The Redox-Sensitive Na/K-ATPase Signaling in Uremic Cardiomyopathy

 , and
, and
Abstract
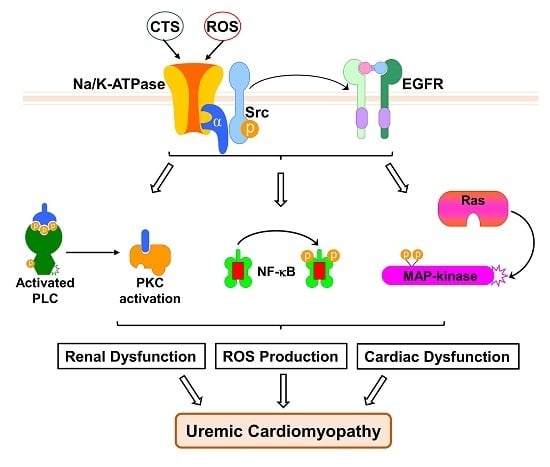
:
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Oxidative Stress and Oxidative Modification in Regulation of Na/K-ATPase Activity
3. The Na/K-ATPase Signaling Function is Redox-Sensitive
4. The Redox-Sensitive Na/K-ATPase Signaling and Na/K-ATPase Signaling-Mediated Oxidant Amplification Loop
5. The Na/K-ATPase Signaling and Oxidative Stress in Uremic Cardiomyopathy
6. Implications and the Possible Role of (p)Naktide
Funding
Conflicts of Interest
Abbreviations
| CKD | chronic kidney disease |
| CTS | cardiotonic steroids |
| IP3Rs | inositol 1,4,5-trisphosphate receptors |
| MBG | marinobufagenin |
| NCX | Na+/Ca2+ exchanger |
| PLC-γ | phospholipase C–γ |
| PNx | partial (5/6th) nephrectomy |
| RNS | reactive nitrogen species |
| ROS | reactive oxygen species |
References
- Schoner, W. Endogenous cardiac glycosides, a new class of steroid hormones. Eur. J. Biochem. 2002, 269, 2440–2448. [Google Scholar] [CrossRef] [PubMed]
- Aperia, A. New roles for an old enzyme: Na,K-ATPase emerges as an interesting drug target. J. Intern. Med. 2007, 261, 44–52. [Google Scholar] [CrossRef] [PubMed]
- Bagrov, A.Y.; Shapiro, J.I. Endogenous digitalis: Pathophysiologic roles and therapeutic applications. Nat. Clin. Pract. Nephrol. 2008, 4, 378–392. [Google Scholar] [CrossRef] [PubMed]
- Bagrov, A.Y.; Shapiro, J.I.; Fedorova, O.V. Endogenous cardiotonic steroids: Physiology, pharmacology, and novel therapeutic targets. Pharmacol. Rev. 2009, 61, 9–38. [Google Scholar] [CrossRef] [PubMed]
- Ellis, D.Z.; Rabe, J.; Sweadner, K.J. Global loss of Na,K-ATPase and its nitric oxide-mediated regulation in a transgenic mouse model of amyotrophic lateral sclerosis. J. Neurosci. 2003, 23, 43–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, M.S.; Akera, T. O2 free radicals: Cause of ischemia-reperfusion injury to cardiac Na+-K+-ATPase. Am. J. Physiol. Heart Circ. Physiol. 1987, 252, H252–H257. [Google Scholar] [CrossRef]
- Xie, Z.J.; Wang, Y.H.; Askari, A.; Huang, W.H.; Klaunig, J.E. Studies on the specificity of the effects of oxygen metabolites on cardiac sodium pump. J. Mol. Cell. Cardiol. 1990, 22, 911–920. [Google Scholar] [CrossRef]
- Huang, W.H.; Wang, Y.; Askari, A. (Na+ + K+)-ATPase: Inactivation and degradation induced by oxygen radicals. Int. J. Biochem. 1992, 24, 621–626. [Google Scholar]
- Huang, W.-H.; Wang, Y.; Askari, A.; Zolotarjova, N.; Ganjeizadeh, M. Different sensitivities of the Na+/K+-ATPase isoforms to oxidants. Biochim. Biophys. Acta Biomembr. 1994, 1190, 108–114. [Google Scholar] [CrossRef]
- Zolotarjova, N.; Ho, C.; Mellgren, R.L.; Askari, A.; Huang, W.H. Different sensitivities of native and oxidized forms of Na+/K(+)-ATPase to intracellular proteinases. Biochim. Biophys. Acta 1994, 1192, 125–131. [Google Scholar] [CrossRef]
- Xie, Z.; Jack-Hays, M.; Wang, Y.; Periyasamy, S.M.; Blanco, G.; Huang, W.H.; Askari, A. Different oxidant sensitivities of the alpha 1 and alpha 2 isoforms of Na+/K(+)-ATPase expressed in baculovirus-infected insect cells. Biochem. Biophys. Res. Commun. 1995, 207, 155–159. [Google Scholar] [CrossRef] [PubMed]
- Mense, M.; Stark, G.; Apell, H.J. Effects of free radicals on partial reactions of the Na,K-ATPase. J. Membr. Biol. 1997, 156, 63–71. [Google Scholar] [CrossRef] [PubMed]
- Thévenod, F.; Friedmann, J.M. Cadmium-mediated oxidative stress in kidney proximal tubule cells induces degradation of Na+/K+-ATPase through proteasomal and endo-/lysosomal proteolytic pathways. FASEB J. 1999, 13, 1751–1761. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- White, C.N.; Figtree, G.A.; Liu, C.-C.; Garcia, A.; Hamilton, E.J.; Chia KK, M.; Rasmussen, H.H. Angiotensin II inhibits the Na+-K+ pump via PKC-dependent activation of NADPH oxidase. Am. J. Physiol. Cell Physiol. 2009, 296, C693–C700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Figtree, G.A.; Liu, C.-C.; Bibert, S.; Hamilton, E.J.; Garcia, A.; White, C.N.; Chia KK, M.; Cornelius, F.; Geering, K.; Rasmussen, H.H. Reversible Oxidative Modification: A Key Mechanism of Na+-K+ Pump Regulation. Circ. Res. 2009, 105, 185–193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bibert, S.; Liu, C.-C.; Figtree, G.A.; Garcia, A.; Hamilton, E.J.; Marassi, F.M.; Sweadner, K.J.; Cornelius, F.; Geering, K.; Rasmussen, H.H. FXYD Proteins Reverse Inhibition of the Na+-K+ Pump Mediated by Glutathionylation of Its β1 Subunit. J. Biol. Chem. 2011, 286, 18562–18572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bogdanova, A.; Petrushanko, I.Y.; Hernansanz-Agustin, P.; Martinez-Ruiz, A. “Oxygen Sensing” by Na,K-ATPase: These Miraculous Thiols. Front. Physiol. 2016, 7, 314. [Google Scholar] [CrossRef]
