y.; Xi, J.J.; Cheung, P.C.F. TAK1, more than just innate immunity. IUBMB Life 2012, 64, 825–834. [Google Scholar] [CrossRef] [PubMed]
- Ajibade, A.A.; Wang, H.Y.; Wang, R.F. Cell type-specific function of TAK1 in innate immune signaling. Trends Immunol. 2013, 34, 307–316. [Google Scholar] [CrossRef] [PubMed]
- Shirakabe, K.; Yamaguchi, K.; Shibuya, H.; Irie, K.; Matsuda, S.; Moriguchi, T.; Gotoh, Y.; Matsumoto, K.; Nishida, E. TAK1 mediates the ceramide signaling to stress-activated protein kinase/c-Jun N-terminal kinase. J. Biol. Chem. 1997, 272, 8141–8144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inagaki, M.; Omori, E.; Kim, J.Y.; Komatsu, Y.; Scott, G.; Ray, M.K.; Yamada, G.; Matsumoto, K.; Mishina, Y.; Ninomiya-Tsuji, J. TAK1-binding Protein 1, TAB1, Mediates Osmotic Stress-induced TAK1 Activation but Is Dispensable for TAK1-mediated Cytokine Signaling. J. Biol. Chem. 2008, 283, 33080–33086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hinz, M.; Stilmann, M.; Arslan, S.C.; Khanna, K.K.; Dittmar, G.; Scheidereit, C. A cytoplasmic ATM-TRAF6-cIAP1 module links nuclear DNA damage signaling to ubiquitin-mediated NF-kappaB activation. Mol. Cell 2010, 40, 63–74. [Google Scholar] [CrossRef] [PubMed]
- Blanco, S.; Santos, C.; Lazo, P.A. Vaccinia-related kinase 2 modulates the stress response to hypoxia mediated by TAK1. Mol. Cell Biol. 2007, 27, 7273–7283. [Google Scholar] [CrossRef] [Green Version]
- Onodera, Y.; Teramura, T.; Takehara, T.; Shigi, K.; Fukuda, K. Reactive oxygen species induce Cox-2 expression via TAK1 activation in synovial fibroblast cells. FEBS Open Biol. 2015, 5, 492–501. [Google Scholar] [CrossRef] [Green Version]
- Omori, E.; Matsumoto, K.; Zhu, S.; Smart, R.C.; Ninomiya-Tsuji, J. Ablation of TAK1 upregulates reactive oxygen species and selectively kills tumor cells. Cancer Res. 2010, 70, 8417–8425. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.S.; Wu, D.; Huang, D.Y.; Lin, W.W. TAK1 inhibition-induced RIP1-dependent apoptosis in murine macrophages relies on constitutive TNF-α signaling and ROS production. J. Biomed. Sci. 2015, 22, 76. [Google Scholar] [CrossRef] [Green Version]
- Sato, S.; Sanjo, H.; Takeda, K.; Ninomiya-Tsuji, J.; Yamamoto, M.; Kawai, T.; Matsumoto, K.; Takeuchi, O.; Akira, S. Essential function for the kinase TAK1 in innate and adaptive immune responses. Nat. Immunol. 2005, 6, 1087–1095. [Google Scholar] [CrossRef] [PubMed]
- Jadrich, J.L.; O’Connor, M.B.; Coucouvanis, E. The TGF beta activated kinase TAK1 regulates vascular development in vivo. Development 2006, 133, 1529–1541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ajibade, A.A.; Wang, Q.; Cui, J.; Zou, J.; Xia, X.; Wang, M.; Tong, Y.; Hui, W.; Liu, D.; Su, B.; et al. TAK1 negatively regulates NF-κB and p38 MAP kinase activation in Gr-1+CD11b+ neutrophils. Immunity 2012, 36, 43–54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakamachi, Y.; Morioka, S.; Mihaly, S.R.; Takaesu, G.; Foley, J.F.; Fessler, M.B.; Ninomiya-Tsuji, J. TAK1 regulates resident macrophages by protecting lysosomal integrity. Cell Death Dis. 2017, 8, e2598. [Google Scholar] [CrossRef] [Green Version]
