Junctional Adhesion Molecule-C Mediates the Recruitment of Embryonic-Endothelial Progenitor Cells to the Perivascular Niche during Tumor Angiogenesis

 , ,
, , 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
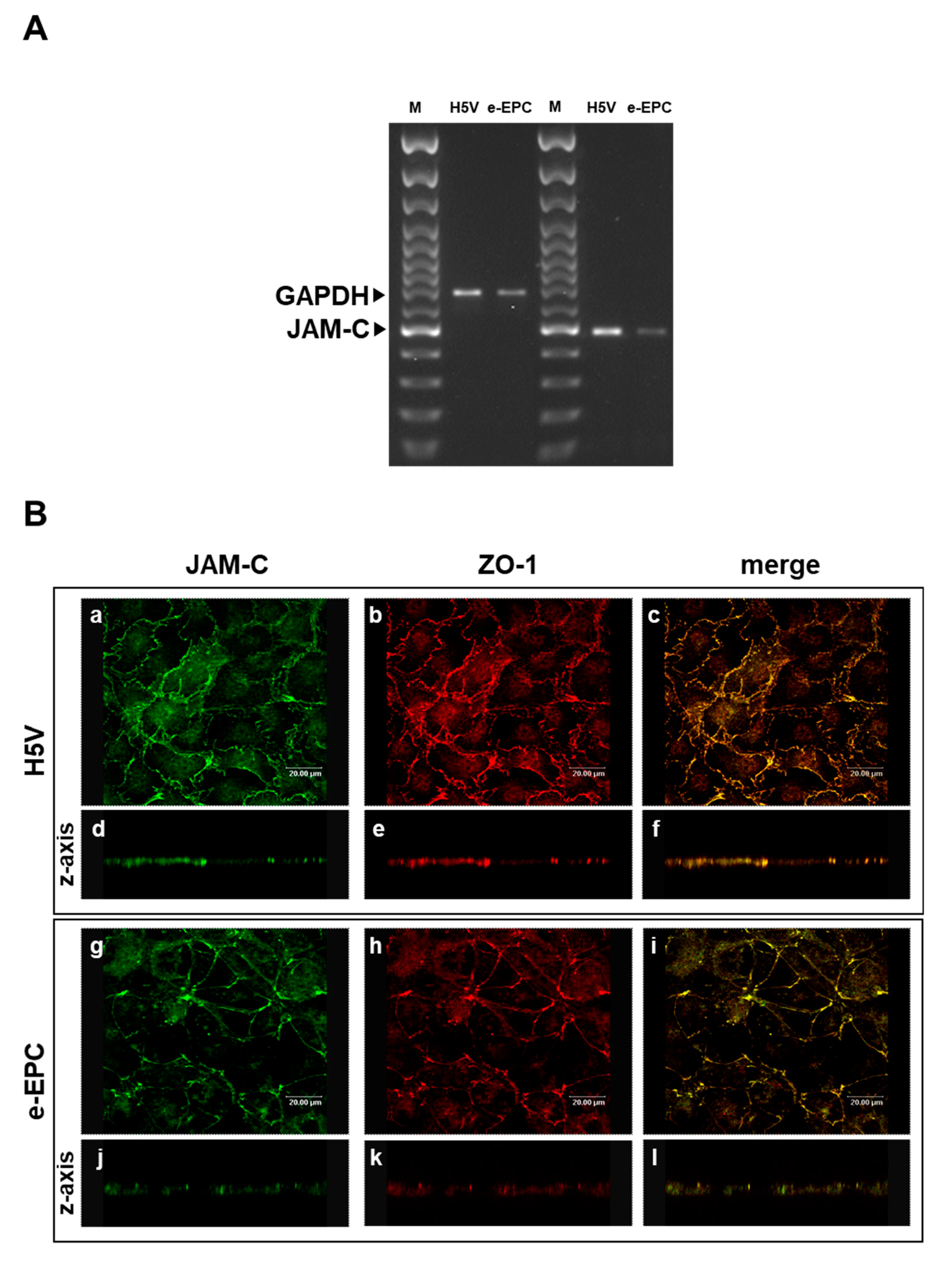
2.1. JAM-C is Expressed by e-EPCs and Localizes at Tight Junctions
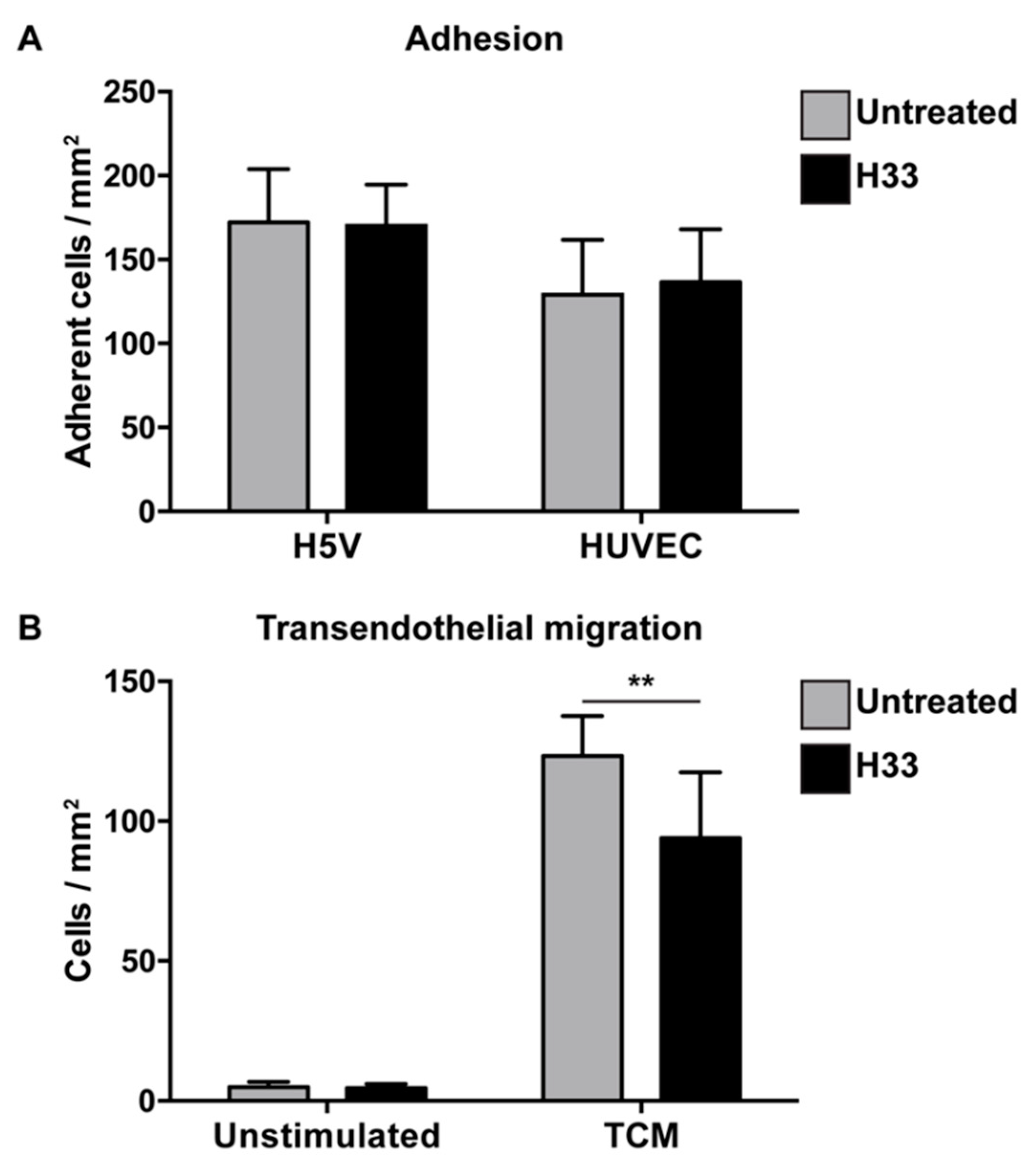
2.2. Role of JAM-C on e-EPC Adhesion and Transendothelial Migration In Vitro
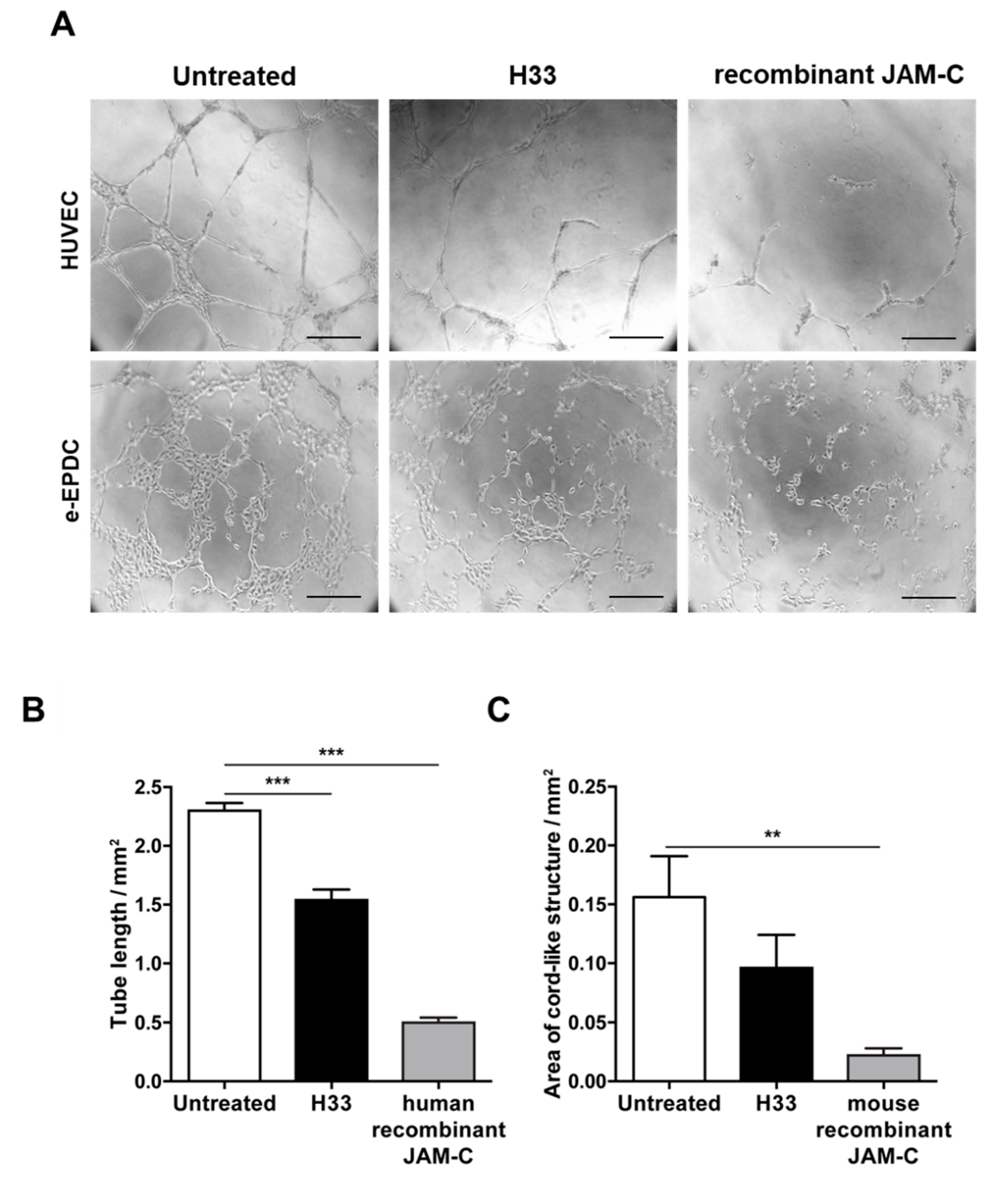
2.3. Inhibition of JAM-C Reduces the Formation of Cord-Like Structures on MatrigelTM In Vitro
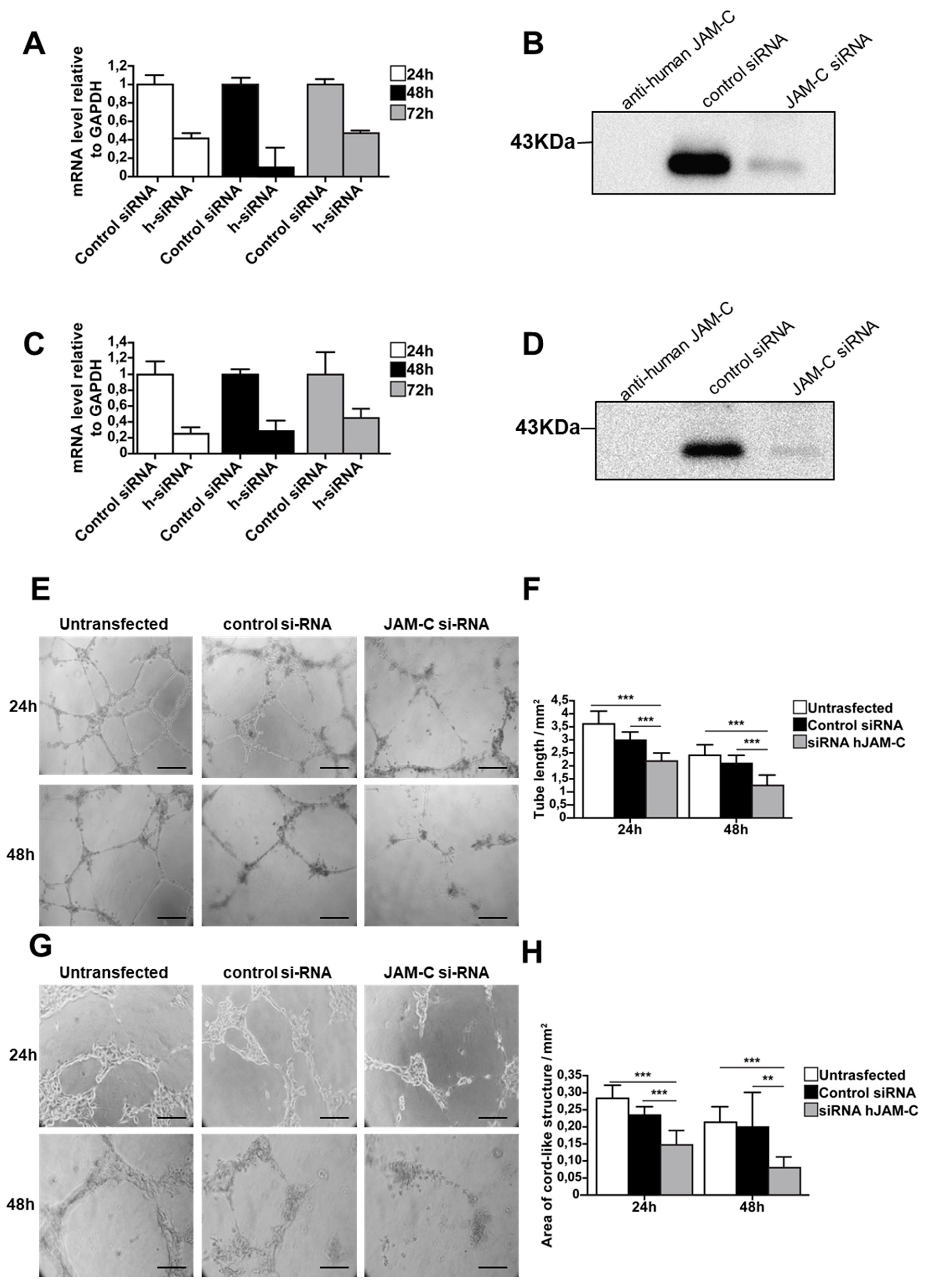
2.4. Knockdown of JAM-C Reduces in Vitro Cord-Like Structures on MatrigelTM
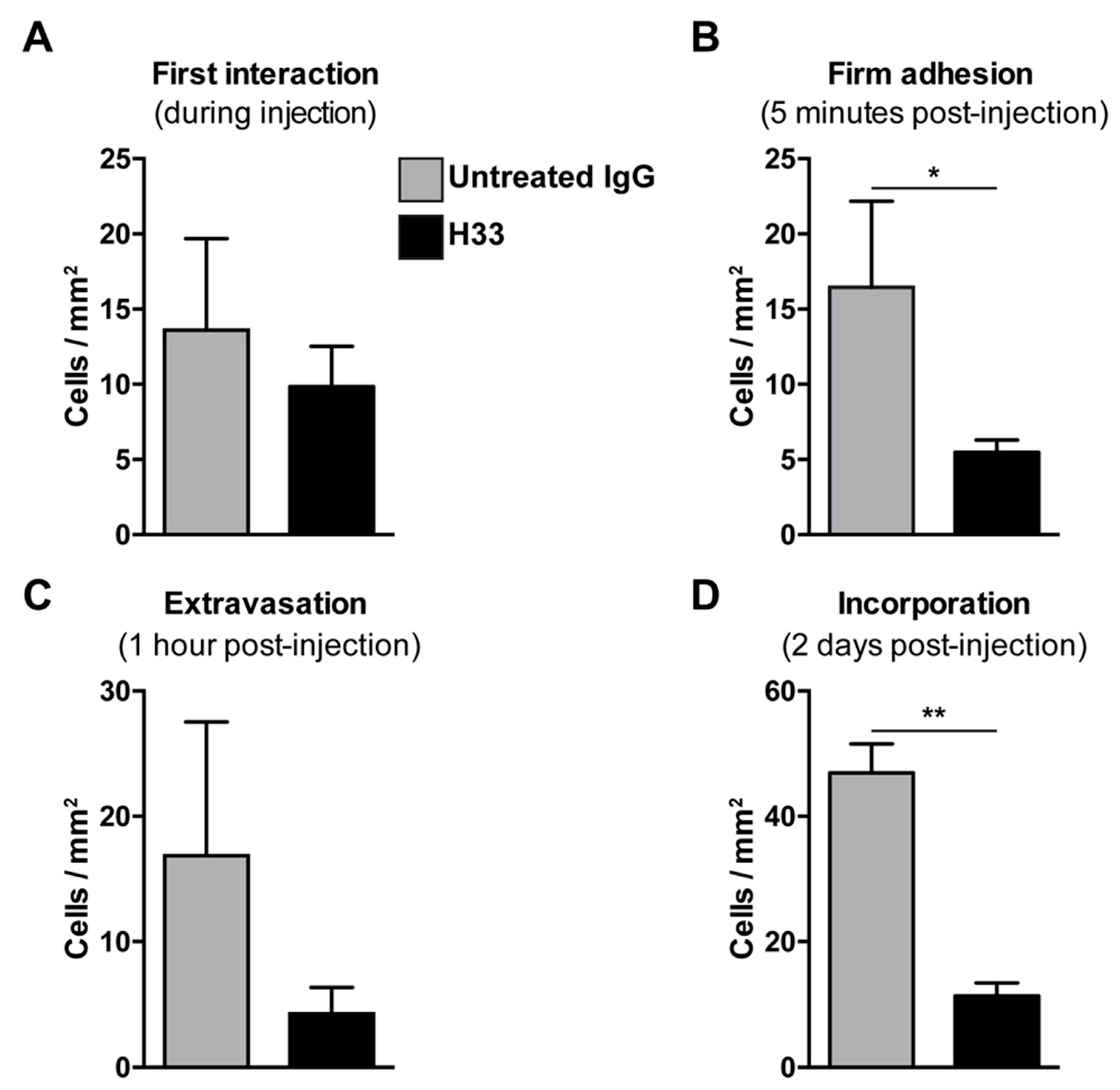
2.5. Role of JAM-C on e-EPC Adhesion and Extravasation In Vivo
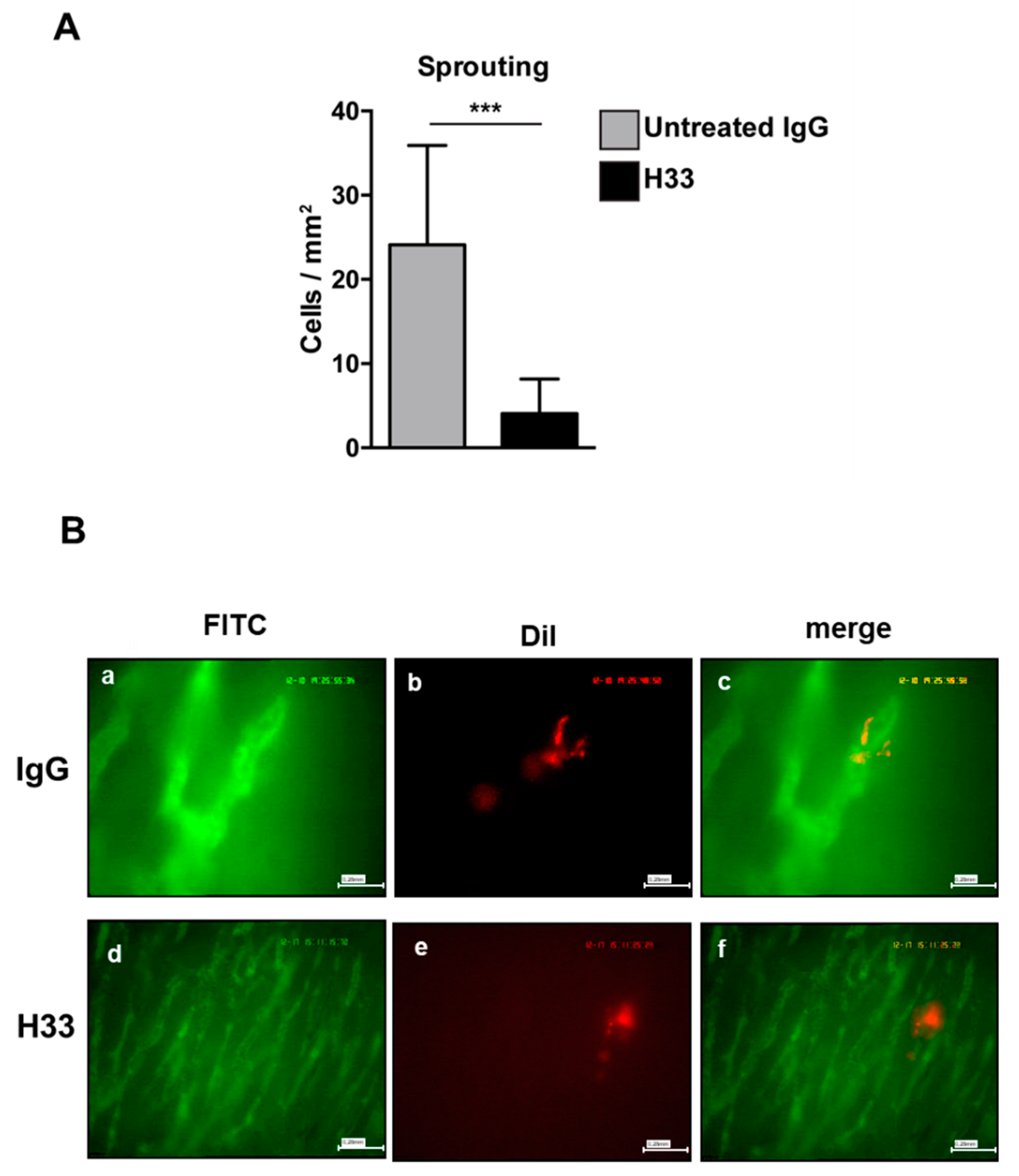
2.6. Anti-JAM-C H33 Monoclonal Antibody Reduces the Recruitment of e-EPCs into the Perivascular Niche and Leads to Loss of e-EPCs in the Tumor Interstitium
3. Discussion
4. Materials and Methods
4.1. Cells and Cell Culture
4.2. Immunofluorescence Staining
4.3. siRNA and Transfection
4.4. RT-PCR and Analysis
4.5. Cell-Surface Biotinylation/JAM-C Immunoprecipitation
4.6. In Vitro Cell–Cell Adhesion Assay
4.7. Invasion Assay
4.8. In Vitro Transendothelial Migration assay
4.9. In Vitro Tube Formation Assay
4.10. Animals
4.11. LLC1 Tumor in the Dorsal Skin-Fold Chamber Model
4.12. Intravital Fluorescence Videomicroscopy
4.13. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| EPCs | Endothelial Progenitor Cells |
| JAM | Junctional Adhesion Molecule |
| e-EPDCs | Embryonic-Endothelial Progenitor-Derived Cells |
| IP | Intraperitoneally |
| IV | Intravenous |
References
- Ding, Y.-T.; Kumar, S.; Yu, D.-C. The role of endothelial progenitor cells in tumour vasculogenesis. Pathobiology 2008, 75, 265–273. [Google Scholar] [CrossRef] [PubMed]
- Roncalli, J.G.; Tongers, J.; Renault, M.-A.; Losordo, D.W. Endothelial progenitor cells in regenerative medicine and cancer: A decade of research. Trends Biotechnol. 2008, 26, 276–283. [Google Scholar] [CrossRef] [PubMed]