- Comellas, A.P.; Dada, L.A.; Lecuona, E.; Pesce, L.M.; Chandel, N.S.; Quesada, N.; Budinger, G.S.; Strous, G.J.; Ciechanover, A.; Sznajder, J.I. Hypoxia-Mediated Degradation of Na,K-ATPase via Mitochondrial Reactive Oxygen Species and the Ubiquitin-Conjugating System. Circ. Res. 2006, 98, 1314–1322. [Google Scholar] [CrossRef] [Green Version]
- Dada, L.A.; Chandel, N.S.; Ridge, K.M.; Pedemonte, C.; Bertorello, A.M.; Sznajder, J.I. Hypoxia-induced endocytosis of Na,K-ATPase in alveolar epithelial cells is mediated by mitochondrial reactive oxygen species and PKC-ζ. J. Clin. Investig. 2003, 111, 1057–1064. [Google Scholar] [CrossRef] [Green Version]
- Figtree, G.A.; Keyvan Karimi, G.; Liu, C.C.; Rasmussen, H.H. Oxidative regulation of the Na(+)-K(+) pump in the cardiovascular system. Free Radic. Biol. Med. 2012, 53, 2263–2268. [Google Scholar] [CrossRef]
- Yan, Y.; Shapiro, A.P.; Haller, S.; Katragadda, V.; Liu, L.; Tian, J.; Basrur, V.; Malhotra, D.; Xie, Z.J.; Abraham, N.G.; et al. Involvement of reactive oxygen species in a feed-forward mechanism of Na/K-ATPase-mediated signaling transduction. J. Biol. Chem. 2013, 288, 34249–34258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, Y.; Shapiro, A.P.; Mopidevi, B.R.; Chaudhry, M.A.; Maxwell, K.; Haller, S.T.; Drummond, C.A.; Kennedy, D.J.; Tian, J.; Malhotra, D.; et al. Protein Carbonylation of an Amino Acid Residue of the Na/K-ATPase alpha1 Subunit Determines Na/K-ATPase Signaling and Sodium Transport in Renal Proximal Tubular Cells. J. Am. Heart Assoc. 2016, 5, e003675. [Google Scholar] [CrossRef] [PubMed]
- Reifenberger, M.S.; Arnett, K.L.; Gatto, C.; Milanick, M.A. The reactive nitrogen species peroxynitrite is a potent inhibitor of renal Na-K-ATPase activity. Am. J. Physiol. Renal Physiol. 2008, 295, F1191–F1198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, C.; Imam, S.Z.; Ali, S.F.; Mayeux, P.R. Peroxynitrite and the regulation of Na+,K+-ATPase activity by angiotensin II in the rat proximal tubule. Nitric Oxide 2002, 7, 30–35. [Google Scholar] [CrossRef]
- McKenna, M.J.; Medved, I.; Goodman, C.A.; Brown, M.J.; Bjorksten, A.R.; Murphy, K.T.; Petersen, A.C.; Sostaric, S.; Gong, X. N-acetylcysteine attenuates the decline in muscle Na+,K+-pump activity and delays fatigue during prolonged exercise in humans. J. Physiol. 2006, 576, 279–288. [Google Scholar] [CrossRef]
- Petrushanko, I.Y.; Yakushev, S.; Mitkevich, V.A.; Kamanina, Y.V.; Ziganshin, R.H.; Meng, X.; Anashkina, A.A.; Makhro, A.; Lopina, O.D.; Gassmann, M.; et al. S-glutathionylation of the Na,K-ATPase catalytic alpha subunit is a determinant of the enzyme redox sensitivity. J. Biol. Chem. 2012, 287, 32195–32205. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.-C.; Garcia, A.; Mahmmoud, Y.A.; Hamilton, E.J.; Galougahi, K.K.; Fry, N.A.; Figtree, G.A.; Cornelius, F.; Clarke, R.J.; Rasmussen, H.H. Susceptibility of β1 Na+-K+ Pump Subunit to Glutathionylation and Oxidative Inhibition Depends on Conformational State of Pump. J. Biol. Chem. 2012, 287, 12353–12364. [Google Scholar] [CrossRef] [Green Version]
- Petrushanko, I.; Bogdanov, N.; Bulygina, E.; Grenacher, B.; Leinsoo, T.; Boldyrev, A.; Gassmann, M.; Bogdanova, A. Na-K-ATPase in rat cerebellar granule cells is redox sensitive. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2006, 290, R916–R925. [Google Scholar] [CrossRef] [Green Version]
- Boldyrev, A.; Kurella, E. Mechanism of oxidative damage of dog kidney Na/K-ATPase. Biochem. Biophys. Res. Commun. 1996, 222, 483–487. [Google Scholar] [CrossRef]