- Fujino, G.; Noguchi, T.; Takeda, K.; Ichijo, H. Thioredoxin and protein kinases in redox signaling. Semin. Cancer Biol. 2006, 16, 427–435. [Google Scholar] [CrossRef]
- Hashimoto, K.; Simmons, A.N.; Kajino-Sakamoto, R.; Tsuji, Y.; Ninomiya-Tsuji, J. TAK1 Regulates the Nrf2 Antioxidant System Through Modulating p62/SQSTM1. Antioxid. Redox. Signal. 2016, 25, 953–964. [Google Scholar] [CrossRef] [Green Version]
- Li, K.; Yang, B.; Zhao, C. Transforming growth factor-β-activated kinase 1 enhances H2O2-induced apoptosis independently of reactive oxygen species in cardiomyocytes. J. Cardiovasc. Med. (Hagerstown) 2014, 15, 565–571. [Google Scholar] [CrossRef]
- Chen, Z.; Shen, X.; Shen, F.; Zhong, W.; Wu, H.; Liu, S.; Lai, J. TAK1 activates AMPK-dependent cell death pathway in hydrogen peroxide-treated cardiomyocytes, inhibited by heat shock protein-70. Mol. Cell Biochem. 2013, 377, 35–44. [Google Scholar] [CrossRef]
- Noguchi, T.; Ishii, K.; Fukutomi, H.; Naguro, I.; Matsuzawa, A.; Takeda, K.; Ichijo, H. Requirement of reactive oxygen species-dependent activation of ASK1-p38 MAPK pathway for extracellular ATP-induced apoptosis in macrophage. J. Biol. Chem. 2008, 283, 7657–7665. [Google Scholar] [CrossRef] [Green Version]
- Noguchi, T.; Takeda, K.; Matsuzawa, A.; Saegusa, K.; Nakano, H.; Gohda, J.; Inoue, J.; Ichijo, H. Recruitment of tumor necrosis factor receptor-associated factor family proteins to apoptosis signal-regulating kinase 1 signalosome is essential for oxidative stress-induced cell death. J. Biol. Chem. 2005, 280, 37033–37040. [Google Scholar] [CrossRef] [Green Version]
- Hirata, Y.; Katagiri, K.; Nagaoka, K.; Morishita, T.; Kudoh, Y.; Hatta, T.; Naguro, I.; Kano, K.; Udagawa, T.; Natsume, T.; et al. TRIM48 Promotes ASK1 Activation and Cell Death through Ubiquitination-Dependent Degradation of the ASK1-Negative Regulator PRMT1. Cell Rep. 2017, 21, 2447–2457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, R.N. Cefotaxime and desacetylcefotaxime antimicrobial interactions. The clinical relevance of enhanced activity: A review. Diagn. Microbiol. Infect. Dis. 1995, 22, 19–33. [Google Scholar] [CrossRef]
- Kalghatgi, S.; Spina, C.S.; Costello, J.C.; Liesa, M.; Morones-Ramirez, J.R.; Slomovic, S.; Molina, A.; Shirihai, O.S.; Collins, J.J. Bactericidal antibiotics induce mitochondrial dysfunction and oxidative damage in Mammalian cells. Sci. Transl. Med. 2013, 5, 192ra185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kohanski, M.A.; Dwyer, D.J.; Hayete, B.; Lawrence, C.A.; Collins, J.J. A common mechanism of cellular death induced by bactericidal antibiotics. Cell 2007, 130, 797–810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamada, M.; Suzuki, M.; Noguchi, T.; Yokosawa, T.; Sekiguchi, Y.; Mutoh, N.; Toyama, T.; Hirata, Y.; Hwang, G.-W.; Matsuzawa, A. The Antibiotic Cefotaxime Works as Both an Activator of Nrf2 and an Inducer of HSP70 in Mammalian Cells. BPB Rep. 2020, 3, 16–21. [Google Scholar] [CrossRef]
