Small Molecules Acting on Myofilaments as Treatments for Heart and Skeletal Muscle Diseases


Abstract
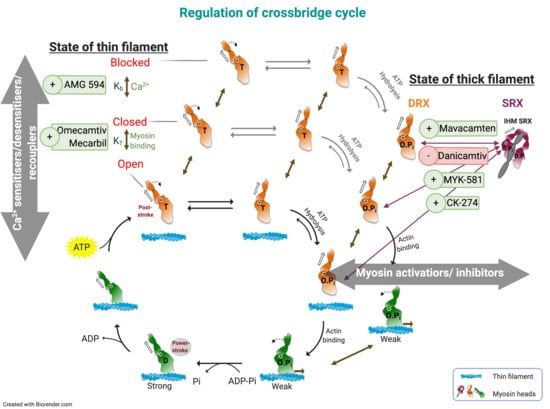
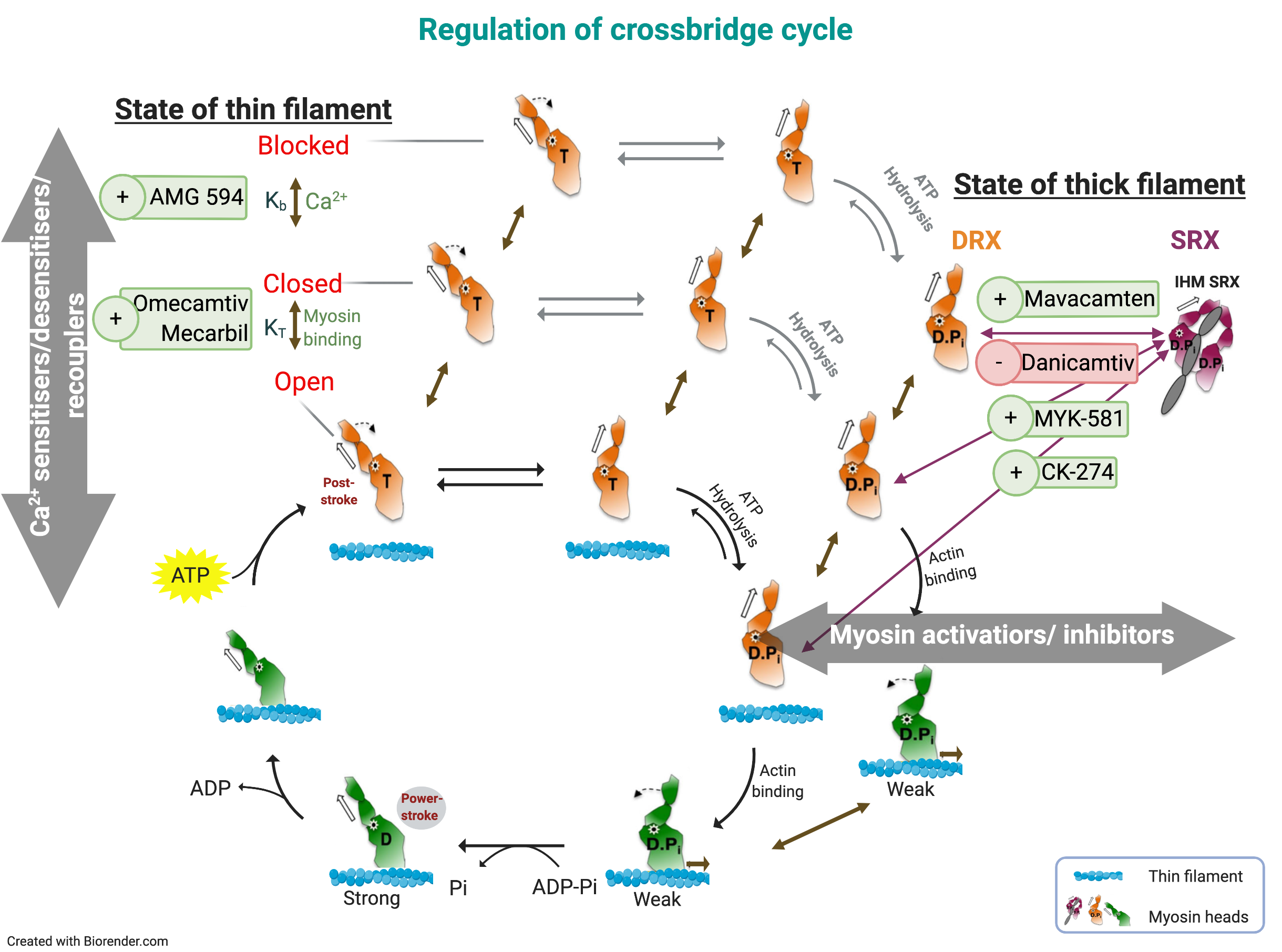
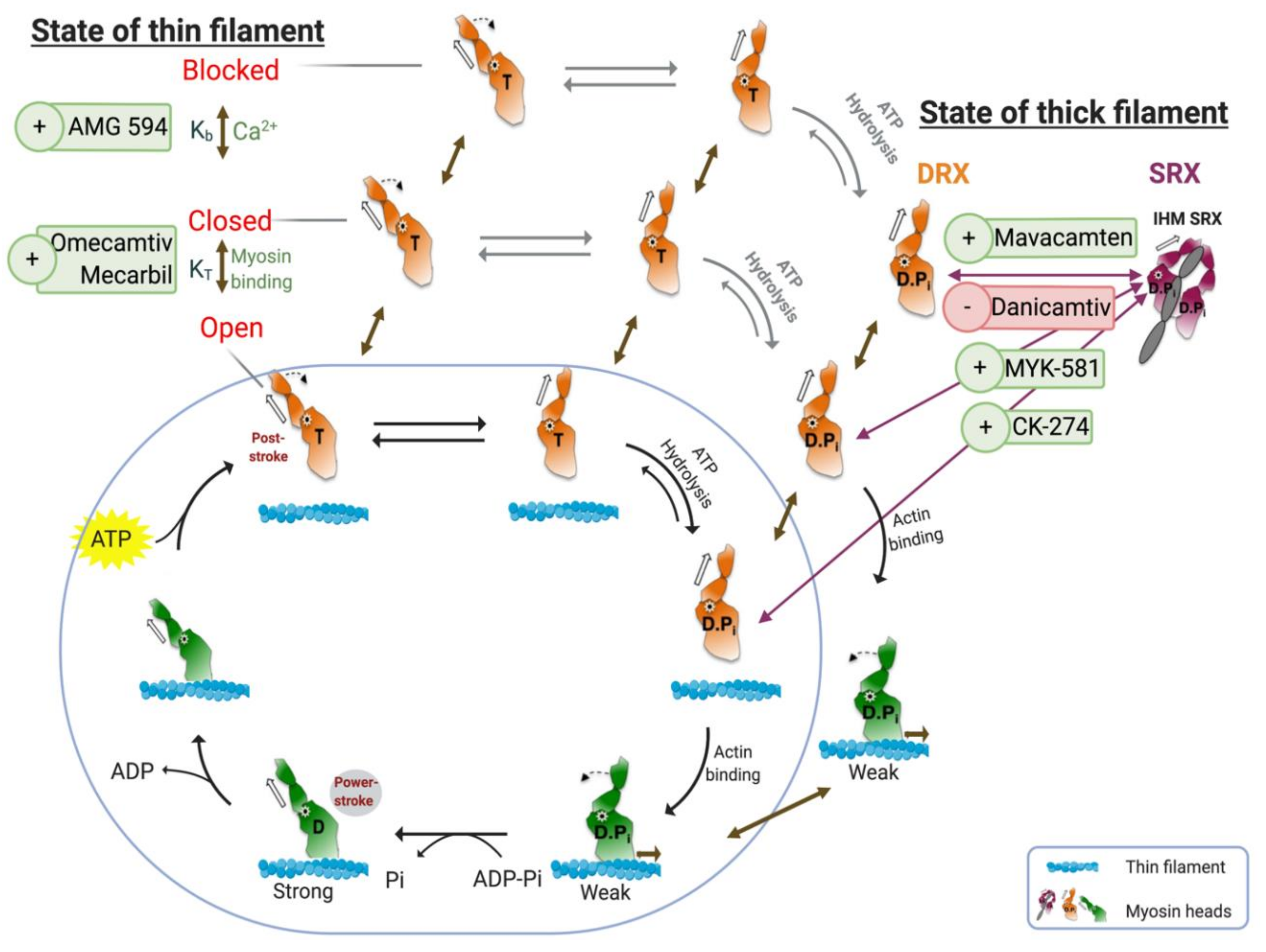
:
1. Introduction
2. Contractile Activators as Treatments for Heart Failure and Muscular Myopathies
2.1. Cardiac Muscle Ca2+-Sensitisers (or Positive Inotropes)
2.2. Cardiac Myosin Activators
2.2.1. Omecamtiv Mecarbil
2.2.2. Danicamtiv
2.2.3. EMD57033
2.2.4. Assessment of Myosin Activators
2.3. Skeletal Muscle Activators and Ca2+-sensitisers
2.3.1. Tirasemtiv
2.3.2. Reldesemtiv
2.3.3. Piperine
3. Contractile Inhibitors as a Treatment for Hypertrophic Cardiomyopathy (HCM)
3.1. Ca2+-Desensitisers
3.1.1. Green Tea Catechins (EGCg and ECg)
3.1.2. Nebivolol
3.2. Recouplers
3.3. Myosin Inhibitors
3.3.1. Blebbistatin and Its Analogues
3.3.2. Mavacamten
3.3.3. CK-3773274 (or CK-274)
3.3.4. Assessment of Cardiac Myosin Inhibitors
4. Discussion
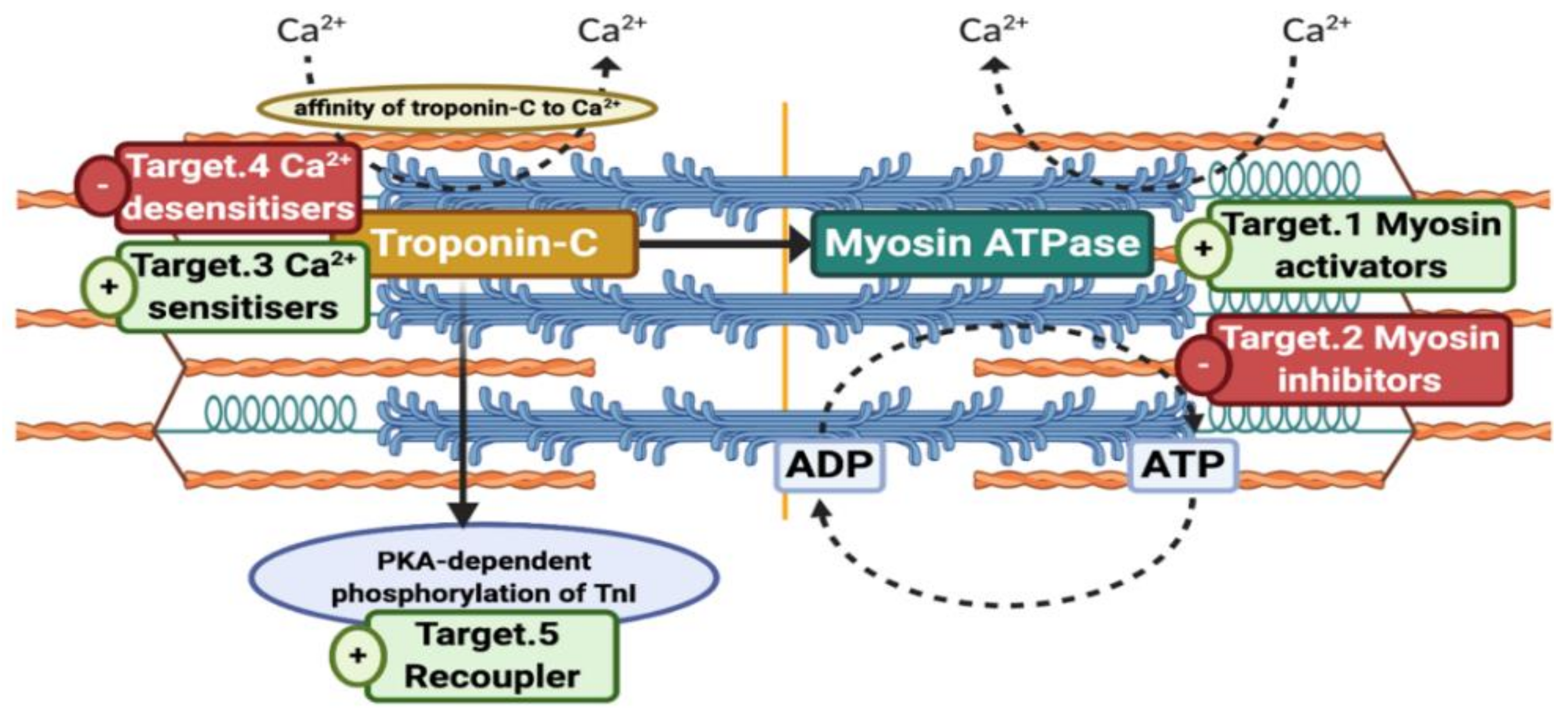
4.1. Targets within the Contractile Apparatus
4.2. Selected Small Molecules with Potential Therapeutic Value
4.3. Limitations and Difficulties
4.4. Future Prospects
Funding
Acknowledgments
Conflicts of Interest
References
- World Health Organization (WHO). Cardiovascular Diseases. 2020. Available online: https://www.who.int/health-topics/cardiovascular-diseases/ (accessed on 2 June 2020).
- Seferović, P.M.; Polovina, M.; Bauersachs, J.; Arad, M.; Gal, T.B.; Lund, L.H.; Felix, S.B.; Arbustini, E.; Caforio, A.L.P.; Farmakis, D.; et al. Heart failure in cardiomyopathies: A position paper from the Heart Failure Association of the European Society of Cardiology. Eur. J. Heart Fail. 2019, 21, 553–576. [Google Scholar] [CrossRef] [Green Version]
- Maron, B.J.; Jeffrey, T.A.; Gaetano, T.; Charles, A.; Domenico, C.; Donna, A.; Moss, A.J.; Seidman, C.E.; Young, B.J. Contemporary Definitions and Classification of the Cardiomyopathies. Circulation 2006, 113, 1807–1816. [Google Scholar] [CrossRef] [Green Version]
- Hershberger, R.E.; Hedges, D.J.; Morales, A. Dilated cardiomyopathy: The complexity of a diverse genetic architecture. Nat. Rev. Cardiol. 2013, 10, 531–547. [Google Scholar] [CrossRef] [PubMed]
- Elliott, P.; McKenna, W.J. Hypertrophic cardiomyopathy. Lancet 2004, 363, 1881–1891. [Google Scholar] [CrossRef]
- Ashrafian, H.; Redwood, C.; Blair, E.; Watkins, H. Hypertrophic cardiomyopathy: A paradigm for myocardial energy depletion. Trends Genet. 2003, 19, 263–268. [Google Scholar] [CrossRef]
- Tardiff, J.C.; Carrier, L.; Bers, D.M.; Poggesi, C.; Ferrantini, C.; Coppini, R.; Maier, L.S.; Ashrafian, H.; Huke, S.; Van der Velden, J. Targets for therapy in sarcomeric cardiomyopathies. Cardiovasc. Res. 2015, 105, 457–470. [Google Scholar] [CrossRef] [Green Version]
- Poggesi, C.; Ho, C.Y. Muscle dysfunction in hypertrophic cardiomyopathy: What is needed to move to translation? J. Muscle Res. Cell Motil. 2014, 35, 37–45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sewry, C.A.; Laitila, J.M.; Wallgren-Pettersson, C. Nemaline myopathies: A current view. J. Muscle Res. Cell Motil. 2019, 40, 111–126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papadaki, M.; Vikhorev, P.G.; Marston, S.B.; Messer, A.E. Uncoupling of myofilament Ca2+ sensitivity from troponin I phosphorylation by mutations can be reversed by epigallocatechin-3-gallate. Cardiovasc. Res. 2015, 108, 99–110. [Google Scholar] [CrossRef] [Green Version]