- Dobrota, D.; Matejovicova, M.; Kurella, E.G.; Boldyrev, A.A. Na/K-ATPase under oxidative stress: Molecular mechanisms of injury. Cell. Mol. Neurobiol. 1999, 19, 141–149. [Google Scholar] [CrossRef]
- Kurella, E.G.; Tyulina, O.V.; Boldyrev, A.A. Oxidative resistance of Na/K-ATPase. Cell. Mol. Neurobiol. 1999, 19, 133–140. [Google Scholar] [CrossRef]
- Bogdanova, A.; Petrushanko, I.; Boldyrev, A.; Gassmann, M. Oxygen- and Redox-Induced Regulation of the Na/K ATPase. Curr. Enzyme Inhib. 2006, 2, 37–59. [Google Scholar] [CrossRef]
- Soares-da-Silva, P.; Silva, E. Renal Redox Balance and Na+,K+-ATPase Regulation: Role in Physiology and Pathophysiology; Faculty of Medicine: Porto, Portugal, 2012. [Google Scholar]
- Liu, L.; Li, J.; Liu, J.; Yuan, Z.; Pierre, S.V.; Qu, W.; Zhao, X.; Xie, Z. Involvement of Na+/K+-ATPase in hydrogen peroxide-induced hypertrophy in cardiac myocytes. Free Radic. Biol. Med. 2006, 41, 1548–1556. [Google Scholar] [CrossRef]
- Goldshleger, R.; Karlish, S.J. Fe-catalyzed cleavage of the alpha subunit of Na/K-ATPase: Evidence for conformation-sensitive interactions between cytoplasmic domains. Proc. Natl. Acad. Sci. USA 1997, 94, 9596–9601. [Google Scholar] [CrossRef] [Green Version]
- Barry, W.H.; Hasin, Y.; Smith, T.W. Sodium pump inhibition, enhanced calcium influx via sodium-calcium exchange, and positive inotropic response in cultured heart cells. Circ. Res. 1985, 56, 231–241. [Google Scholar] [CrossRef] [Green Version]
- Reuter, H.; Pott, C.; Goldhaber, J.I.; Henderson, S.A.; Philipson, K.D.; Schwinger, R.H. Na(+)--Ca2+ exchange in the regulation of cardiac excitation-contraction coupling. Cardiovasc. Res. 2005, 67, 198–207. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Tian, J.; Haas, M.; Shapiro, J.I.; Askari, A.; Xie, Z. Ouabain interaction with cardiac Na+/K+-ATPase initiates signal cascades independent of changes in intracellular Na+ and Ca2+ concentrations. J. Biol. Chem. 2000, 275, 27838–27844. [Google Scholar]
- Tian, J.; Gong, X.; Xie, Z. Signal-transducing function of Na+-K+-ATPase is essential for ouabain’s effect on [Ca2+]i in rat cardiac myocytes. Am. J. Physiol. Heart Circ. Physiol. 2001, 281, H1899–H1907. [Google Scholar] [CrossRef] [Green Version]
- Tian, J.; Liu, J.; Garlid, K.D.; Shapiro, J.I.; Xie, Z. Involvement of mitogen-activated protein kinases and reactive oxygen species in the inotropic action of ouabain on cardiac myocytes. A potential role for mitochondrial K(ATP) channels. Mol. Cell Biochem. 2003, 242, 181–187. [Google Scholar] [CrossRef]
- Schoner, W.; Scheiner-Bobis, G. Endogenous and exogenous cardiac glycosides: Their roles in hypertension, salt metabolism, and cell growth. Am. J. Physiol. Cell Physiol. 2007, 293, C509–C536. [Google Scholar] [CrossRef]
- Schoner, W.; Scheiner-Bobis, G. Role of endogenous cardiotonic steroids in sodium homeostasis. Nephrol. Dial. Transplant. 2008, 23, 2723–2729. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aizman, O.; Uhlen, P.; Lal, M.; Brismar, H.; Aperia, A. Ouabain, a steroid hormone that signals with slow calcium oscillations. Proc. Natl. Acad. Sci. USA 2001, 98, 13420–13424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyakawa-Naito, A.; Uhlen, P.; Lal, M.; Aizman, O.; Mikoshiba, K.; Brismar, H.; Zelenin, S.; Aperia, A. Cell signaling microdomain with Na,K-ATPase and inositol 1,4,5-trisphosphate receptor generates calcium oscillations. J. Biol. Chem. 2003, 278, 50355–50361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, Z.; Cai, T.; Tian, J.; Ivanov, A.V.; Giovannucci, D.R.; Xie, Z. Na/K-ATPase tethers phospholipase C and IP3 receptor into a calcium-regulatory complex. Mol. Biol. Cell 2005, 16, 4034–4045. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Cai, T.; Yang, C.; Turner, D.A.; Giovannucci, D.R.; Xie, Z. Regulation of Inositol 1,4,5-Trisphosphate Receptor-mediated Calcium Release by the Na/K-ATPase in Cultured Renal Epithelial Cells. J. Biol. Chem. 2008, 283, 1128–1136. [Google Scholar] [CrossRef] [Green Version]
- Haas, M.; Askari, A.; Xie, Z. Involvement of Src and epidermal growth factor receptor in the signal-transducing function of Na+/K+-ATPase. J. Biol. Chem. 2000, 275, 27832–27837. [Google Scholar] [CrossRef] [Green Version]