- Noguchi, T.; Suzuki, M.; Mutoh, N.; Hirata, Y.; Tsuchida, M.; Miyagawa, S.; Hwang, G.W.; Aoki, J.; Matsuzawa, A. Nuclear-accumulated SQSTM1/p62-based ALIS act as microdomains sensing cellular stresses and triggering oxidative stress-induced parthanatos. Cell Death Dis. 2018, 9, 1193. [Google Scholar] [CrossRef]
- Hirata, Y.; Takahashi, M.; Kudoh, Y.; Kano, K.; Kawana, H.; Makide, K.; Shinoda, Y.; Yabuki, Y.; Fukunaga, K.; Aoki, J.; et al. Fatty acids promote proinflammatory signaling and cell death by stimulating the apoptosis signal-regulating kinase 1 (ASK1)-p38 pathway. J. Biol. Chem. 2017, 292, 8174–8185. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Shi, P.; Chen, Q.; Huang, Z.; Zou, D.; Zhang, J.; Gao, X.; Lin, Z. Mitochondrial ROS promote macrophage pyroptosis by inducing GSDMD oxidation. J. Mol. Cell Biol. 2019, 11, 1069–1082. [Google Scholar] [CrossRef] [Green Version]
- Smiley, S.T.; Reers, M.; Mottola-Hartshorn, C.; Lin, M.; Chen, A.; Smith, T.W.; Steele, G.D., Jr.; Chen, L.B. Intracellular heterogeneity in mitochondrial membrane potentials revealed by a J-aggregate-forming lipophilic cation JC-1. Proc. Natl. Acad. Sci. USA 1991, 88, 3671–3675. [Google Scholar] [CrossRef] [Green Version]
- Arthur, J.S.; Ley, S.C. Mitogen-activated protein kinases in innate immunity. Nat. Rev. Immunol. 2013, 13, 679–692. [Google Scholar] [CrossRef]
- Papadakis, E.; Finegan, K.; Wang, X.; Robinson, A.; Guo, C.; Kayahara, M.; Tournier, C. The regulation of Bax by c-Jun N-terminal protein kinase (JNK) is a prerequisite to the mitochondrial-induced apoptotic pathway. FEBS Lett. 2006, 580, 1320–1326. [Google Scholar] [CrossRef] [PubMed]
- Hindi, S.M.; Sato, S.; Xiong, G.; Bohnert, K.R.; Gibb, A.A.; Gallot, Y.S.; McMillan, J.D.; Hill, B.G.; Uchida, S.; Kumar, A. TAK1 regulates skeletal muscle mass and mitochondrial function. JCI Insight. 2018, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vaarmann, A.; Mandel, M.; Zeb, A.; Wareski, P.; Liiv, J.; Kuum, M.; Antsov, E.; Liiv, M.; Cagalinec, M.; Choubey, V.; et al. Mitochondrial biogenesis is required for axonal growth. Development 2016, 143, 1981–1992. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luong, N.C.; Abiko, Y.; Shibata, T.; Uchida, K.; Warabi, E.; Suzuki, M.; Noguchi, T.; Matsuzawa, A.; Kumagai, Y. Redox cycling of 9,10-phenanthrenequinone activates epidermal growth factor receptor signaling through S-oxidation of protein tyrosine phosphatase 1B. J. Toxicol. Sci. 2020, 45, 349–363. [Google Scholar] [CrossRef] [PubMed]
- Hirata, Y.; Nada, Y.; Yamada, Y.; Toyama, T.; Fukunaga, K.; Hwang, G.W.; Noguchi, T.; Matsuzawa, A. Elaidic Acid Potentiates Extracellular ATP-Induced Apoptosis via the P2X. Biol. Pharm. Bull. 2020, 43, 1562–1569. [Google Scholar] [CrossRef] [PubMed]