- Messer, A.E.; Marston, S.B. Investigating the role of uncoupling of troponin I phosphorylation from changes in myofibrillar Ca2+-sensitivity in the pathogenesis of cardiomyopathy. Front. Physiol. 2014, 5, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Metra, M.; Teerlink, J.R. Heart failure. Lancet 2017, 390, 1981–1995. [Google Scholar] [CrossRef]
- Hartupee, J.; Mann, D.L. Neurohormonal activation in heart failure with reduced ejection fraction. Nat. Rev. Cardiol. 2016, 14, 30–38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmad, T.; Miller, P.E.; McCullough, M.; Desai, N.R.; Riello, R.; Psotka, M.; Böhm, M.; Allen, L.A.; Teerlink, J.R.; Rosano, G.M.C.; et al. Why has positive inotropy failed in chronic heart failure? Lessons from prior inotrope trials. Eur. J. Heart Fail. 2019, 21, 1064–1078. [Google Scholar] [CrossRef] [PubMed]
- Ponikowski, P.; Voors, A.A.; Anker, S.D.; Bueno, H.; Cleland, J.G.F.; Coats, A.J.S.; Falk, V.; González-Juanatey, J.R.; Harjola, V.P.; Jankowska, E.A.; et al. 2016 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure. Eur. J. Heart Fail. 2019, 21, 1064–1078. [Google Scholar]
- Yancy, C.W.; Jessup, M.; Bozkurt, B.; Butler, J.; Casey, D.E.; Colvin, M.M.; Drazner, M.H.; Filippatos, G.; Fonarow, G.C.; Givertz, M.M.; et al. 2016 ACC/AHA/HFSA focused update on new pharmacological therapy for heart failure: An update of the 2013 ACCF/AHA guideline for the management of heart failure: A report of the American College of Cardiology/American Heart Association Task Force on Clinic. Circulation 2016, 134, e282–e293. [Google Scholar] [CrossRef] [PubMed]
- Papp, Z.; Édes, I.; Fruhwald, S.; De Hert, S.G.; Salmenperä, M.; Leppikangas, H.; Mebazaa, A.; Landoni, G.; Grossini, E.; Caimmi, P.; et al. Levosimendan: Molecular mechanisms and clinical implications: Consensus of experts on the mechanisms of action of levosimendan. Int. J. Cardiol. 2012, 159, 82–87. [Google Scholar] [CrossRef] [Green Version]
- Li, M.X.; Robertson, I.M.; Sykes, B.D. Interaction of cardiac troponin with cardiotonic drugs: A structural perspective. Biochem. Biophys. Res. Commun. 2008, 369, 88–99. [Google Scholar] [CrossRef] [Green Version]
- Sorsa, T.; Heikkinen, S.; Abbott, M.B.; Abusamhadneh, E.; Laakso, T.; Tilgmann, C.; Serimaa, R.; Annila, A.; Rosevear, P.R.; Drakenberg, T.; et al. Binding of Levosimendan, a Calcium Sensitizer, to Cardiac Troponin C. J. Biol. Chem. 2001, 276, 9337–9343. [Google Scholar] [CrossRef] [Green Version]
- Bokník, P.; Neumann, J.; Kaspareit, G.; Schmitz, W.; Scholz, H.; Vahlensieck, U.; Zimmermann, N. Mechanisms of the contractile effects of levosimendan in the mammalian heart. J. Pharmacol. Exp. Ther. 1997, 280, 277–283. [Google Scholar]
- Lubsen, J. Effect of pimobendan on exercise capacity in patients with heart failure: Main results from the Pimobendan in Congestive Heart Failure (PICO) trial. Heart 1996, 76, 223–231. [Google Scholar] [CrossRef]
- AdisInsight. Pimobendan–AdisInsight. 2009. Available online: https://adisinsight.springer.com/drugs/800000190 (accessed on 8 September 2020).