- Aydemir-Koksoy, A.; Abramowitz, J.; Allen, J.C. Ouabain-induced signaling and vascular smooth muscle cell proliferation. J. Biol. Chem. 2001, 276, 46605–46611. [Google Scholar] [CrossRef] [Green Version]
- Haas, M.; Wang, H.; Tian, J.; Xie, Z. Src-mediated inter-receptor cross-talk between the Na+/K+-ATPase and the epidermal growth factor receptor relays the signal from ouabain to mitogen-activated protein kinases. J. Biol. Chem. 2002, 277, 18694–18702. [Google Scholar] [CrossRef] [Green Version]
- Xie, Z.; Kometiani, P.; Liu, J.; Li, J.; Shapiro, J.I.; Askari, A. Intracellular reactive oxygen species mediate the linkage of Na+/K+-ATPase to hypertrophy and its marker genes in cardiac myocytes. J. Biol. Chem. 1999, 274, 19323–19328. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Ye, Q.; Liu, C.; Xie, J.X.; Yan, Y.; Lai, F.; Duan, Q.; Li, X.; Tian, J.; Xie, Z. Involvement of Na/K-ATPase in hydrogen peroxide-induced activation of the Src/ERK pathway in LLC-PK1 cells. Free Radic. Biol. Med. 2014, 71, 415–426. [Google Scholar] [CrossRef]
- Kajikawa, M.; Fujimoto, S.; Tsuura, Y.; Mukai, E.; Takeda, T.; Hamamoto, Y.; Takehiro, M.; Fujita, J.; Yamada, Y.; Seino, Y. Ouabain Suppresses Glucose-Induced Mitochondrial ATP Production and Insulin Release by Generating Reactive Oxygen Species in Pancreatic Islets. Diabetes 2002, 51, 2522–2529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Y.T.; Chueh, S.C.; Teng, C.M.; Guh, J.H. Investigation of ouabain-induced anticancer effect in human androgen-independent prostate cancer PC-3 cells. Biochem. Pharmacol. 2004, 67, 727–733. [Google Scholar] [CrossRef] [PubMed]
- Boldyrev, A.; Bulygina, E.; Yuneva, M.; Schoner, W. Na/K-ATPase regulates intracellular ROS level in cerebellum neurons. Ann. N. Y. Acad. Sci. 2003, 986, 519–521. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, D.J.; Vetteth, S.; Periyasamy, S.M.; Kanj, M.; Fedorova, L.; Khouri, S.; Kahaleh, M.B.; Xie, Z.; Malhotra, D.; Kolodkin, N.I.; et al. Central role for the cardiotonic steroid marinobufagenin in the pathogenesis of experimental uremic cardiomyopathy. Hypertension 2006, 47, 488–495. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, D.J.; Vetteth, S.; Xie, M.; Periyasamy, S.M.; Xie, Z.; Han, C.; Basrur, V.; Mutgi, K.; Fedorov, V.; Malhotra, D.; et al. Ouabain decreases sarco(endo)plasmic reticulum calcium ATPase activity in rat hearts by a process involving protein oxidation. Am. J. Physiol. Heart Circ. Physiol. 2006, 291, H3003–H3011. [Google Scholar] [CrossRef] [PubMed]
- Zhang, A.Y.; Yi, F.; Zhang, G.; Gulbins, E.; Li, P.-L. Lipid Raft Clustering and Redox Signaling Platform Formation in Coronary Arterial Endothelial Cells. Hypertension 2006, 47, 74–80. [Google Scholar] [CrossRef]
- Zuo, L.; Ushio-Fukai, M.; Ikeda, S.; Hilenski, L.; Patrushev, N.; Alexander, R.W. Caveolin-1 Is Essential for Activation of Rac1 and NAD(P)H Oxidase After Angiotensin II Type 1 Receptor Stimulation in Vascular Smooth Muscle Cells: Role in Redox Signaling and Vascular Hypertrophy. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 1824–1830. [Google Scholar] [CrossRef] [Green Version]
- Touyz, R.M. Lipid Rafts Take Center Stage in Endothelial Cell Redox Signaling by Death Receptors. Hypertension 2006, 47, 16–18. [Google Scholar] [CrossRef] [Green Version]
- Han, W.; Li, H.; Villar, V.A.M.; Pascua, A.M.; Dajani, M.I.; Wang, X.; Natarajan, A.; Quinn, M.T.; Felder, R.A.; Jose, P.A.; et al. Lipid Rafts Keep NADPH Oxidase in the Inactive State in Human Renal Proximal Tubule Cells. Hypertension 2008, 51, 481–487. [Google Scholar] [CrossRef] [Green Version]
- Seshiah, P.N.; Weber, D.S.; Rocic, P.; Valppu, L.; Taniyama, Y.; Griendling, K.K. Angiotensin II Stimulation of NAD(P)H Oxidase Activity. Circ. Res. 2002, 91, 406–413. [Google Scholar] [CrossRef] [Green Version]