- Asahara, T.; Murohara, T.; Sullivan, A.; Silver, M.; van der Zee, R.; Li, T.; Witzenbichler, B.; Schatteman, G.; Isner, J.M. Isolation of putative progenitor endothelial cells for angiogenesis. Science 1997, 275, 964–967. [Google Scholar] [CrossRef]
- Urbich, C.; Heeschen, C.; Aicher, A.; Sasaki, K.-I.; Bruhl, T.; Farhadi, M.R.; Vajkoczy, P.; Hofmann, W.K.; Peters, C.; Pennacchio, L.A.; et al. Cathepsin L is required for endothelial progenitor cell-induced neovascularization. Nat. Med. 2005, 11, 206–213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lyden, D.; Hattori, K.; Dias, S.; Costa, C.; Blaikie, P.; Butros, L.; Chadburn, A.; Heissig, B.; Marks, W.; Witte, L.; et al. Impaired recruitment of bone-marrow-derived endothelial and hematopoietic precursor cells blocks tumor angiogenesis and growth. Nat. Med. 2001, 7, 1194–1201. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Sharpe, E.E.; Maupin, A.B.; Teleron, A.A.; Pyle, A.L.; Carmeliet, P.; Young, P.P. VEGF and PlGF promote adult vasculogenesis by enhancing EPC recruitment and vessel formation at the site of tumor neovascularization. FASEB J. 2006, 20, 1495–1497. [Google Scholar] [CrossRef] [Green Version]
- Kupatt, C.; Horstkotte, J.; Vlastos, G.A.; Pfosser, A.; Lebherz, C.; Semisch, M.; Thalgott, M.; Büttner, K.; Browarzyk, C.; Mages, J.; et al. Embryonic endothelial progenitor cells expressing a broad range of proangiogenic and remodeling factors enhance vascularization and tissue recovery in acute and chronic ischemia. FASEB J. 2005, 19, 1576–1578. [Google Scholar] [CrossRef] [Green Version]
- Vajkoczy, P.; Blum, S.; Lamparter, M.; Mailhammer, R.; Erber, R.; Engelhardt, B.; Vestweber, D.; Hatzopoulos, A.K. Multistep nature of microvascular recruitment of ex vivo-expanded embryonic endothelial progenitor cells during tumor angiogenesis. J. Exp. Med. 2003, 197, 1755–1765. [Google Scholar] [CrossRef] [Green Version]
- Machein, M.R.; Renninger, S.; de Lima-Hahn, E.; Plate, K.H. Minor contribution of bone marrow-derived endothelial progenitors to the vascularization of murine gliomas. Brain Pathol. 2003, 13, 582–597. [Google Scholar] [CrossRef]
- Peters, B.A.; Diaz, L.A.; Polyak, K.; Meszler, L.; Romans, K.; Guinan, E.C.; Antin, J.H.; Myerson, D.; Hamilton, S.R.; Vogelstein, B.; et al. Contribution of bone marrow-derived endothelial cells to human tumor vasculature. Nat. Med. 2005, 11, 261–262. [Google Scholar] [CrossRef]
