Regulatory Mechanisms of Mitochondrial Function and Cardiac Aging

{kind=link}
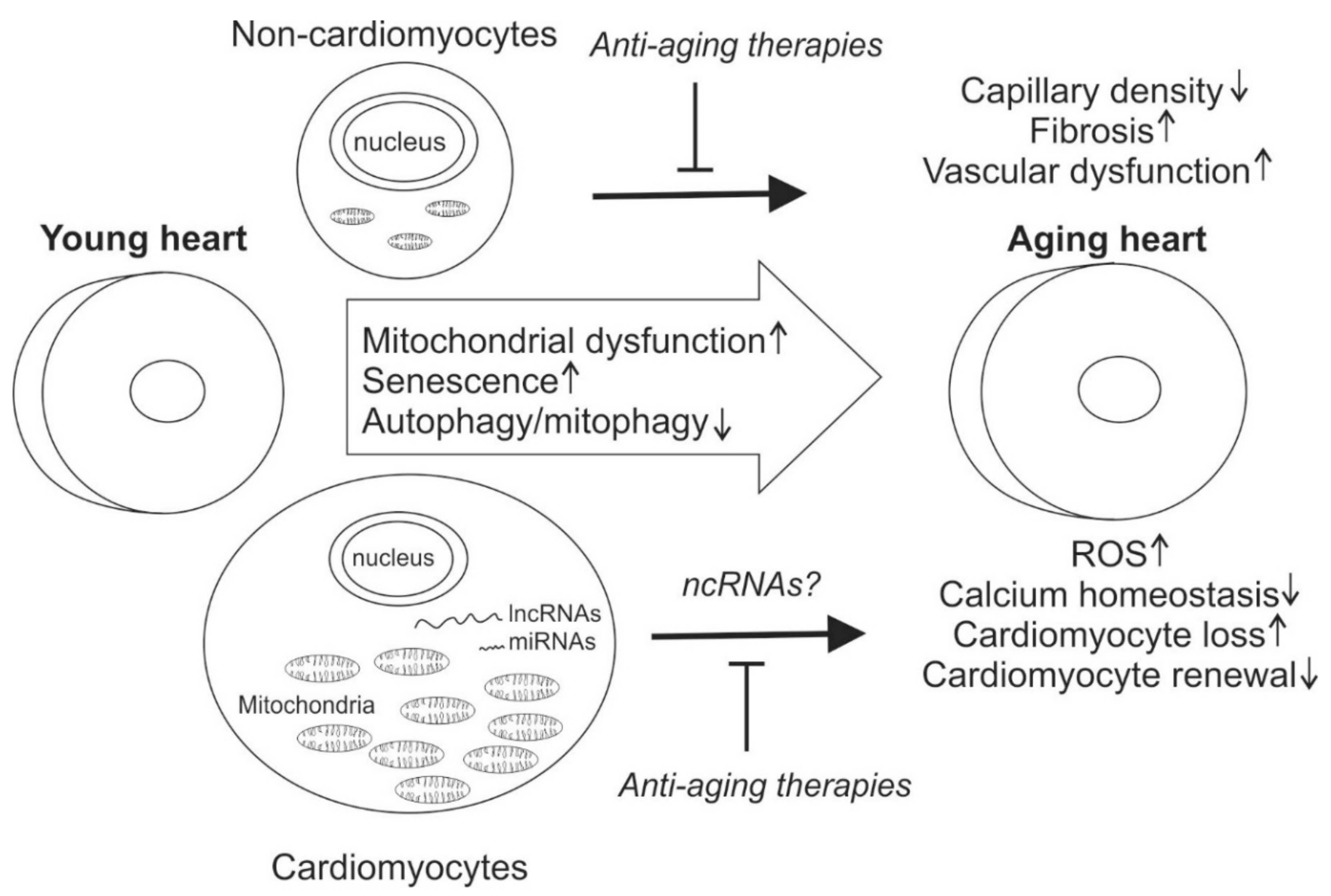
Abstract
:1. Introduction
2. Multi-Steps of Senescence
3. Anti-Aging Therapies
4. Cardiac Senescence
5. Mitochondria in the Aging Heart
6. Mitophagy
6.1. PGC-1α
6.2. Sirtuins
7. Cardiomyocyte Cell Cycle and Reprogramming
8. Concluding Remarks
Author Contributions
Funding
Conflicts of Interest
References
- Gude, N.A.; Broughton, K.M.; Firouzi, F.; Sussman, M.A. Cardiac Ageing: Extrinsic and Intrinsic Factors in Cellular Renewal and Senescence. Nat. Rev. Cardiol. 2018, 15, 523–542. [Google Scholar] [CrossRef] [PubMed]
- Booth, L.N.; Brunet, A. The Aging Epigenome. Mol. Cell 2016, 62, 728–744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sessions, A.O.; Engler, A.J. Mechanical Regulation of Cardiac Aging in Model Systems. Circ. Res. 2016, 118, 1553–1562. [Google Scholar] [CrossRef] [Green Version]
- Paneni, F.; Cañestro, C.D.; Libby, P.; Lüscher, T.F.; Camici, G.G. The Aging Cardiovascular System: Understanding it at the Cellular and Clinical Levels. J. Am. Coll. Cardiol. 2017, 69, 1952–1967. [Google Scholar] [CrossRef]
- Chiao, Y.A.; Rabinovitch, P.S. The Aging Heart. Cold Spring Harb. Perspect. Med. 2015, 5, a025148. [Google Scholar] [CrossRef]
- Lähteenvuo, J.; Rosenzweig, A. Effects of Aging on Angiogenesis. Circ. Res. 2012, 110, 1252–1264. [Google Scholar] [CrossRef] [Green Version]
- Bergmann, O.; Bhardwaj, R.D.; Bernard, S.; Zdunek, S.; Barnabe-Heider, F.; Walsh, S.; Zupicich, J.; Alkass, K.; Buchholz, B.A.; Druid, H.; et al. Evidence for Cardiomyocyte Renewal in Humans. Science 2009, 324, 98–102. [Google Scholar] [CrossRef] [Green Version]
- Lesnefsky, E.J.; Chen, Q.; Hoppel, C.L. Mitochondrial Metabolism in Aging Heart. Circ. Res. 2016, 118, 1593–1611. [Google Scholar] [CrossRef] [Green Version]
- Hernandez-Segura, A.; Nehme, J.; Demaria, M. Hallmarks of Cellular Senescence. Trends Cell Biol. 2018, 28, 436–453. [Google Scholar] [CrossRef]
- Hayflick, L. The Limited in Vitro Lifetime of Human Diploid Cell Strains. Exp. Cell Res. 1965, 37, 614–636. [Google Scholar] [CrossRef]
- Campisi, J.; d’Adda di Fagagna, F. Cellular Senescence: When Bad Things Happen to Good Cells. Nat. Rev. Mol. Cell Biol. 2007, 8, 729–740. [Google Scholar] [CrossRef] [PubMed]