- Takahashi, R.; Endoh, M. Increase in myofibrillar Ca2+ sensitivity induced by UD-CG 212 Cl, an active metabolite of pimobendan, in canine ventricular myocardium. J. Cardiovasc. Pharmacol. 2001, 37, 209–218. [Google Scholar] [CrossRef] [PubMed]
- Bowles, D.; Fry, D. Pimobendan and its use in treating canine congestive heart failure. Compend. Contin. Educ. Vet. 2011, 33, E1. [Google Scholar] [PubMed]
- AdisInsight. Bepridil-AdisInsight. 2000. Available online: https://adisinsight.springer.com/drugs/800014884 (accessed on 15 July 2020).
- AdisInsight. Senazodan-AdisInsight. 2010. Available online: https://adisinsight.springer.com/drugs/800000316 (accessed on 7 September 2020).
- Brixius, K.; Reicke, S.; Reuter, H.; Schwinger, R.H.G. Effects of the Ca2+ sensitizers EMD 57033 and CGP 48506 on myocardial contractility and Ca2+ transients in human ventricular and atrial myocardium. Z. Kardiol. 2001, 91, 312–318. [Google Scholar] [CrossRef] [PubMed]
- Baudenbacher, F.; Schober, T.; Pinto, J.R.; Sidorov, V.Y.; Hilliard, F.; Solaro, R.J.; Potter, J.D.; Knollmann, B.C. Myofilament Ca2+ sensitization causes susceptibility to cardiac arrhythmia in mice. J. Clin. Investig. 2008, 118, 3893–3903. [Google Scholar] [CrossRef] [Green Version]
- AdisInsight. AMG 594-AdisInsight. 2019. Available online: https://adisinsight.springer.com/drugs/800054180#:~:text=AMG (accessed on 24 July 2020).
- Reagan, J.D.; Hartman, J.J.; Motani, A.S.; Sutherland, W.; Poppe, L.; Hoagland, K.; Rock, B.; Lobenhofer, E.; Nguyen, K.K.; Liu, Q. The novel myotrope, AMG 594, is a small-molecule cardiac troponin activator that increases cardiac contractility in vitro and in vivo. In Proceedings of the Keystone Symposia on Molecular and Cellular Biology, Keystone, CO, USA, 1–5 March 2020; Available online: https://cytokinetics.com/wp-content/uploads/2020/03/Keystone_HF_2020_AMG594.pdf (accessed on 24 July 2020).
- Packer, M.; Colucci, W.; Fisher, L.; Massie, B.M.; Teerlink, J.R.; Young, J.; Padley, R.J.; Thakkar, R.; Delgado-Herrera, L.; Salon, J.; et al. Effect of levosimendan on the short-term clinical course of patients with acutely decompensated heart failure. JACC Heart Fail. 2013, 1, 103–111. [Google Scholar] [CrossRef]
- Moiseyev, V.S.; Põder, P.; Andrejevs, N.; Ruda, M.Y.; Golikov, A.P.; Lazebnik, L.B.; Kobalava, Z.D.; Lehtonen, L.A.; Laine, T.; Nieminen, M.S.; et al. Safety and efficacy of a novel calcium sensitizer, levosimendan, in patients with left ventricular failure due to an acute myocardial infarction: A randomized, placebo-controlled, double-blind study (RUSSLAN). Eur. Heart J. 2002, 23, 1422–1432. [Google Scholar] [CrossRef] [Green Version]
- Follath, F.; Cleland, J.G.F.; Just, H.; Papp, J.G.Y.; Scholz, H.; Peuhkurinen, K.; Harjola, V.P.; Mitrovic, V.; Abdalla, M.; Sandell, E.P.; et al. Efficacy and safety of intravenous levosimendan compared with dobutamine in severe low-output heart failure (the LIDO study): A randomised double-blind trial. Lancet 2002, 360, 196–202. [Google Scholar] [CrossRef]
- Mebazaa, A.; Nieminen, M.S.; Packer, M.; Cohen-Solal, A.; Kleber, F.X.; Pocock, S.J.; Thakkar, R.; Padley, R.J.; Põder, P.; Kivikko, M. Levosimendan vs. dobutamine for patients with acute decompensated heart failure: The SURVIVE randomized trial. J. Am. Med. Assoc. 2007, 297, 1883–1891. [Google Scholar] [CrossRef] [Green Version]
- Malik, F.I.; Hartman, J.J.; Elias, K.A.; Morgan, B.P.; Rodriguez, H.; Brejc, K.; Anderson, R.L.; Sueoka, S.H.; Lee, K.H.; Finer, J.T.; et al. Cardiac myosin activation: A potential therapeutic approach for systolic heart failure. Science 2011, 331, 1439–1443. [Google Scholar] [CrossRef] [Green Version]
- Malik, F.I.; Morgan, B.P. Cardiac myosin activation part 1: From concept to clinic. J. Mol. Cell Cardiol. 2011, 51, 454–461. [Google Scholar] [CrossRef]
- Packer, M.; Kukin, M.L.; Sollano, J.A.; Carver, J.R.; Rodeheffer, R.J.; Ivanhoe, R.J.; Dibianco, R.; Zeldis, S.M.; Hendrix, G.H.; Bommer, W.J.; et al. Effect of Oral Milrinone on Mortality in Severe Chronic Heart Failure. N. Engl. J. Med. 1991, 325, 1468–1475. [Google Scholar] [CrossRef] [PubMed]
- O’Connor, C.M.; Gattis, W.A.; Uretsky, B.F.; Adams, K.F., Jr.; McNulty, S.E.; Grossman, S.H.; McKenna, W.J.; Zannad, F.; Swedberg, K.; Gheorghiade, M.; et al. Continuous intravenous dobutamine is associated with an increased risk of death in patients with advanced heart failure: Insights from the Flolan International Randomized Survival Trial (FIRST). Am. Heart J. 1999, 138, 78–86. [Google Scholar] [CrossRef]
- Morgan, B.P.; Muci, A.; Lu, P.P.; Qian, X.; Tochimoto, T.; Smith, W.W.; Garard, M.; Kraynack, E.; Collibee, S.; Suehiro, I.; et al. Discovery of omecamtiv mecarbil the first, selective, small molecule activator of cardiac myosin. ACS Med. Chem. Lett. 2010, 1, 472–477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swenson, A.M.; Tang, X.W.; Blair, C.A.; Fetrow, C.M.; Unrath, W.C.; Previs, M.J.; Campbell, K.S.; Yengo, C.M. Omecamtiv mecarbil enhances the duty ratio of human β-cardiac myosin resulting in increased calcium sensitivity and slowed force development in cardiac muscle. J. Biol. Chem. 2017, 292, 3768–3778. [Google Scholar] [CrossRef] [Green Version]
- Nagy, L.; Kovács, A.; Bõdi, B.; Pásztor, E.T.; Fülöp, G.; Tõth, A.; Édes, I.; Papp, Z. The novel cardiac myosin activator omecamtiv mecarbil increases the calcium sensitivity of force production in isolated cardiomyocytes and skeletal muscle fibres of the rat. Br. J. Pharmacol. 2015, 172, 4506–4518. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; White, H.D.; Belknap, B.; Winkelmann, D.A.; Forgacs, E. Omecamtiv Mecarbil Modulates the Kinetic and Motile Properties of Porcine β-Cardiac Myosin. Biochemistry 2015, 54, 1963–1975. [Google Scholar] [CrossRef]
- Woody, M.S.; Greenberg, M.J.; Barua, B.; Winkelmann, D.A.; Goldman, Y.E.; Ostap, E.M. Positive cardiac inotrope omecamtiv mecarbil activates muscle despite suppressing the myosin working stroke. Nat. Commun. 2018, 9, 3838. [Google Scholar] [CrossRef] [Green Version]
- Lehrer, S.S.; Geeves, M.A. The muscle thin filament as a classical cooperative/allosteric regulatory system11Edited by P. E. Wright. J. Mol. Biol. 1998, 277, 1081–1089. [Google Scholar] [CrossRef]
- Bakkehaug, J.P.; Kildal, A.B.; Engstad, E.T.; Boardman, N.; Næsheim, T.; Rønning, L.; Aasum, E.; Larsen, T.S.; Myrmel, T.; How, O.J. Myosin activator omecamtiv mecarbil increases myocardial oxygen consumption and impairs cardiac efficiency mediated by resting myosin ATPase activity. Circ. Heart Fail. 2015, 8, 766–775. [Google Scholar] [CrossRef] [Green Version]
- Teerlink, J.R.; Malik, F.I.; Kass, D.A. Letter by Teerlink et al Regarding Article, “Myosin Activator Omecamtiv Mecarbil Increases Myocardial Oxygen Consumption and Impairs Cardiac Efficiency Mediated by Resting Myosin ATPase Activity”. Circ. Heart Fail. 2015, 8, 1141. [Google Scholar] [CrossRef] [Green Version]
- Teerlink, J.R.; Clarke, C.P.; Saikali, K.G.; Lee, J.H.; Chen, M.M.; Escandon, R.D.; Elliott, L.; Bee, R.; Habibzadeh, M.R.; Goldman, J.H.; et al. Dose-dependent augmentation of cardiac systolic function with the selective cardiac myosin activator, omecamtiv mecarbil: A first-in-man study. Lancet 2011, 378, 667–675. [Google Scholar] [CrossRef]