- Garcia-Barros, M.; Paris, F.; Cordon-Cardo, C.; Lyden, D.; Rafii, S.; Haimovitz-Friedman, A.; Fuks, Z.; Kolesnick, R. Tumor response to radiotherapy regulated by endothelial cell apoptosis. Science 2003, 300, 1155–1159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bieback, K.; Vinci, M.; Elvers-Hornung, S.; Bartol, A.; Gloe, T.; Czabanka, M.; Klüter, H.; Augustin, H.; Vajkoczy, P. Recruitment of human cord blood-derived endothelial colony-forming cells to sites of tumor angiogenesis. Cytotherapy 2013, 15, 726–739. [Google Scholar] [CrossRef] [PubMed]
- Corada, M.; Chimenti, S.; Cera, M.R.; Vinci, M.; Salio, M.; Fiordaliso, F.; De Angelis, N.; Villa, A.; Bossi, M.; Staszewsky, L.I.; et al. Junctional adhesion molecule-A-deficient polymorphonuclear cells show reduced diapedesis in peritonitis and heart ischemia-reperfusion injury. Proc. Natl. Acad. Sci. USA 2005, 102, 10634–10639. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zen, K.; Liu, Y.; McCall, I.C.; Wu, T.; Lee, W.; Babbin, B.A.; Nusrat, A.; Parkos, C.A. Neutrophil migration across tight junctions is mediated by adhesive interactions between epithelial coxsackie and adenovirus receptor and a junctional adhesion molecule-like protein on neutrophils. Mol. Biol. Cell 2005, 16, 2694–2703. [Google Scholar] [CrossRef] [Green Version]
- Wegmann, F.; Petri, B.; Khandoga, A.G.; Moser, C.; Khandoga, A.; Volkery, S.; Li, H.; Nasdala, I.; Brandau, O.; Fässler, R.; et al. ESAM supports neutrophil extravasation, activation of Rho, and VEGF-induced vascular permeability. J. Exp. Med. 2006, 203, 1671–1677. [Google Scholar] [CrossRef] [Green Version]
- Cooke, V.G.; Naik, M.U.; Naik, U.P. Fibroblast growth factor-2 failed to induce angiogenesis in junctional adhesion molecule-A-deficient mice. Arteriosclerosis, Thrombosis, and Vascular Biology 2006, 26, 2005–2011. [Google Scholar] [CrossRef] [Green Version]
- Weber, C.; Fraemohs, L.; Dejana, E. The role of junctional adhesion molecules in vascular inflammation. Nat. Rev. Immunol. 2007, 7, 467–477. [Google Scholar] [CrossRef]