- Touyz, R.M.; Yao, G.; Schiffrin, E.L. c-Src induces phosphorylation and translocation of p47phox: Role in superoxide generation by angiotensin II in human vascular smooth muscle cells. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 981–987. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Touyz, R.M. Reactive oxygen species, vascular oxidative stress, and redox signaling in hypertension: What is the clinical significance? Hypertension 2004, 44, 248–252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Munzel, T.; Gori, T.; Bruno, R.M.; Taddei, S. Is oxidative stress a therapeutic target in cardiovascular disease? Eur. Heart J. 2010, 31, 2741–2748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, H.-Y.; Caballero, B.; Chang, S.; Alberg, A.J.; Semba, R.D.; Schneyer, C.R.; Wilson, R.F.; Cheng, T.-Y.; Vassy, J.; Prokopowicz, G.; et al. The Efficacy and Safety of Multivitamin and Mineral Supplement Use To Prevent Cancer and Chronic Disease in Adults: A Systematic Review for a National Institutes of Health State-of-the-Science Conference. Ann. Intern. Med. 2006, 145, 372–385. [Google Scholar] [CrossRef] [Green Version]
- Quintas, L.E.; Pierre, S.V.; Liu, L.; Bai, Y.; Liu, X.; Xie, Z.J. Alterations of Na+/K+-ATPase function in caveolin-1 knockout cardiac fibroblasts. J. Mol. Cell. Cardiol. 2010, 49, 525–531. [Google Scholar] [CrossRef] [Green Version]
- Bai, Y.; Wu, J.; Li, D.; Morgan, E.E.; Liu, J.; Zhao, X.; Walsh, A.; Saikumar, J.; Tinkel, J.; Joe, B.; et al. Differential roles of caveolin-1 in ouabain-induced Na+/K+-ATPase cardiac signaling and contractility. Physiol. Genom. 2016, 48, 739–748. [Google Scholar] [CrossRef] [Green Version]
- Liu, L.; Zhao, X.; Pierre, S.V.; Askari, A. Association of PI3K-Akt signaling pathway with digitalis-induced hypertrophy of cardiac myocytes. Am. J. Physiol. Cell Physiol. 2007, 293, C1489–C1497. [Google Scholar] [CrossRef]
- Bai, Y.; Morgan, E.E.; Giovannucci, D.R.; Pierre, S.V.; Philipson, K.D.; Askari, A.; Liu, L. Different roles of the cardiac Na+/Ca2+-exchanger in ouabain-induced inotropy, cell signaling, and hypertrophy. Am. J. Physiol. Heart Circ. Physiol. 2013, 304, H427–H435. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.; Li, D.; Du, L.; Baldawi, M.; Gable, M.E.; Askari, A.; Liu, L. Ouabain prevents pathological cardiac hypertrophy and heart failure through activation of phosphoinositide 3-kinase alpha in mouse. Cell Biosci. 2015, 5, 64. [Google Scholar] [CrossRef] [Green Version]
- Pratt, R.D.; Brickman, C.R.; Cottrill, C.L.; Shapiro, J.I.; Liu, J. The Na/K-ATPase Signaling: From Specific Ligands to General Reactive Oxygen Species. Int. J. Mol. Sci. 2018, 19, 2600. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Kesiry, R.; Periyasamy, S.M.; Malhotra, D.; Xie, Z.; Shapiro, J.I. Ouabain induces endocytosis of plasmalemmal Na/K-ATPase in LLC-PK1 cells by a clathrin-dependent mechanism. Kidney Int. 2004, 66, 227–241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Liang, M.; Liu, L.; Malhotra, D.; Xie, Z.; Shapiro, J.I. Ouabain-induced endocytosis of the plasmalemmal Na/K-ATPase in LLC-PK1 cells requires caveolin-1. Kidney Int. 2005, 67, 1844–1854. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, H.; Wu, L.; Qu, W.; Malhotra, D.; Xie, Z.; Shapiro, J.I.; Liu, J. Regulation of apical NHE3 trafficking by ouabain-induced activation of the basolateral Na+-K+-ATPase receptor complex. Am. J. Physiol. Cell Physiol. 2008, 294, C555–C563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bock, J.S.; Gottlieb, S.S. Cardiorenal syndrome: New perspectives. Circulation 2010, 121, 2592–2600. [Google Scholar] [CrossRef] [Green Version]
- Ronco, C.; Haapio, M.; House, A.A.; Anavekar, N.; Bellomo, R. Cardiorenal syndrome. J. Am. Coll. Cardiol. 2008, 52, 1527–1539. [Google Scholar] [CrossRef] [Green Version]
- House, A.A.; Anand, I.; Bellomo, R.; Cruz, D.; Bobek, I.; Anker, S.D.; Aspromonte, N.; Bagshaw, S.; Berl, T.; Daliento, L.; et al. Definition and classification of Cardio-Renal Syndromes: Workgroup statements from the 7th ADQI Consensus Conference. Nephrol. Dial. Transplant. 2010, 25, 1416–1420. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Shapiro, J.I. Evolving concepts in the pathogenesis of uraemic cardiomyopathy. Nat. Rev. Nephrol. 2019, 15, 159–175. [Google Scholar] [CrossRef]
- Winchester, J.F.; Audia, P.F. Extracorporeal strategies for the removal of middle molecules. Semin. Dial. 2006, 19, 110–114. [Google Scholar] [CrossRef]