- Teerlink, J.R.; Felker, G.M.; McMurray, J.J.V.; Ponikowski, P.; Metra, M.; Filippatos, G.S.; Ezekowitz, J.A.; Dickstein, K.; Cleland, J.G.F.; Kim, J.B.; et al. Acute Treatment with Omecamtiv Mecarbil to Increase Contractility in Acute Heart Failure: The ATOMIC-AHF Study. J. Am. Coll. Cardiol. 2016, 67, 1444–1455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Starling, R.C. Cardiac Myosin Activators for the Treatment of Heart Failure: Stop Now or Push Ahead? J. Am. Coll. Cardiol. 2016, 67, 1456–1458. [Google Scholar] [CrossRef] [PubMed]
- Teerlink, J.R.; Diaz, R.; Felker, G.M.; McMurray, J.J.V.V.; Metra, M.; Solomon, S.D.; Adams, K.F.; Anand, I.; Arias-Mendoza, A.; Biering-Sørensen, T.; et al. Cardiac Myosin Activation with Omecamtiv Mecarbil in Systolic Heart Failure. N. Engl. J. Med. 2020. [Google Scholar] [CrossRef]
- Fernandes, S.; Oikonomopoulos, A.; Jimenez-MacInnes, S.K.; Aschar-Sobbi, R.; Henze, M.; Sumandea, M.; Gan, Q.F.; Anderson, R.L.; Del Rio, C.L. MYK-491, a Novel Small-Molecule Cardiac Myosin Activator Increases Cardiac Systolic Function and Preserves Mechanical Efficiency: Pre-Clinical in vivo and in vitro Evidence. Circulation 2019, 140 (Suppl. S1). [Google Scholar] [CrossRef]
- Tamby, J.F.; Fang, L.; Lickliter, J.; Hegde, S.; Surks, H.; Reele, S.; Teichman, S.; Yang, C.; Fernandes, S.; Lambing, J.; et al. MYK-491, a Novel Cardiac Myosin Activator, Increases Cardiac Contractility in Healthy Volunteers. Eur. J. Heart Fail. 2019, 21, 423. [Google Scholar]
- Voors, A.A.; Tamby, J.F.; Cleland, J.G.; Koren, M.; Forgosh, L.B.; Gupta, D.; Lund, L.H.; Camacho, A.; Karra, R.; Swart, H.P.; et al. Effects of danicamtiv, a novel cardiac myosin activator, in heart failure with reduced ejection fraction: Experimental data and clinical results from a phase 2a trial. Eur. J. Heart Fail. 2020, 22, 1649–1658. [Google Scholar] [CrossRef]
- Cleland, J.G.F.; Teerlink, J.R.; Senior, R.; Nifontov, E.M.; Mc Murray, J.J.V.; Lang, C.C.; Tsyrlin, V.A.; Greenberg, B.H.; Mayet, J.; Francis, D.P.; et al. The effects of the cardiac myosin activator, omecamtiv mecarbil, on cardiac function in systolic heart failure: A double-blind, placebo-controlled, crossover, dose-ranging phase 2 trial. Lancet 2011, 378, 676–683. [Google Scholar] [CrossRef]
- Greenberg, B.H.; Chou, W.; Saikali, K.G.; Escandón, R.; Lee, J.H.; Chen, M.M.; Treshkur, T.; Megreladze, I.; Wasserman, S.M.; Eisenberg, P.; et al. Safety and Tolerability of Omecamtiv Mecarbil During Exercise in Patients with Ischemic Cardiomyopathy and Angina. JACC Heart Fail. 2015, 3, 22–29. [Google Scholar] [CrossRef] [Green Version]
- Teerlink, J.R.; Felker, G.M.; McMurray, J.J.V.; Solomon, S.D.; Adams, K.F.; Cleland, J.G.F.; Ezekowitz, J.A.; Goudev, A.; Macdonald, P.; Metra, M.; et al. Chronic Oral Study of Myosin Activation to Increase Contractility in Heart Failure (COSMIC-HF): A phase 2, pharmacokinetic, randomised, placebo-controlled trial. Lancet 2016, 388, 2895–2903. [Google Scholar] [CrossRef] [Green Version]
- Teerlink, J.R.; Felker, G.M.; McMurray, J.; Solomon, S.; Cleland, J.; Goldsmith, S.; Kurtz, C.; Buchele, G.; Legg, J.; Malik, F.; et al. Effect of Omecamtiv Mecarbil in Patients With Atrial Fibrillation and Heart Failure With Reduced Ejection Fraction: Results From Cosmic-Hf. J. Am. Coll. Cardiol. 2019, 73, 691. [Google Scholar] [CrossRef]
- Teerlink, J.R.; Diaz, R.; Felker, G.M.; McMurray, J.J.V.; Metra, M.; Solomon, S.D.; Legg, J.C.; Büchele, G.; Varin, C.; Kurtz, C.E.; et al. Omecamtiv Mecarbil in Chronic Heart Failure With Reduced Ejection Fraction: Rationale and Design of GALACTIC-HF. JACC Heart Fail. 2020, 8, 329–340. [Google Scholar] [CrossRef] [PubMed]
- Teerlink, J.R.; Diaz, R.; Felker, G.M.; McMurray, J.J.V.; Metra, M.; Solomon, S.D.; Legg, J.C.; Büchele, G.; Varin, C.; Kurtz, C.E.; et al. Baseline characteristics from the cardiovascular outcomes trial of omecamtiv mecarbil (GALACTIC-HF). J. Am. Coll. Cardiol. 2020, 75, 754. [Google Scholar] [CrossRef]
- Lewis, G.; Böhm, M.; Cohen-Solal, A.; Ezekowitz, J.; Metra, M.; Ponikowski, P.; Teerlink, J.; Voors, A.; Whellan, D.; Legg, J. Multicenter Exercise Tolerance Evaluation of Omecamtiv Mecarbil Related to Increased Contractility in Heart Failure (METEORIC-HF). In Proceedings of the Heart Failure Society of America 23rd Annual Scientific Meeting, Philadelphia, PA, USA, 13–16 September 2019; Available online: https://cytokinetics.com/wp-content/uploads/2019/09/CY006-19-METEORIC-HFSA19-Study-Design-Poster_MT11_FINAL_UPLOAD.pdf (accessed on 14 August 2020).
- Radke, M.B.; Taft, M.H.; Stapel, B.; Hilfiker-Kleiner, D.; Preller, M.; Manstein, D.J. Small molecule-mediated refolding and activation of myosin motor function. eLife 2014, 3, e01603. [Google Scholar] [CrossRef] [PubMed]
- Amgen. FDA Grants Fast Track Designation for Omecamtiv Mecarbil in Heart Failure. Available online: https://www.amgen.com/media/news-releases/2020/05/fda-grants-fast-track-designation-for-omecamtiv-mecarbil-in-heart-failure/ (accessed on 19 August 2020).
- Silber, E.N.; Katz, L.N. Therapeutic strategies for managing heart failure. In Heart Failure: Pathophysiology, Molecular Biology, and Clinical Management; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2000; pp. 309–339. [Google Scholar]
- Russell, A.J.; Hartman, J.J.; Hinken, A.C.; Muci, A.R.; Kawas, R.; Driscoll, L.; Godinez, G.; Lee, K.H.; Marquez, D.; Browne Iv, W.F.; et al. Activation of fast skeletal muscle troponin as a potential therapeutic approach for treating neuromuscular diseases. Nat. Med. 2012, 18, 452–455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marston, S. Small molecule studies: The fourth wave of muscle research. J. Muscle Res. Cell Motil. 2019, 40, 69–76. [Google Scholar] [CrossRef] [Green Version]
- Hansen, R.; Saikali, K.G.; Chou, W.; Russell, A.J.; Chen, M.M.; Vijayakumar, V.; Stoltz, R.R.; Baudry, S.; Enoka, R.M.; Morgans, D.J.; et al. Tirasemtiv amplifies skeletal muscle response to nerve activation in humans. Muscle Nerve 2014, 50, 925–931. [Google Scholar] [CrossRef] [Green Version]
- Cytokinetics. Cytokinetics Announces Orphan Drug Designation Granted to CK-2017357 for the Treatment of Amyotrophic Lateral Sclerosis | Cytokinetics, Inc. Available online: http://ir.cytokinetics.com/news-releases/news-release-details/cytokinetics-announces-orphan-drug-designation-granted-ck (accessed on 25 July 2020).
- Cytokinetics. Cytokinetics Announces Negative Results From VITALITY-ALS|Cytokinetics, Inc. Available online: http://ir.cytokinetics.com/news-releases/news-release-details/cytokinetics-announces-negative-results-vitality-als (accessed on 24 July 2020).
- AdisInsight. Reldesemtiv-Astellas Pharma/Cytokinetics-AdisInsight. 2013. Available online: https://adisinsight.springer.com/drugs/800037810 (accessed on 26 July 2020).
- Andrews, J.A.; Miller, T.M.; Vijayakumar, V.; Stoltz, R.; James, J.K.; Meng, L.; Wolff, A.A.; Malik, F.I. CK-2127107 amplifies skeletal muscle response to nerve activation in humans. Muscle Nerve 2018, 57, 729–734. [Google Scholar] [CrossRef]
- CytoKinetics. Cytokinetics Announces Results of FORTITUDE-ALS, a Phase 2 Clinical Trial of Reldesemtiv in Patients With ALS, Presented at American Academy of Neurology Annual Meeting | Cytokinetics, Inc. Available online: http://ir.cytokinetics.com/news-releases/news-release-details/cytokinetics-announces-results-fortitude-als-phase-2-clinical (accessed on 27 July 2020).