- Garrido-Urbani, S.; Bradfield, P.F.; Lee, B.P.-L.; Imhof, B.A. Vascular and epithelial junctions: A barrier for leucocyte migration. Biochem. Soc. Trans. 2008, 36, 203–211. [Google Scholar] [CrossRef] [Green Version]
- Aurrand-Lions, M.; Duncan, L.; Ballestrem, C.; Imhof, B.A. JAM-2, a novel immunoglobulin superfamily molecule, expressed by endothelial and lymphatic cells. J. Biol. Chem. 2001, 276, 2733–2741. [Google Scholar] [CrossRef] [Green Version]
- Johnson-Léger, C.A.; Aurrand-Lions, M.; Beltraminelli, N.; Fasel, N.; Imhof, B.A. Junctional adhesion molecule-2 (JAM-2) promotes lymphocyte transendothelial migration. Blood 2002, 100, 2479–2486. [Google Scholar] [CrossRef] [Green Version]
- Liang, T.W.; Chiu, H.H.; Gurney, A.; Sidle, A.; Tumas, D.B.; Schow, P.; Foster, J.; Klassen, T.; Dennis, K.; DeMarco, R.A.; et al. Vascular endothelial-junctional adhesion molecule (VE-JAM)/JAM 2 interacts with T, NK, and dendritic cells through JAM 3. J. Immunol. 2002, 168, 1618–1626. [Google Scholar] [CrossRef] [Green Version]
- Santoso, S.; Sachs, U.J.H.; Kroll, H.; Linder, M.; Ruf, A.; Preissner, K.T.; Chavakis, T. The junctional adhesion molecule 3 (JAM-3) on human platelets is a counterreceptor for the leukocyte integrin Mac-1. J. Exp. Med. 2002, 196, 679–691. [Google Scholar] [CrossRef] [PubMed]
- Zen, K.; Babbin, B.A.; Liu, Y.; Whelan, J.B.; Nusrat, A.; Parkos, C.A. JAM-C is a component of desmosomes and a ligand for CD11b/CD18-mediated neutrophil transepithelial migration. Mol. Biol. Cell 2004, 15, 3926–3937. [Google Scholar] [CrossRef] [PubMed]
- Ebnet, K.; Aurrand-Lions, M.; Kuhn, A.; Kiefer, F.; Butz, S.; Zander, K.; Meyer zu Brickwedde, M.-K.; Suzuki, A.; Imhof, B.A.; Vestweber, D. The junctional adhesion molecule (JAM) family members JAM-2 and JAM-3 associate with the cell polarity protein PAR-3: A possible role for JAMs in endothelial cell polarity. J. Cell Sci. 2003, 116, 3879–3891. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bradfield, P.F.; Scheiermann, C.; Nourshargh, S.; Ody, C.; Luscinskas, F.W.; Rainger, G.E.; Nash, G.B.; Miljkovic-Licina, M.; Aurrand-Lions, M.; Imhof, B.A. JAM-C regulates unidirectional monocyte transendothelial migration in inflammation. Blood 2007, 110, 2545–2555. [Google Scholar] [CrossRef] [PubMed]