- Lekawanvijit, S.; Kompa, A.R.; Manabe, M.; Wang, B.H.; Langham, R.G.; Nishijima, F.; Kelly, D.J.; Krum, H. Chronic kidney disease-induced cardiac fibrosis is ameliorated by reducing circulating levels of a non-dialysable uremic toxin, indoxyl sulfate. PLoS ONE 2012, 7, e41281. [Google Scholar] [CrossRef] [Green Version]
- Mohmand, B.; Malhotra, D.K.; Shapiro, J.I. Uremic cardiomyopathy: Role of circulating digitalis like substances. Front. Biosci. 2005, 10, 2036–2044. [Google Scholar] [CrossRef] [Green Version]
- Kolmakova, E.V.; Haller, S.T.; Kennedy, D.J.; Isachkina, A.N.; Budny, G.V.; Frolova, E.V.; Piecha, G.; Nikitina, E.R.; Malhotra, D.; Fedorova, O.V.; et al. Endogenous cardiotonic steroids in chronic renal failure. Nephrol. Dial. Transplant. 2011, 26, 2912–2919. [Google Scholar] [CrossRef] [Green Version]
- Komiyama, Y.; Dong, X.H.; Nishimura, N.; Masaki, H.; Yoshika, M.; Masuda, M.; Takahashi, H. A novel endogenous digitalis, telocinobufagin, exhibits elevated plasma levels in patients with terminal renal failure. Clin. Biochem. 2005, 38, 36–45. [Google Scholar] [CrossRef] [PubMed]
- Manunta, P.; Stella, P.; Rivera, R.; Ciurlino, D.; Cusi, D.; Ferrandi, M.; Hamlyn, J.M.; Bianchi, G. Left ventricular mass, stroke volume, and ouabain-like factor in essential hypertension. Hypertension 1999, 34, 450–456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kennedy, D.J.; Shrestha, K.; Sheehey, B.; Li, X.S.; Guggilam, A.; Wu, Y.; Finucan, M.; Gabi, A.; Medert, C.M.; Westfall, K.; et al. Elevated Plasma Marinobufagenin, An Endogenous Cardiotonic Steroid, Is Associated With Right Ventricular Dysfunction and Nitrative Stress in Heart Failure. Circ. Heart Fail. 2015, 8, 1068–1076. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bagrov, A.Y.; Fedorova, O.V.; Dmitrieva, R.I.; Howald, W.N.; Hunter, A.P.; Kuznetsova, E.A.; Shpen, V.M. Characterization of a urinary bufodienolide Na+,K+-ATPase inhibitor in patients after acute myocardial infarction. Hypertension 1998, 31, 1097–1103. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Tian, J.; Chaudhry, M.; Maxwell, K.; Yan, Y.; Wang, X.; Shah, P.T.; Khawaja, A.A.; Martin, R.; Robinette, T.J.; et al. Attenuation of Na/K-ATPase Mediated Oxidant Amplification with pNaKtide Ameliorates Experimental Uremic Cardiomyopathy. Sci. Rep. 2016, 6, 34592. [Google Scholar] [CrossRef]
- Drummond, C.A.; Sayed, M.; Evans, K.L.; Shi, H.; Wang, X.; Haller, S.T.; Liu, J.; Cooper, C.J.; Xie, Z.; Shapiro, J.I.; et al. Reduction of Na/K-ATPase affects cardiac remodeling and increases c-kit cell abundance in partial nephrectomized mice. Am. J. Physiol. Heart Circ. Physiol. 2014, 306, H1631–H1643. [Google Scholar] [CrossRef] [Green Version]
- Priyadarshi, S.; Valentine, B.; Han, C.; Fedorova, O.V.; Bagrov, A.Y.; Liu, J.; Periyasamy, S.M.; Kennedy, D.; Malhotra, D.; Xie, Z.; et al. Effect of green tea extract on cardiac hypertrophy following 5/6 nephrectomy in the rat. Kidney Int. 2003, 63, 1785–1790. [Google Scholar] [CrossRef] [Green Version]
- Elkareh, J.; Kennedy, D.J.; Yashaswi, B.; Vetteth, S.; Shidyak, A.; Kim, E.G.; Smaili, S.; Periyasamy, S.M.; Hariri, I.M.; Fedorova, L.; et al. Marinobufagenin stimulates fibroblast collagen production and causes fibrosis in experimental uremic cardiomyopathy. Hypertension 2007, 49, 215–224. [Google Scholar] [CrossRef] [Green Version]
- Periyasamy, S.M.; Chen, J.; Cooney, D.; Carter, P.; Omran, E.; Tian, J.; Priyadarshi, S.; Bagrov, A.; Fedorova, O.; Malhotra, D.; et al. Effects of uremic serum on isolated cardiac myocyte calcium cycling and contractile function. Kidney Int. 2001, 60, 2367–2376. [Google Scholar] [CrossRef] [Green Version]
- Kennedy, D.J.; Elkareh, J.; Shidyak, A.; Shapiro, A.P.; Smaili, S.; Mutgi, K.; Gupta, S.; Tian, J.; Morgan, E.; Khouri, S.; et al. Partial nephrectomy as a model for uremic cardiomyopathy in the mouse. Am. J. Physiol. Renal Physiol. 2008, 294, F450–F454. [Google Scholar] [CrossRef] [PubMed]