- Noguchi, T.; Tsuchida, M.; Kogue, Y.; Spadini, C.; Hirata, Y.; Matsuzawa, A. Brefeldin A-Inhibited Guanine Nucleotide-Exchange Factor 1 (BIG1) Governs the Recruitment of Tumor Necrosis Factor Receptor-Associated Factor 2 (TRAF2) to Tumor Necrosis Factor Receptor 1 (TNFR1) Signaling Complexes. Int. J. Mol. Sci. 2016, 17, 1869. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kudoh, Y.; Noguchi, T.; Ishii, C.; Maeda, K.; Nishidate, A.; Hirata, Y.; Matsuzawa, A. Antibiotic Vancomycin Promotes the Gene Expression of NOD-Like Receptor Families in Macrophages. BPB Rep. 2018, 1, 6–10. [Google Scholar] [CrossRef]
- Sekiguchi, Y.; Yamada, M.; Noguchi, T.; Noomote, C.; Tsuchida, M.; Kudoh, Y.; Hirata, Y.; Matsuzawa, A. The anti-cancer drug gefitinib accelerates Fas-mediated apoptosis by enhancing caspase-8 activation in cancer cells. J. Toxicol. Sci. 2019, 44, 435–440. [Google Scholar] [CrossRef] [Green Version]
- Tsuchida, M.; Yokosawa, T.; Noguchi, T.; Shimada, T.; Yamada, M.; Sekiguchi, Y.; Hirata, Y.; Matsuzawa, A. Pro-apoptotic functions of TRAF2 in p53-mediated apoptosis induced by cisplatin. J. Toxicol. Sci. 2020, 45, 219–226. [Google Scholar] [CrossRef] [Green Version]
- Yokosawa, T.; Yamada, M.; Noguchi, T.; Suzuki, S.; Hirata, Y.; Matsuzawa, A. Pro-caspase-3 protects cells from polymyxin B-induced cytotoxicity by preventing ROS accumulation. J. Antibiot. 2019, 72, 848–852. [Google Scholar] [CrossRef]
- Hirata, Y.; Inoue, A.; Suzuki, S.; Takahashi, M.; Matsui, R.; Kono, N.; Noguchi, T.; Matsuzawa, A. Trans-Fatty acids facilitate DNA damage-induced apoptosis through the mitochondrial JNK-Sab-ROS positive feedback loop. Sci. Rep. 2020, 10, 2743. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Suzuki, M.; Asai, Y.; Kagi, T.; Noguchi, T.; Yamada, M.; Hirata, Y.; Matsuzawa, A. TAK1 Mediates ROS Generation Triggered by the Specific Cephalosporins through Noncanonical Mechanisms. Int. J. Mol. Sci. 2020, 21, 9497. https://doi.org/10.3390/ijms21249497
Suzuki M, Asai Y, Kagi T, Noguchi T, Yamada M, Hirata Y, Matsuzawa A. TAK1 Mediates ROS Generation Triggered by the Specific Cephalosporins through Noncanonical Mechanisms. International Journal of Molecular Sciences. 2020; 21(24):9497. https://doi.org/10.3390/ijms21249497
Chicago/Turabian StyleSuzuki, Midori, Yukino Asai, Tomohiro Kagi, Takuya Noguchi, Mayuka Yamada, Yusuke Hirata, and Atsushi Matsuzawa. 2020. "TAK1 Mediates ROS Generation Triggered by the Specific Cephalosporins through Noncanonical Mechanisms" International Journal of Molecular Sciences 21, no. 24: 9497. https://doi.org/10.3390/ijms21249497
APA StyleSuzuki, M., Asai, Y., Kagi, T., Noguchi, T., Yamada, M., Hirata, Y., & Matsuzawa, A. (2020). TAK1 Mediates ROS Generation Triggered by the Specific Cephalosporins through Noncanonical Mechanisms. International Journal of Molecular Sciences, 21(24), 9497. https://doi.org/10.3390/ijms21249497