- Keiper, T.; Al-Fakhri, N.; Chavakis, E.; Athanasopoulos, A.N.; Isermann, B.; Herzog, S.; Saffrich, R.; Hersemeyer, K.; Bohle, R.M.; Haendeler, J.; et al. The role of junctional adhesion molecule-C (JAM-C) in oxidized LDL-mediated leukocyte recruitment. FASEB J. 2005, 19, 2078–2080. [Google Scholar] [CrossRef] [PubMed]
- Aurrand-Lions, M.; Lamagna, C.; Dangerfield, J.P.; Wang, S.; Herrera, P.; Nourshargh, S.; Imhof, B.A. Junctional adhesion molecule-C regulates the early influx of leukocytes into tissues during inflammation. J. Immunol. 2005, 174, 6406–6415. [Google Scholar] [CrossRef] [Green Version]
- Chavakis, T.; Keiper, T.; Matz-Westphal, R.; Hersemeyer, K.; Sachs, U.J.; Nawroth, P.P.; Preissner, K.T.; Santoso, S. The junctional adhesion molecule-C promotes neutrophil transendothelial migration in vitro and in vivo. J. Biol. Chem. 2004, 279, 55602–55608. [Google Scholar] [CrossRef] [Green Version]
- Orlova, V.V.; Economopoulou, M.; Lupu, F.; Santoso, S.; Chavakis, T. Junctional adhesion molecule-C regulates vascular endothelial permeability by modulating VE-cadherin-mediated cell-cell contacts. J. Exp. Med. 2006, 203, 2703–2714. [Google Scholar] [CrossRef] [Green Version]
- Lamagna, C.; Hodivala-Dilke, K.M.; Imhof, B.A.; Aurrand-Lions, M. Antibody against junctional adhesion molecule-C inhibits angiogenesis and tumor growth. Cancer Res. 2005, 65, 5703–5710. [Google Scholar] [CrossRef] [Green Version]
- Rabquer, B.J.; Amin, M.A.; Teegala, N.; Shaheen, M.K.; Tsou, P.-S.; Ruth, J.H.; Lesch, C.A.; Imhof, B.A.; Koch, A.E. Junctional adhesion molecule-C is a soluble mediator of angiogenesis. J. Immunol. 2010, 185, 1777–1785. [Google Scholar] [CrossRef] [PubMed]
- Hatzopoulos, A.K.; Folkman, J.; Vasile, E.; Eiselen, G.K.; Rosenberg, R.D. Isolation and characterization of endothelial progenitor cells from mouse embryos. Development 1998, 125, 1457–1468. [Google Scholar] [PubMed]
- Arcangeli, M.-L.; Bardin, F.; Frontera, V.; Bidaut, G.; Obrados, E.; Adams, R.H.; Chabannon, C.; Aurrand-Lions, M. Function of Jam-B/Jam-C interaction in homing and mobilization of human and mouse hematopoietic stem and progenitor cells. Stem Cells 2014, 32, 1043–1054. [Google Scholar] [CrossRef]