- Haller, S.T.; Kennedy, D.J.; Shidyak, A.; Budny, G.V.; Malhotra, D.; Fedorova, O.V.; Shapiro, J.I.; Bagrov, A.Y. Monoclonal antibody against marinobufagenin reverses cardiac fibrosis in rats with chronic renal failure. Am. J. Hypertens. 2012, 25, 690–696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elkareh, J.; Periyasamy, S.M.; Shidyak, A.; Vetteth, S.; Schroeder, J.; Raju, V.; Hariri, I.M.; El-Okdi, N.; Gupta, S.; Fedorova, L.; et al. Marinobufagenin induces increases in procollagen expression in a process involving protein kinase C and Fli-1: Implications for uremic cardiomyopathy. Am. J. Physiol. Renal Physiol. 2009, 296, F1219–F1226. [Google Scholar] [CrossRef] [PubMed]
- Tian, J.; Shidyak, A.; Periyasamy, S.M.; Haller, S.; Taleb, M.; El-Okdi, N.; Elkareh, J.; Gupta, S.; Gohara, S.; Fedorova, O.V.; et al. Spironolactone attenuates experimental uremic cardiomyopathy by antagonizing marinobufagenin. Hypertension 2009, 54, 1313–1320. [Google Scholar] [CrossRef] [Green Version]
- El-Okdi, N.; Smaili, S.; Raju, V.; Shidyak, A.; Gupta, S.; Fedorova, L.; Elkareh, J.; Periyasamy, S.; Shapiro, A.P.; Kahaleh, M.B.; et al. Effects of cardiotonic steroids on dermal collagen synthesis and wound healing. J. Appl. Physiol. 2008, 105, 30–36. [Google Scholar] [CrossRef] [Green Version]
- Fedorova, L.V.; Raju, V.; El-Okdi, N.; Shidyak, A.; Kennedy, D.J.; Vetteth, S.; Giovannucci, D.R.; Bagrov, A.Y.; Fedorova, O.V.; Shapiro, J.I.; et al. The cardiotonic steroid hormone marinobufagenin induces renal fibrosis: Implication of epithelial-to-mesenchymal transition. Am. J. Physiol. Renal Physiol. 2009, 296, F922–F934. [Google Scholar] [CrossRef] [Green Version]
- Haller, S.T.; Yan, Y.; Drummond, C.A.; Xie, J.; Tian, J.; Kennedy, D.J.; Shilova, V.Y.; Xie, Z.; Liu, J.; Cooper, C.J.; et al. Rapamycin Attenuates Cardiac Fibrosis in Experimental Uremic Cardiomyopathy by Reducing Marinobufagenin Levels and Inhibiting Downstream Pro-Fibrotic Signaling. J. Am. Heart Assoc. 2016, 5, e004106. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.; Bai, Y.; Chen, Y.; Wang, Y.; Sottejeau, Y.; Liu, L.; Li, X.; Lingrel, J.B.; Malhotra, D.; Cooper, C.J.; et al. Reduction of Na/K-ATPase Potentiates Marinobufagenin-induced Cardiac Dysfunction and Myocyte Apoptosis. J. Biol. Chem. 2012, 287, 16390–16398. [Google Scholar] [CrossRef] [Green Version]
- Drummond, C.A.; Hill, M.C.; Shi, H.; Fan, X.; Xie, J.X.; Haller, S.T.; Kennedy, D.J.; Liu, J.; Garrett, M.R.; Xie, Z.; et al. Na/K-ATPase signaling regulates collagen synthesis through microRNA-29b-3p in cardiac fibroblasts. Physiol. Genom. 2016, 48, 220–229. [Google Scholar] [CrossRef] [Green Version]
- Drummond, C.A.; Fan, X.; Haller, S.T.; Kennedy, D.J.; Liu, J.; Tian, J. Na/K-ATPase signaling mediates miR-29b-3p regulation and cardiac fibrosis formation in mice with chronic kidney disease. PLoS ONE 2018, 13, e0197688. [Google Scholar] [CrossRef] [Green Version]
- Taylor, D.; Bhandari, S.; Seymour, A.M. Mitochondrial dysfunction in uremic cardiomyopathy. Am. J. Physiol. Renal Physiol. 2015, 308, F579–F587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Small, D.M.; Coombes, J.S.; Bennett, N.; Johnson, D.W.; Gobe, G.C. Oxidative stress, anti-oxidant therapies and chronic kidney disease. Nephrology 2012, 17, 311–321. [Google Scholar] [CrossRef] [PubMed]
- Ferrandi, M.; Molinari, I.; Barassi, P.; Minotti, E.; Bianchi, G.; Ferrari, P. Organ hypertrophic signaling within caveolae membrane subdomains triggered by ouabain and antagonized by PST 2238. J. Biol. Chem. 2004, 279, 33306–33314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, L.; Li, H.; Xie, Z. Ouabain-induced hypertrophy in cultured cardiac myocytes is accompanied by changes in expression of several late response genes. J. Mol. Cell. Cardiol. 1997, 29, 429–437. [Google Scholar] [CrossRef] [PubMed]
- Norgaard, A.; Bagger, J.P.; Bjerregaard, P.; Baandrup, U.; Kjeldsen, K.; Thomsen, P.E. Relation of left ventricular function and Na,K-pump concentration in suspected idiopathic dilated cardiomyopathy. Am. J. Cardiol. 1988, 61, 1312–1315. [Google Scholar] [CrossRef]
- Schwinger, R.H.; Bundgaard, H.; Muller-Ehmsen, J.; Kjeldsen, K. The Na, K-ATPase in the failing human heart. Cardiovasc. Res. 2003, 57, 913–920. [Google Scholar] [CrossRef] [Green Version]