- Cheng, A.J.; Hwee, D.T.; Kim, L.H.; Durham, N.; Yang, H.T.; Hinken, A.C.; Kennedy, A.R.; Terjung, R.L.; Jasper, J.R.; Malik, F.I.; et al. Fast skeletal muscle troponin activator CK-2066260 increases fatigue resistance by reducing the energetic cost of muscle contraction. J. Physiol. 2019, 597, 4615–4625. [Google Scholar] [CrossRef] [Green Version]
- Lee, E.J.; De Winter, J.M.; Buck, D.; Jasper, J.R.; Malik, F.I.; Labeit, S.; Ottenheijm, C.A.; Granzier, H. Fast Skeletal Muscle Troponin Activation Increases Force of Mouse Fast Skeletal Muscle and Ameliorates Weakness Due to Nebulin-Deficiency. PLoS ONE 2013, 8, e55861. [Google Scholar] [CrossRef]
- Nogara, L.; Naber, N.; Pate, E.; Canton, M.; Reggiani, C.; Cooke, R. Piperine’s mitigation of obesity and diabetes can be explained by its up-regulation of the metabolic rate of resting muscle. Proc. Natl. Acad. Sci. USA 2016, 113, 13009–13014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cooke, R. The role of the myosin ATPase activity in adaptive thermogenesis by skeletal muscle. Biophys. Rev. 2011, 3, 33–45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tiwari, A.; Mahadik, K.R.; Gabhe, S.Y. Piperine: A comprehensive review of methods of isolation, purification, and biological properties. Med. Drug Discov. 2020, 7, 100027. [Google Scholar] [CrossRef]
- Shefner, J.M.; Wolff, A.A.; Meng, L.; Bian, A.; Lee, J.; Barragan, D.; Andrews, J.A. A randomized, placebo-controlled, double-blind phase IIb trial evaluating the safety and efficacy of tirasemtiv in patients with amyotrophic lateral sclerosis. Amyotroph. Later. Scler. Frontotempo. Degener. 2016, 17, 426–435. [Google Scholar] [CrossRef]
- Cytokinetics. Cytokinetics Announces Top-Line Results From BENEFIT-ALS|Cytokinetics, Inc. 2014. Available online: https://adisinsight.springer.com/trials/700221015 (accessed on 4 July 2020).
- Andrews, J.A.; Cudkowicz, M.E.; Hardiman, O.; Meng, L.; Bian, A.; Lee, J.; Wolff, A.A.; Malik, F.I.; Shefner, J.M. VITALITY-ALS, a phase III trial of tirasemtiv, a selective fast skeletal muscle troponin activator, as a potential treatment for patients with amyotrophic lateral sclerosis: Study design and baseline characteristics. Amyotroph. Later. Scler. Frontotempo. Degener. 2018, 19, 259–266. [Google Scholar] [CrossRef] [Green Version]
- Shefner, J.M.; Cudkowicz, M.E.; Hardiman, O.; Cockroft, B.M.; Lee, J.H.; Malik, F.I.; Meng, L.; Rudnicki, S.A.; Wolff, A.A.; Andrews, J.A.; et al. A phase III trial of Tirasemtiv as a potential treatment for amyotrophic lateral sclerosis. Amyotroph. Later. Scler. Frontotempo. Degener. 2019, 20, 584–594. [Google Scholar] [CrossRef] [Green Version]
- AdisInsight. Tirasemtiv-Cytokinetics–AdisInsight. 2009. Available online: https://adisinsight.springer.com/drugs/800030378 (accessed on 24 July 2020).
- Spudich, J.A. The myosin mesa and a possible unifying hypothesis for the molecular basis of human hypertrophic cardiomyopathy. Biochem. Soc. Trans. 2015, 43, 64–72. [Google Scholar] [CrossRef] [Green Version]
- Spudich, J.A. Three perspectives on the molecular basis of hypercontractility caused by hypertrophic cardiomyopathy mutations. Pflug. Arch. Eur. J. Physiol. 2019, 471, 701–717. [Google Scholar] [CrossRef] [Green Version]
- Buvoli, M.; Hamady, M.; Leinwand, L.A.; Knight, R. Bioinformatics Assessment of β-Myosin Mutations Reveals Myosin’s High Sensitivity to Mutations. Trends Cardiovasc. Med. 2008, 18, 141–149. [Google Scholar] [CrossRef] [Green Version]
- Walsh, R.; Rutland, C.; Thomas, R.; Loughna, S. Cardiomyopathy: A systematic review of disease-causing mutations in myosin heavy chain 7 and their phenotypic manifestations. Cardiology 2009, 115, 49–60. [Google Scholar] [CrossRef]
- Spudich, J.A. Hypertrophic and Dilated Cardiomyopathy: Four Decades of Basic Research on Muscle Lead to Potential Therapeutic Approaches to These Devastating Genetic Diseases. Biophys. J. 2014, 106, 1236–1249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gersh, B.J.; Maron, B.J.; Bonow, R.O.; Dearani, J.A.; Fifer, M.A.; Link, M.S.; Naidu, S.S.; Nishimura, R.A.; Ommen, S.R.; Rakowski, H.; et al. 2011 ACCF/AHA guideline for the diagnosis and treatment of hypertrophic cardiomyopathy: A report of the American College of cardiology foundation/American heart association task force on practice guidelines. Circulation 2011, 124, 783–831. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zamorano, J.L.; Anastasakis, A.; Borger, M.A.; Borggrefe, M.; Cecchi, F.; Charron, P.; Hagege, A.A.; Lafont, A.; Limongelli, G.; Mahrholdt, H.; et al. 2014 ESC guidelines on diagnosis and management of hypertrophic cardiomyopathy: The task force for the diagnosis and management of hypertrophic cardiomyopathy of the European Society of Cardiology (ESC). Eur. Heart J. 2014, 35, 2733–2779. [Google Scholar] [CrossRef]
- Arts, I.C.W.; Hollman, P.C.H.; Feskens, E.J.M.; Bueno de Mesquita, H.B.; Kromhout, D. Catechin intake might explain the inverse relation between tea consumption and ischemic heart disease: The Zutphen Elderly Study. Am. J. Clin. Nutr. 2001, 74, 227–232. [Google Scholar] [CrossRef]
- Kokubo, Y.; Iso, H.; Saito, I.; Yamagishi, K.; Yatsuya, H.; Ishihara, J.; Inoue, M.; Tsugane, S. The impact of green tea and coffee consumption on the reduced risk of stroke incidence in japanese population: The Japan public health center-based study cohort. Stroke 2013, 44, 1369–1374. [Google Scholar] [CrossRef]
- Harbowy, M.E.; Balentine, D.A.; Davies, A.P.; Cai, Y. Tea Chemistry. Crit. Rev. Plant Sci. 1997, 16, 415–480. [Google Scholar] [CrossRef]
- Tadano, N.; Du, C.K.; Yumoto, F.; Morimoto, S.; Ohta, M.; Xie, M.F.; Nagata, K.; Zhan, D.Y.; Lu, Q.W.; Miwa, Y.; et al. Biological actions of green tea catechins on cardiac troponin C. Br. J. Pharmacol. 2010, 161, 1034–1043. [Google Scholar] [CrossRef] [Green Version]
- Robinson, P.J.; Patel, S.; Liu, X.; Zhang, Y.-H.; Khandelwal, A.; Blagg, B.; Casadei, B.; Watkins, H.; Redwood, C. Novel Potential Treatment of Familial Hypertrophic Cardiomyopathy with Analogues of the Green Tea Polyphenol Epigallocatechin-3-Gallate. Biophys. J. 2016, 110, 125A. [Google Scholar] [CrossRef]
- Zeitz, O.; Rahman, A.; Hasenfuss, G.; Janssen, P.M.L. Impact of β-adrenoceptor antagonists on myofilament calcium sensitivity of rabbit and human myocardium. J. Cardiovas. Pharmacol. 2000, 36, 126–131. [Google Scholar] [CrossRef]
- Stücker, S.; Kresin, N.; Carrier, L.; Friedrich, F.W. Nebivolol desensitizes myofilaments of a hypertrophic cardiomyopathy mouse model. Front. Physiol. 2017, 8, 558. [Google Scholar] [CrossRef] [Green Version]
- Wright, P.T.; Tsui, S.F.; Francis, A.J.; MacLeod, K.T.; Marston, S.B. Approaches to High-Throughput Analysis of Cardiomyocyte Contractility. Front. Physiol. 2020, 11, 612. [Google Scholar] [CrossRef] [PubMed]
- Sheehan, A.; Messer, A.E.; Papadaki, M.; Choudhry, A.; Kren, V.; Biedermann, D.; Blagg, B.; Khandelwal, A.; Marston, S.B. Molecular defects in cardiac myofilament Ca2+-regulation due to cardiomyopathy-linked mutations can be reversed by small molecules binding to troponin. Front. Physiol. 2018, 9, 243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baell, J.; Walters, M.A. Chemical con artists foil drug discovery. Nature 2014, 513, 481–483. [Google Scholar] [CrossRef] [PubMed]
- Ingólfsson, H.I.; Thakur, P.; Herold, K.F.; Hobart, E.A.; Ramsey, N.B.; Periole, X.; De Jong, D.H.; Zwama, M.; Yilmaz, D.; Hall, K.; et al. Phytochemicals perturb membranes and promiscuously alter protein function. ACS Chem. Biol. 2014, 9, 1788–1798. [Google Scholar] [CrossRef] [PubMed]
- Memo, M.; Leung, M.C.; Ward, D.G.; Dos Remedios, C.; Morimoto, S.; Zhang, L.; Ravenscroft, G.; McNamara, E.; Nowak, K.J.; Marston, S.B.; et al. Familial dilated cardiomyopathy mutations uncouple troponin I phosphorylation from changes in myofibrillar Ca2+ sensitivity. Cardiovasc. Res. 2013, 99, 65–73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vikhorev, P.G.; Song, W.; Wilkinson, R.; Copeland, O.N.; Messer, A.E.; Ferenczi, M.A.; Marston, S.B. The dilated cardiomyopathy-causing mutation ACTC E361G in cardiac muscle myofibrils specifically abolishes modulation of Ca2+ regulation by phosphorylation of troponin I. Biophys. J. 2014, 107, 2369–2380. [Google Scholar] [CrossRef] [Green Version]