- Santoso, S.; Orlova, V.V.; Song, K.; Sachs, U.J.; Andrei-Selmer, C.L.; Chavakis, T. The homophilic binding of junctional adhesion molecule-C mediates tumor cell-endothelial cell interactions. J. Biol. Chem. 2005, 280, 36326–36333. [Google Scholar] [CrossRef] [Green Version]
- Hecht, N.; Schneider, U.C.; Czabanka, M.; Vinci, M.; Hatzopoulos, A.K.; Vajkoczy, P.; Woitzik, J. Endothelial progenitor cells augment collateralization and hemodynamic rescue in a model of chronic cerebral ischemia. J. Cereb. Blood Flow Metab. 2014, 34, 1297–1305. [Google Scholar] [CrossRef] [PubMed]
- Garrido-Urbani, S.; Vonlaufen, A.; Stalin, J.; De Grandis, M.; Ropraz, P.; Jemelin, S.; Bardin, F.; Scheib, H.; Aurrand-Lions, M.; Imhof, B.A. Junctional adhesion molecule C (JAM-C) dimerization aids cancer cell migration and metastasis. Biochim. Biophys. Acta Mol. Cell Res. 2018, 1865, 638–649. [Google Scholar] [CrossRef]
- Aurrand-Lions, M.; Johnson-Leger, C.; Wong, C.; Pasquier, D.L.; Imhof, B.A. Heterogeneity of endothelial junctions is reflected by differential expression and specific subcellular localization of the three JAM family members. Blood 2001, 98, 3699–3707. [Google Scholar] [CrossRef]
- Satohisa, S.; Chiba, H.; Osanai, M.; Ohno, S.; Kojima, T.; Saito, T.; Sawada, N. Behavior of tight-junction, adherens-junction and cell polarity proteins during HNF-4alpha-induced epithelial polarization. Exp. Cell Res. 2005, 310, 66–78. [Google Scholar] [CrossRef]
- Gliki, G.; Ebnet, K.; Aurrand-Lions, M.; Imhof, B.A.; Adams, R.H. Spermatid differentiation requires the assembly of a cell polarity complex downstream of junctional adhesion molecule-C. Nature 2004, 431, 320–324. [Google Scholar] [CrossRef]
- Sacharidou, A.; Koh, W.; Stratman, A.N.; Mayo, A.M.; Fisher, K.E.; Davis, G.E. Endothelial lumen signaling complexes control 3D matrix-specific tubulogenesis through interdependent Cdc42- and MT1-MMP-mediated events. Blood 2010, 115, 5259–5269. [Google Scholar] [CrossRef] [Green Version]