- Kennedy, D.; Omran, E.; Periyasamy, S.M.; Nadoor, J.; Priyadarshi, A.; Willey, J.C.; Malhotra, D.; Xie, Z.; Shapiro, J.I. Effect of chronic renal failure on cardiac contractile function, calcium cycling, and gene expression of proteins important for calcium homeostasis in the rat. J. Am. Soc. Nephrol. 2003, 14, 90–97. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Chaudhry, M.A.; Nie, Y.; Xie, Z.; Shapiro, J.I.; Liu, J. A Mouse 5/6th Nephrectomy Model That Induces Experimental Uremic Cardiomyopathy. JoVE 2017, 129, e55825. [Google Scholar]
- Li, Z.; Cai, T.; Tian, J.; Xie, J.X.; Zhao, X.; Liu, L.; Shapiro, J.I.; Xie, Z. NaKtide, a Na/K-ATPase-derived peptide Src inhibitor, antagonizes ouabain-activated signal transduction in cultured cells. J. Biol. Chem. 2009, 284, 21066–21076. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Xie, Z. The Na/K-ATPase/Src complex and cardiotonic steroid-activated protein kinase cascades. Pflug. Arch. 2009, 457, 635–644. [Google Scholar] [CrossRef]
- Li, Z.; Zhang, Z.; Xie, J.X.; Li, X.; Tian, J.; Cai, T.; Cui, H.; Ding, H.; Shapiro, J.I.; Xie, Z. Na/K-ATPase mimetic pNaKtide peptide inhibits the growth of human cancer cells. J. Biol. Chem. 2011, 286, 32394–32403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pratt, R.D.; Brickman, C.; Nawab, A.; Cottrill, C.; Snoad, B.; Lakhani, H.V.; Jelcick, A.; Henderson, B.; Bhardwaj, N.N.; Sanabria, J.R.; et al. The Adipocyte Na/K-ATPase Oxidant Amplification Loop is the Central Regulator of Western Diet-Induced Obesity and Associated Comorbidities. Sci. Rep. 2019, 9, 7927. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sodhi, K.; Nichols, A.; Mallick, A.; Klug, R.L.; Liu, J.; Wang, X.; Srikanthan, K.; Goguet-Rubio, P.; Nawab, A.; Pratt, R.; et al. The Na/K-ATPase Oxidant Amplification Loop Regulates Aging. Sci. Rep. 2018, 8, 9721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sodhi, K.; Srikanthan, K.; Goguet-Rubio, P.; Nichols, A.; Mallick, A.; Nawab, A.; Martin, R.; Shah, P.T.; Chaudhry, M.; Sigdel, S.; et al. pNaKtide Attenuates Steatohepatitis and Atherosclerosis by Blocking Na/K-ATPase/ROS Amplification in C57Bl6 and ApoE Knockout Mice Fed a Western Diet. Sci. Rep. 2017, 7, 193. [Google Scholar] [CrossRef]
- Bartlett, D.E.; Miller, R.B.; Thiesfeldt, S.; Lakhani, H.V.; Khanal, T.; D Pratt, R.; Cottrill, C.L.; Klug, R.L.; Adkins, N.S.; Bown, P.C.; et al. Uremic Toxins Activates Na/K-ATPase Oxidant Amplification Loop Causing Phenotypic Changes in Adipocytes in In Vitro Models. Int. J. Mol. Sci. 2018, 19, 2685. [Google Scholar] [CrossRef] [Green Version]
- Boldyrev, A.A.; Bulygina, E.R.; Kramarenko, G.G.; Vanin, A.F. Effect of nitroso compounds on Na/K-ATPase. Biochim. Biophys. Acta Bioenerg. 1997, 1321, 243–251. [Google Scholar] [CrossRef] [Green Version]
- Petrushanko, I.Y.; Mitkevich, V.A.; Lakunina, V.A.; Anashkina, A.A.; Spirin, P.V.; Rubtsov, P.M.; Prassolov, V.S.; Bogdanov, N.B.; Hanggi, P.; Fuller, W.; et al. Cysteine residues 244 and 458–459 within the catalytic subunit of Na,K-ATPase control the enzyme’s hydrolytic and signaling function under hypoxic conditions. Redox Biol. 2017, 13, 310–319. [Google Scholar] [CrossRef] [Green Version]


© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, J.; Nie, Y.; Chaudhry, M.; Bai, F.; Chuang, J.; Sodhi, K.; Shapiro, J.I. The Redox-Sensitive Na/K-ATPase Signaling in Uremic Cardiomyopathy. Int. J. Mol. Sci. 2020, 21, 1256. https://doi.org/10.3390/ijms21041256
Liu J, Nie Y, Chaudhry M, Bai F, Chuang J, Sodhi K, Shapiro JI. The Redox-Sensitive Na/K-ATPase Signaling in Uremic Cardiomyopathy. International Journal of Molecular Sciences. 2020; 21(4):1256. https://doi.org/10.3390/ijms21041256
Chicago/Turabian StyleLiu, Jiang, Ying Nie, Muhammad Chaudhry, Fang Bai, Justin Chuang, Komal Sodhi, and Joseph I. Shapiro. 2020. "The Redox-Sensitive Na/K-ATPase Signaling in Uremic Cardiomyopathy" International Journal of Molecular Sciences 21, no. 4: 1256. https://doi.org/10.3390/ijms21041256
APA StyleLiu, J., Nie, Y., Chaudhry, M., Bai, F., Chuang, J., Sodhi, K., & Shapiro, J. I. (2020). The Redox-Sensitive Na/K-ATPase Signaling in Uremic Cardiomyopathy. International Journal of Molecular Sciences, 21(4), 1256. https://doi.org/10.3390/ijms21041256