- Wilkinson, R.; Song, W.; Smoktunowicz, N.; Marston, S. A dilated cardiomyopathy mutation blunts adrenergic response and induces contractile dysfunction under chronic angiotensin II stress. Am. J. Physiol. Circ. Physiol. 2015, 309, H1936–H1946. [Google Scholar] [CrossRef] [Green Version]
- Wilson, I.B.; Ginsburg, S. A powerful reactivator of alkylphosphate-inhibited acetylcholinesterase. BBA Biochim. Biophys. Acta 1955, 18, 168–170. [Google Scholar] [CrossRef]
- Cheung, A.; Dantzig, J.A.; Hollingworth, S.; Baylor, S.M.; Goldman, Y.E.; Mitchinson, T.J.; Straight, A.F. A small-molecule inhibitor of skeletal muscle myosin II. Nat. Cell Biol. 2002, 4, 83–88. [Google Scholar] [CrossRef]
- Straight, A.F.; Cheung, A.; Limouze, J.; Chen, I.; Westwood, N.J.; Sellers, J.R.; Mitchison, T.J. Dissecting temporal and spatial control of cytokinesis with a myosin II inhibitor. Science 2003, 299, 1743–1747. [Google Scholar] [CrossRef] [Green Version]
- Abi-Gerges, N.; Pointon, A.; Pullen, G.F.; Morton, M.J.; Oldman, K.L.; Armstrong, D.; Valentin, J.P.; Pollard, C.E. Preservation of cardiomyocytes from the adult heart. J. Mol. Cell Cardiol. 2013, 64, 108–119. [Google Scholar] [CrossRef] [PubMed]
- Rauscher, A.; Gyimesi, M.; Kovács, M.; Málnási-Csizmadia, A. Targeting Myosin by Blebbistatin Derivatives: Optimization and Pharmacological Potential. Trends Biochem. Sci. 2018, 43, 700–713. [Google Scholar] [CrossRef] [PubMed]
- Wilson, C.; Naber, N.; Pate, E.; Cooke, R. The myosin inhibitor blebbistatin stabilizes the super-relaxed state in skeletal muscle. Biophys. J. 2014, 107, 1637–1646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roman, B.I.; Verhasselt, S.; Stevens, C.V. Medicinal Chemistry and Use of Myosin II Inhibitor (S)-Blebbistatin and Its Derivatives. J. Med. Chem. 2018, 61, 9410–9428. [Google Scholar] [CrossRef]
- Spudich, J.A.; Aksel, T.; Bartholomew, S.R.; Nag, S.; Kawana, M.; Yu, E.C.; Sarkar, S.S.; Sung, J.; Sommese, R.F.; Sutton, S.; et al. Effects of hypertrophic and dilated cardiomyopathy mutations on power output by human β-cardiac myosin. J. Exp. Biol. 2016, 219, 161–167. [Google Scholar] [CrossRef] [Green Version]
- Tekakirikul, P.; Eminaga, S.; Toka, O.; Alcalai, R.; Wang, L.; Wakimoto, H.; Nayor, M.; Konno, T.; Gorham, J.M.; Wolf, C.M.; et al. Cardiac fibrosis in mice with hypertrophic cardiomyopathy is mediated by non-myocyte proliferation and requires Tgf-β. J. Clin. Investig. 2010, 120, 3520–3529. [Google Scholar] [CrossRef] [Green Version]
- Green, E.M.; Wakimoto, H.; Anderson, R.L.; Evanchik, M.J.; Gorham, J.M.; Harrison, B.C.; Henze, M.; Kawas, R.; Oslob, J.D.; Rodriguez, H.M.; et al. Heart disease: A small-molecule inhibitor of sarcomere contractility suppresses hypertrophic cardiomyopathy in mice. Science 2016, 351, 617–621. [Google Scholar] [CrossRef] [Green Version]
- Anderson, R.L.; Trivedi, D.V.; Sarkar, S.S.; Henze, M.; Ma, W.; Gong, H.; Rogers, C.S.; Gorham, J.M.; Wong, F.L.; Morck, M.M.; et al. Deciphering the super relaxed state of human β-cardiac myosin and the mode of action of mavacamten from myosin molecules to muscle fibers. Proc. Natl. Acad. Sci. USA 2018, 115, E8143–E8152. [Google Scholar] [CrossRef] [Green Version]
- Rohde, J.A.; Roopnarine, O.; Thomas, D.D.; Muretta, J.M. Mavacamten stabilizes an autoinhibited state of two-headed cardiac myosin. Proc. Natl. Acad. Sci. USA 2018, 115, E7486–E7494. [Google Scholar] [CrossRef] [Green Version]
- Stern, J.A.; Markova, S.; Ueda, Y.; Kim, J.B.; Pascoe, P.J.; Evanchik, M.J.; Green, E.M.; Harris, S.P. A small molecule inhibitor of sarcomere contractility acutely relieves left ventricular outflow tract obstruction in feline hypertrophic cardiomyopathy. PLoS ONE 2016, 11, e0168407–e0168413. [Google Scholar] [CrossRef] [Green Version]
- Del Rio, C.L.; Yukie, U.; Baker, D.C.; Dalton, R.S.; Laurence, L.; Philip, J.; Bari, O.; Joseph, L.; Evanchik, M.J.; Green, E.M. Abstract 20593: In vivo Cardiac Effects of Mavacamten (MYK-461): Evidence for Negative Inotropy and Improved Compliance. Circulation 2017, 136 (Suppl. S1), A20593. [Google Scholar]
- Grillo, M.P.; Erve, J.C.L.L.; Dick, R.; Driscoll, J.P.; Haste, N.; Markova, S.; Brun, P.; Carlson, T.J.; Evanchik, M. In vitro and in vivo pharmacokinetic characterization of mavacamten, a first-in-class small molecule allosteric modulator of beta cardiac myosin. Xenobiotica 2019, 49, 718–733. [Google Scholar] [CrossRef] [PubMed]
- Heitner, S.B.; Jacoby, D.; Lester, S.J.; Owens, A.; Wang, A.; Zhang, D.; Lambing, J.; Lee, J.; Semigran, M.; Sehnert, A.J. Mavacamten treatment for obstructive hypertrophic cardiomyopathy a clinical trial. Ann. Intern. Med. 2019, 170, 741–748. [Google Scholar] [CrossRef] [PubMed]
- Heitner, S.B.; Lester, S.; Wang, A.; Hegde, S.M.; Fang, L.; Balaratnam, G.; Sehnert, A.J.; Jacoby, D. Precision Pharmacological Treatment for Obstructive Hypertrophic Cardiomyopathy With Mavacamten: One-Year Results From PIONEER-OLE. Circulation 2019, 140, A13962. [Google Scholar]
- Ho, C.Y.; Mealiffe, M.E.; Bach, R.G.; Bhattacharya, M.; Choudhury, L.; Edelberg, J.M.; Hegde, S.M.; Jacoby, D.; Lakdawala, N.K.; Lester, S.J.; et al. Evaluation of Mavacamten in Symptomatic Patients With Nonobstructive Hypertrophic Cardiomyopathy. J. Am. Coll. Cardiol. 2020, 75, 2649–2660. [Google Scholar] [CrossRef]
- Olivotto, I.; Oreziak, A.; Barriales-villa, R.; Abraham, T.P.; Masri, A.; Garcia-pavia, P.; Saberi, S.; Lakdawala, N.K.; Hegde, S.M.; Solomon, S.D.; et al. Mavacamten for treatment of symptomatic obstructive hypertrophic cardiomyopathy (EXPLORER-HCM): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2020, 396, 759–769. [Google Scholar] [CrossRef]
- Ho, C.Y.; Olivotto, I.; Jacoby, D.; Lester, S.J.; Roe, M.; Wang, A.; Waldman, C.B.; Zhang, D.; Sehnert, A.J.; Heitner, S.B. Study Design and Rationale of EXPLORER-HCM: Evaluation of Mavacamten in Adults with Symptomatic Obstructive Hypertrophic Cardiomyopathy. Circ. Heart Fail. 2020, 13, 59–67. [Google Scholar] [CrossRef]
- Del Rio, C.L.; Aprajita, Y.; S, F.B.; Christopher, Z.; Trisha, S.; Frank, R.; John, S.; Lee-Jae, G.; Allison, H.; Julie, G.; et al. Abstract 14585: Chronic Treatment With a Mavacamten-Like Myosin-Modulator (MYK-581) Blunts Disease Progression in a Mini-Pig Genetic Model of Non-Obstructed Hypertrophic Cardiomyopathy: In Vivo Evidence for Improved Relaxation and Functional Reserve. Circulation 2019, 140, A14585. [Google Scholar] [CrossRef]
- MyoKardia Programs|MyoKardia. 2020. Available online: https://myokardia.com/programs (accessed on 22 August 2020).
- AdisInsight. A Phase 1 Randomized, Placebo-Controlled, Single and Multiple-Ascending Dose Study of MYK-224 in Healthy Volunteers—AdisInsight. Available online: https://adisinsight.springer.com/trials/700301316 (accessed on 1 September 2020).
- NIH. Study Evaluating the Safety, Tolerability and Preliminary Pharmacokinetics and Pharmacodynamics of MYK-461-Full Text View—ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT02329184 (accessed on 30 August 2020).
- Jacoby, D.; Lester, S.; Owens, A.; Wang, A.; Young, D.; Tripuraneni, R.; Semigran, M.; Heitner, S. Reduction in left ventricular outflow tract gradient with mavacamten (myk-461) in symptomatic obstructive hypertrophic cardiomyopathy patients (PIONEER-HCM). J. Am. Coll. Cardiol. 2018, 71, A644. [Google Scholar] [CrossRef]
- Heitner, S.B.; Jacoby, D.; Lester, S.; Owens, A.T.; Wang, A.; Shah, A.; Hegde, S.; Fang, L.; Sehnert, A.J.; Semigran, M. Mavacamten Improves Left Ventricular Relaxation and Compliance in Obstructive Hypertrophic Cardiomyopathy Through Direct Myosin Modulation. Circulation 2018, 138 (Suppl. S1), A17141. [Google Scholar] [CrossRef]
- Ho, C.Y. Safety and Efficacy of Mavacamten in Symptomatic Non-Obstructive Hypertrophic Cardiomyopathy: The MAVERICK-HCM Study. In Proceedings of the American College of Cardiology (ACC)/World Congress of Cardiology’s (WCC) Virtual Scientific Sessions, Boston, MA, USA, 15 June 2020; pp. 2–4. Available online: https://www.emjreviews.com/cardiology/symposium/safety-and-efficacy-of-mavacamten-in-patients-with-symptomatic-non-obstructive-hypertrophic-cardiomyopathy-the-maverick-hcm-study/ (accessed on 30 August 2020).