- Stellos, K.; Langer, H.; Gnerlich, S.; Panagiota, V.; Paul, A.; Schönberger, T.; Ninci, E.; Menzel, D.; Mueller, I.; Bigalke, B.; et al. Junctional adhesion molecule A expressed on human CD34+ cells promotes adhesion on vascular wall and differentiation into endothelial progenitor cells. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 1127–1136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Palma, M.; Venneri, M.A.; Roca, C.; Naldini, L. Targeting exogenous genes to tumor angiogenesis by transplantation of genetically modified hematopoietic stem cells. Nat. Med. 2003, 9, 789–795. [Google Scholar] [CrossRef] [PubMed]
- Grunewald, M.; Avraham, I.; Dor, Y.; Bachar-Lustig, E.; Itin, A.; Jung, S.; Yung, S.; Chimenti, S.; Landsman, L.; Abramovitch, R.; et al. VEGF-induced adult neovascularization: Recruitment, retention, and role of accessory cells. Cell 2006, 124, 175–189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zentilin, L.; Tafuro, S.; Zacchigna, S.; Arsic, N.; Pattarini, L.; Sinigaglia, M.; Giacca, M. Bone marrow mononuclear cells are recruited to the sites of VEGF-induced neovascularization but are not incorporated into the newly formed vessels. Blood 2006, 107, 3546–3554. [Google Scholar] [CrossRef]
- Scheiermann, C.; Meda, P.; Aurrand-Lions, M.; Madani, R.; Yiangou, Y.; Coffey, P.; Salt, T.E.; Ducrest-Gay, D.; Caille, D.; Howell, O.; et al. Expression and function of junctional adhesion molecule-C in myelinated peripheral nerves. Science 2007, 318, 1472–1475. [Google Scholar] [CrossRef] [Green Version]
- Mandicourt, G.; Iden, S.; Ebnet, K.; Aurrand-Lions, M.; Imhof, B.A. JAM-C regulates tight junctions and integrin-mediated cell adhesion and migration. J. Biol. Chem. 2007, 282, 1830–1837. [Google Scholar] [CrossRef] [Green Version]
- Chan, B.M.; Elices, M.J.; Murphy, E.; Hemler, M.E. Adhesion to vascular cell adhesion molecule 1 and fibronectin. Comparison of alpha 4 beta 1 (VLA-4) and alpha 4 beta 7 on the human B cell line JY. J. Biol. Chem. 1992, 267, 8366–8370. [Google Scholar]
- Lehr, H.A.; Leunig, M.; Menger, M.D.; Nolte, D.; Messmer, K. Dorsal skinfold chamber technique for intravital microscopy in nude mice. Am. J. Pathol. 1993, 143, 1055–1062. [Google Scholar]
- Vajkoczy, P.; Ullrich, A.; Menger, M.D. Intravital fluorescence videomicroscopy to study tumor angiogenesis and microcirculation. Neoplasia 2000, 2, 53–61. [Google Scholar] [CrossRef] [Green Version]
- Vajkoczy, P.; Schilling, L.; Ullrich, A.; Schmiedek, P.; Menger, M.D. Characterization of angiogenesis and microcirculation of high-grade glioma: An intravital multifluorescence microscopic approach in the athymic nude mouse. J. Cereb. Blood Flow Metab. 1998, 18, 510–520. [Google Scholar] [CrossRef] [Green Version]
- Chavakis, E.; Hain, A.; Vinci, M.; Carmona, G.; Bianchi, M.E.; Vajkoczy, P.; Zeiher, A.M.; Chavakis, T.; Dimmeler, S. High-mobility group box 1 activates integrin-dependent homing of endothelial progenitor cells. Circ. Res. 2007, 100, 204–212. [Google Scholar] [CrossRef] [PubMed] [Green Version]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Czabanka, M.; Petrilli, L.L.; Elvers-Hornung, S.; Bieback, K.; Albert Imhof, B.; Vajkoczy, P.; Vinci, M. Junctional Adhesion Molecule-C Mediates the Recruitment of Embryonic-Endothelial Progenitor Cells to the Perivascular Niche during Tumor Angiogenesis. Int. J. Mol. Sci. 2020, 21, 1209. https://doi.org/10.3390/ijms21041209
Czabanka M, Petrilli LL, Elvers-Hornung S, Bieback K, Albert Imhof B, Vajkoczy P, Vinci M. Junctional Adhesion Molecule-C Mediates the Recruitment of Embryonic-Endothelial Progenitor Cells to the Perivascular Niche during Tumor Angiogenesis. International Journal of Molecular Sciences. 2020; 21(4):1209. https://doi.org/10.3390/ijms21041209
Chicago/Turabian StyleCzabanka, Marcus, Lucia Lisa Petrilli, Susanne Elvers-Hornung, Karen Bieback, Beat Albert Imhof, Peter Vajkoczy, and Maria Vinci. 2020. "Junctional Adhesion Molecule-C Mediates the Recruitment of Embryonic-Endothelial Progenitor Cells to the Perivascular Niche during Tumor Angiogenesis" International Journal of Molecular Sciences 21, no. 4: 1209. https://doi.org/10.3390/ijms21041209
APA StyleCzabanka, M., Petrilli, L. L., Elvers-Hornung, S., Bieback, K., Albert Imhof, B., Vajkoczy, P., & Vinci, M. (2020). Junctional Adhesion Molecule-C Mediates the Recruitment of Embryonic-Endothelial Progenitor Cells to the Perivascular Niche during Tumor Angiogenesis. International Journal of Molecular Sciences, 21(4), 1209. https://doi.org/10.3390/ijms21041209