- Heitner, S.; Wang, A.; Jacoby, D.; Lester, S.; Carlson, T.; Zhang, D.; Sehnert, A.; Ho, C. Maverick-HCM: Phase 2 randomized, multi-center, double-blind, placebo-controlled concentration-guided study to evaluate mavacamten (MYK-461) in adults with symptomatic non-obstructive hypertrophic cardiomyopathy. Circulation 2018, 138, A17067. [Google Scholar] [CrossRef]
- NIH. A Long-Term Safety Extension Study of Mavacamten in Adults Who Have Completed MAVERICK-HCM or EXPLORER-HCM—Full Text View—ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT03723655 (accessed on 1 September 2020).
- NIH. A Study to Evaluate Mavacamten in Adults With Symptomatic Obstructive HCM Who Are Eligible for Septal Reduction Therapy—Full Text View—ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT04349072 (accessed on 1 September 2020).
- NIH. A Single and Multiple Ascending Dose Study of CK-3773274 in Healthy Adult Subjects—Full Text View—ClinicalTrials.gov. Available online: https://www.clinicaltrials.gov/ct2/show/NCT03767855?term=ck-3773274 (accessed on 31 August 2020).
- CytoKinetics. CytoKinetics Announces Data from Phase 1 Study of CK- 3773274 at the HFSA 23rd Annual Scientific Meeting. Available online: http://ir.cytokinetics.com/news-releases/news-release-details/cytokinetics-announces-data-phase-1-study-ck-3773274-hfsa-23r (accessed on 31 August 2020).
- Robertson, L.A.; Armas, D.R.; Robbie, E.; Osmukhina, A.; Li, H.; Malik, F.I.; Solomon, S.D. A First in Human Study of the Selective Cardiac Myosin Inhibitor, CK-3773274. J. Card. Fail. 2019, 25, S79–S80. [Google Scholar] [CrossRef] [Green Version]
- NIH. REDWOOD-HCM: Randomized Evaluation of Dosing with CK-3773274 in Obstructive Outflow Disease in HCM—Full Text View—ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT04219826 (accessed on 31 August 2020).
- Hartman, J.J.; Hwee, D.T.; Wang, J.; Wu, Y.; Schaletzky, J.; Paliwal, P.; Lee, K.; Taheri, K.D.; Wehri, E.; Ewing, T.J.; et al. Characterization of the Cardiac Myosin Inhibitor CK-3773274: A Potential Therapeutic Approach for Hypertrophic Cardiomyopathy. Biophys. J. 2020, 118, 596a. [Google Scholar] [CrossRef]
- Hwee, D.T.; Hartman, J.J.; Wang, J.; Wu, Y.; Schaletzky, J.; Paliwal, P.; Lee, K.; Taheri, K.D.; Wehri, E.; Chuang, C. Pharmacologic Characterization of the Cardiac Myosin Inhibitor, CK-3773274: A Potential Therapeutic Approach for Hypertrophic Cardiomyopathy. Circ. Res. 2019, 125 (Suppl. S1), A332. [Google Scholar] [CrossRef]
- Hwee, D.T.; Wu, Y.; Cremin, P.; Morgan, B.P.; Malik, F.I.; Chin, E.R. The Cardiac Myosin Inhibitor, CK-3773274, Reduces Contractility in the R403q Mouse Model of Hypertrophic Cardiomyopathy. Circ. Res. 2019, 125 (Suppl. S1), A615. [Google Scholar] [CrossRef]
- AdisInsight. CK 274—AdisInsight. 2020. Available online: https://adisinsight.springer.com/drugs/800053181 (accessed on 31 August 2020).
- Myokardia. MyoKardia Announces Receipt of Breakthrough Therapy Designation from FDA for Mavacamten for the Treatment of Symptomatic, Obstructive Hypertrophic Cardiomyopathy Nasdaq:MYOK. Available online: https://www.globenewswire.com/news-release/2020/07/23/2066908/0/en/MyoKardia-Announces-Receipt-of-Breakthrough-Therapy-Designation-from-FDA-for-Mavacamten-for-the-Treatment-of-Symptomatic-Obstructive-Hypertrophic-Cardiomyopathy.html (accessed on 29 August 2020).
- Ashrafian, H.; Frenneaux, M.P.; Opie, L.H. Metabolic mechanisms in heart failure. Circulation 2007, 116, 434–448. [Google Scholar] [CrossRef]
- MyoKardia. MyoKardia Announces Positive Topline Data from its Phase 2 MAVERICK-HCM Clinical Trial of Mavacamten|Bristol-Myers Squibb. Available online: http://investors.myokardia.com/news-releases/news-release-details/myokardia-announces-positive-topline-data-its-phase-2-maverick (accessed on 10 December 2020).




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drug Name | Trial and Year(s) | Study Design | (n) Targeted Population | Aim | Key Findings | Ref. |
|---|---|---|---|---|---|---|
| Pimobendan | PICO 1996 | Randomised, double blind, placebo controlled trial | (317) Patients with LVEF ≤ 45% | To determine the effects of pimobendan 2.5 and 5 mg daily on exercise capacity in patients with chronic HF | -Increase exercise tolerance, -Pimobendan increased mortality | [21] |
| Levosimendan | RUSSLAN 2002 | Randomised, double-blind, placebo-controlled study | (504) Patients with LV failure complicating AMI | To evaluate the safety and efficacy of levosimendan in patients with left ventricular failure complicating acute myocardial infarction | -Low-dose levosimendan reduced the risk of worsening HF | [32] |
| LIDO 2002 | Multicentre, randomised, double-blind, double-dummy, parallel-group trial | (203) Patients with ADHF | To evaluate the effects of levosimendan vs. dobutamine on haemodynamic performance and clinical outcome in patients with low-output HF | -Improved haemodynamic performance more effectively than dobutamine -Reduced mortality with levosimendan for up to 180 days | [33] | |
| NCT00048425 REVIVE I and II 2004,2013 | Randomised, multicentre, double blind, 2 sequential trials | (700) Patients with ADHF | To evaluate efficacy of iv levosimendan vs. placebo in the short-term treatment of decompensated chronic heart failure | -Rapid and durable symptomatic relief -Increased risk of adverse cardiovascular events and 14-day mortality | [31] | |
| NCT00348504 SURVIVE 2007 | Randomised, double-blind, multicentre, Parallel-group study | (1327) Patients with ADHF | To assess the effect of a short-term IV infusion of levosimendan or dobutamine on long-term survival | -Initial reduction in BNP -No significant reduction of all-cause mortality at 180 days -No effect on any secondary clinical outcomes | [34] |
| Drug Name | Trial, (Phase) and Year(s) | Study Design | (n) Targeted Population | Dose and (Trial Duration) | Primary Endpoint/or Aim | Key Findings | Ref. |
|---|---|---|---|---|---|---|---|
| Omecamtiv Mecarbil (formerly CK-1827452 OR AMG 423) | NCT01380223 (I) 2005–2006 | Double-blind, randomised, four-way crossover, placebo-controlled, dose-escalation, single-centre study | (34) Healthy males | IV 0.005–1.0 mg/kg/h (6 h) | To determine maximum tolerated dose of OM | -OM increases SET, SEF, SV, FS (all p < 0·0001) -Maximum tolerated dose of OM was 0·5 mg/kg/h | [47] |
| NCT00624442 (II) 2007–2009 | Double-blind, randomised, placebo controlled, dose-escalation, multicentre international study | (45) Patients with stable chronic systolic heart failure | IV Loading 0.125–1.0 mg/kg/h; maintenance 0.0625–0.5 mg/kg/h (4 treatments at least 7 days apart) | To assess safety and tolerability of OM | -OM caused a concentration-dependent increases in SET and SV, also a reduction in HR was reported (p < 0.0001) -Cardiac ischaemia was observed in two patients at high plasma concentrations (» 1750–1350 ng/mL) | [54] | |
| NCT00682565 (II) 2008 | Double-blind, randomised, placebo-controlled, multicentre study | (94) Patients with ischemic cardiomyopathy and angina | IV Loading 24–48 mg/h for 2 h maintenance 6–11 mg/h for 18 h (7 days) | To assess the safety and tolerability of OM during symptom-limited exercise in patients with ischemic cardiomyopathy and angina | -Asymptomatic elevation in troponin and CPK-MB levels | [55] | |
| NCT01300013 (IIb) ATOMIC-AHF 2013–2015 | Double-blind, randomised, placebo-controlled, multicentre Study | (613) Patients with acute systolic heart failure (AHF) | IV Loading 7.5–20 mg/h for 4 h maintenance 1.5–4 mg/h for 44 h (48 h) | Dyspnea relief in patients assessed after 6, 24 and 48 h (using the 7-point Likert scale) | -No improvement in primary endpoint or secondary outcomes -Similar rates of adverse events between treatment and placebo groups -OM increased SET and decreased LVESD | [48] | |
| NCT01786512 (IIb) COSMIC-HF 2011–2015 | Double-blind, randomised, placebo-controlled, multicentre, dose-escalation study | (448) Patients with systemic chronic heart failure with LVEF ≤ 40% | Oral 25 mg twice daily or PK-guided titration to 50 mg twice Daily (20 weeks) | To assess safety, tolerability and pharmacokinetics of OM in 20 weeks of treatment | - OM increased SET and SV - OM reduced HR and NT-proBNP - Comparable adverse events between the groups | [56,57] | |
| NCT02929329 (III) GALACTIC-HF 2020 | Double-blind, randomised, placebo-controlled, multicentre international study | (8256) Patients with symptomatic chronic HF with EF ≤ 35% | Oral 25 mg twice daily or PK-guided titration to 50 mg twice Daily in addition to standard HF therapy (21.8 months) | Time to the next cardiovascular death or first HF event whichever occurred first | -Primary-outcome event occurred in 37% of the OM group and in 39.1% of the placebo group (95% CI 0.86 to 0.99; p = 0.03) -10% reduction in the median NT-proBNP level in OM group than placebo group at week 24 compared to Baseline; the median cardiac troponin I level was 4 ng/L higher than baseline. | [50,58,59] | |
| NCT03759392 (III) METEORIC-HF 2021 | Double-blind, randomised, placebo-controlled, multicentre study | (270) Patients with chronic HFrEF | Oral 25 mg twice daily Or PK-guided titration to 50 mg twice Daily (20 weeks) | Change in pVO2 on cardiopulmonary exercise testing from baseline to Week 20 | Ongoing phase III trial | [60] | |
| Danicamtiv (MYK-491) | NCT03062956 (I) 2017 | Randomised, placebo-controlled study of single ascending oral doses | (67) Healthy volunteers | Oral Range 3–550 mg (5 days) | To investigate safety, tolerability, pharmacokinetics and pharmacodynamics of MYK-491 | -Dose and concentration dependent increased contractility -Modest increase in SET and SV -The drug was generally well-tolerated in the range of 3 to 550 mg | [52] |
| NCT03447990 (IIa) 2018–2019 | Randomised, double-blind, placebo-controlled, two-part adaptive design study | (40) Patients with HFrEF | 175–550 mg or placebo (9 days then follow-up for a week) | To further investigate safety, PK/PD and tolerability of MYK-491 | -50 mg BID achieved steady state concentrations at 2000 to 3500 ng/mL -Dose-dependent increase in LVSV, SET -No reports of cardiac ischemia | [53] |
| Drug Name | Trial and Year(s) | Study Design | (n) Targeted Population | Aim | Key Findings | Ref. |
|---|---|---|---|---|---|---|
| Tirasemtiv (formerly known as CK-2017357) | NCT01709 149 BENEFIT-ALS 2012–2014 | Multi-national, double-blind, randomised, placebo-controlled study | (596) Patients with ALS | To evaluate the safety and effectiveness of CK-2017357 when taken with or without riluzole in patients with ALS | -Primary endpoint was not met. -Mixed results were observed for the secondary endpoints. | [77,78] |
| NCT02496767 VITALITY-ALS | Multi-national, double-blind, randomised, placebo-controlled, parallel-group study | (744) Patients with ALS | To confirm and extend results from a large phase IIb trial and maximize tolerability with a slower dose escalation | -Primary and secondary endpoints did not show significant differences. -Dizziness, fatigue, nausea, weight loss, and insomnia occurred more frequently on Tirasemtiv. -Tirasemtiv was poorly tolerated. | [79,80] | |
| NCT02936635 VIGOR-ALS 2016–2018 | Open-label extension study | (280) Patients who Completed VITALITY-ALS | To assess the long-term safety and tolerability of Tirasemtiv in patients with ALS | -No available data | [81] | |
| Reldesemtiv (CK-2127107) | NCT03065959 2017 | Randomised, double-blinded study | (42) Elderly patients with muscle fatigue | To evaluate the effect of Reldesemtiv in elder patients with muscle fatigue | -Trial terminated | [69] |
| NCT03160898 FORTITUDE-ALS 2018–2019 | Double-blind, randomised, dose-ranging, placebo-controlled parallel group study | (458) Patients with ALS | To evaluate effect of CK-2127107 vs. placebo on respiratory function and other measures of skeletal muscle function in patients with ALS | -No statistical significance In the primary endpoint of change from baseline in SVC after 12 weeks of treatment -All Reldesemtiv groups had declined SVC and ALSFRS-R less than patients on placebo | [71] |
| Drug Name | Trial, (Phase) and Year(s) | Study Design | (n) Targeted Population | Dose and (Trial Duration) | Primary Endpoint/or Aim | Key Findings | Ref. |
|---|---|---|---|---|---|---|---|
| Mavacamten (formerly known as MYK-461) | NCT02329184 (I) 2014–2016 | Open-label, First-in-human study | (15) Patients with HCM | No available data (28 days) | To assess safety, tolerability, preliminary pharmacokinetics and pharmacodynamics of single ascending oral doses | -No available data | [126] |
| NCT02842242 PIONEER-HCM (II) 2016–2017 | Open-label, Nonrandomised, Pilot Study | (21) Patients with HOCM with resting LVOT gradients of ≥30 or ≥50 mm Hg of provoked gradient | Cohort A Mava 10–20 mg/day w/o background medications Cohort B Mava 2–5 mg/day with b-blockers allowed (12 weeks) | Change in post-exercise peak LVOT gradient from baseline to week 12 | -In cohort A, Mava reduced mean post-exercise LVOT gradient from 103 to 19 mmHg at week 12 (p = 0.008), reduced resting LVEF and increased Peak VO2 -In cohort B, Mava decreased post-exercise LVOT gradient from 86 to 64 mm Hg (p = 0.020), 6% mean change in resting LVEF and elevated peak VO2 -Most serious AEs are reduced LVEF at higher plasma concentrations and atrial fibrillation | [118,127,128] | |
| NCT03496168 PIONEER-OLE (II) 2018–2020 | Open-label extension study | (13) Patients with HOCM from PIONEER-HCM | After 6-18 months of PIONEER-HCM, Mava was administered in doses of 5,10 or 15 mg (48 weeks) | Frequency and severity of adverse events and serious adverse events | -Interventricular septal thickness was reduced without changes in posterior wall thickness -AEs were mostly mild and transient in nature, no serious adverse events were reported -Mava reduced resting and post-exercise LVOT | [119] | |
| NCT 03442764 MAVERICK-HCM (II) 2018–2020 | Randomised, double-blind, exploratory, placebo-controlled, multicentre, dose-ranging study | (59) Patients with nHCM | Initial dose 5 mg 1 dose titration at week 6 (2.5, 5, 10 or 15 mg) (16 weeks followed by 8 weeks washout) | To assess the safety and tolerability of Mava in patients with systemic nHCM | -SAEs occurred in 10% of participants on Mava and in 21% participants on placebo, indicating significant no difference. -Reversible reduction in LVEF ≤ 45% -NT-proBNP decreased by 53% in the pooled Mava group versus 1% in the placebo group (p = 0.0005), 34% reduction in cardiac troponin I in Mava group (p = 0.009) | [120,129,130] | |
| NCT03470545 EXPLORER-HCM (III) 2018–2020 | Multicentre, randomised, double-blind, placebo-controlled parallel-group study | (250) Patients with HOCM | Starting dose 5 mg (30 weeks) | 1.5 mL/kg per min or greater increase in pVO2 and at least one NYHA class reduction OR 3 mL/kg per min or greater pVO2 increase without NYHA class worsening | -37% of patients on Mava vs. 17% on placebo met the composite primary endpoint (p = 0·0005) -A post-exercise LVOT gradient 50 mmHg was achieved in 74% of patients in Mava group and increased pVO2 -complete ablation of all LVOT was achieved in 57% | [121,122] | |
| NCT03723655 MAVA-LTE (III) 2018–2025 | Randomised, long-term safety extension study | (310) Patients who completed MAVERICK-HCM or EXPLORER-HCM | No available data (252 weeks) | Frequency and severity of treatment-emergent adverse events and serious AEs | Ongoing phase III trial | [131] | |
|
NCT04349072 VALOR-HCM (III) 2020–2024 | Randomised, double-blind, placebo-controlled study | (100) Patients with HOCM who are eligible for septal reduction therapy | No available data (32 weeks) | Septal Reduction Therapy (SRT) Status | Ongoing phase III trial | [132] | |
| CK-274 | NCT03767855 (I) 2018–2020 | Double-Blind, randomised, placebo-controlled, multi-part, single and multiple ascending dose study | (115) healthy volunteers | No available data (Up to 29 days) | To assess safety, PK and PD of CK-274 | - CK-274 was safe and well tolerated in healthy participants. -No serious AEs and clinically meaningful changes in vital signs, ECGs or laboratory tests were observed -Dose-dependent reduction in LVEF | [133,134,135] |
| NCT04219826 REDWOOD-HCM (II) 2020–2021 | Multicentre, randomised, double-blind, placebo-controlled, dose-finding study | Patients with HOCM | Cohort A 5–10 mg [ECG guided] Cohort 3 10–30 mg of oral CK-274 (10 weeks of treatment and 4 weeks of washout) | To determine the safety and tolerability of CK-274 | Ongoing phase II trial | [136] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alsulami, K.; Marston, S. Small Molecules Acting on Myofilaments as Treatments for Heart and Skeletal Muscle Diseases. Int. J. Mol. Sci. 2020, 21, 9599. https://doi.org/10.3390/ijms21249599
Alsulami K, Marston S. Small Molecules Acting on Myofilaments as Treatments for Heart and Skeletal Muscle Diseases. International Journal of Molecular Sciences. 2020; 21(24):9599. https://doi.org/10.3390/ijms21249599
Chicago/Turabian StyleAlsulami, Khulud, and Steven Marston. 2020. "Small Molecules Acting on Myofilaments as Treatments for Heart and Skeletal Muscle Diseases" International Journal of Molecular Sciences 21, no. 24: 9599. https://doi.org/10.3390/ijms21249599
APA StyleAlsulami, K., & Marston, S. (2020). Small Molecules Acting on Myofilaments as Treatments for Heart and Skeletal Muscle Diseases. International Journal of Molecular Sciences, 21(24), 9599. https://doi.org/10.3390/ijms21249599