- Childs, B.G.; Durik, M.; Baker, D.J.; van Deursen, J.M. Cellular Senescence in Aging and Age-Related Disease: From Mechanisms to Therapy. Nat. Med. 2015, 21, 1424–1435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prata, L.G.L.; Ovsyannikova, I.G.; Tchkonia, T.; Kirkland, J.L. Senescent Cell Clearance by the Immune System: Emerging Therapeutic Opportunities. Semin. Immunol. 2019, 40, 101275. [Google Scholar] [CrossRef]
- Bergmann, O.; Zdunek, S.; Felker, A.; Salehpour, M.; Alkass, K.; Bernard, S.; Sjostrom, S.L.; Szewczykowska, M.; Jackowska, T.; Dos Remedios, C. Dynamics of Cell Generation and Turnover in the Human Heart. Cell 2015, 161, 1566–1575. [Google Scholar] [CrossRef]
- Dai, D.F.; Santana, L.F.; Vermulst, M.; Tomazela, D.M.; Emond, M.J.; MacCoss, M.J.; Gollahon, K.; Martin, G.M.; Loeb, L.A.; Ladiges, W.C.; et al. Overexpression of Catalase Targeted to Mitochondria Attenuates Murine Cardiac Aging. Circulation 2009, 119, 2789–2797. [Google Scholar] [CrossRef]
- Folgueras, A.R.; Freitas-Rodríguez, S.; Velasco, G.; López-Otín, C. Mouse Models to Disentangle the Hallmarks of Human Aging. Circ. Res. 2018, 123, 905–924. [Google Scholar] [CrossRef]
- Gebert, L.F.; MacRae, I.J. Regulation of microRNA Function in Animals. Nat. Rev. Mol. Cell Biol. 2019, 20, 21–37. [Google Scholar] [CrossRef]
- Berezikov, E. Evolution of microRNA Diversity and Regulation in Animals. Nat. Rev. Genet. 2011, 12, 846. [Google Scholar] [CrossRef]
- 6 Non-Coding RNA Characterization. Nature 2019. [CrossRef]
- Sun, W.; Yang, Y.; Xu, C.; Guo, J. Regulatory Mechanisms of Long Noncoding RNAs on Gene Expression in Cancers. Cancer Genet. 2017, 216, 105–110. [Google Scholar] [CrossRef]
- He, S.; Sharpless, N.E. Senescence in Health and Disease. Cell 2017, 169, 1000–1011. [Google Scholar] [CrossRef] [PubMed]
- Rayess, H.; Wang, M.B.; Srivatsan, E.S. Cellular Senescence and Tumor Suppressor Gene p16. Int. J. Cancer 2012, 130, 1715–1725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Keizer, P.L. The Fountain of Youth by Targeting Senescent Cells? Trends Mol. Med. 2017, 23, 6–17. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.Y.; Jee, H.J.; Um, J.; Kim, Y.M.; Bae, S.S.; Yun, J. Cooperation between p21 and Akt is Required for p53-dependent Cellular Senescence. Aging Cell 2017, 16, 1094–1103. [Google Scholar] [CrossRef]
- Moiseeva, O.; Bourdeau, V.; Roux, A.; Deschenes-Simard, X.; Ferbeyre, G. Mitochondrial Dysfunction Contributes to Oncogene-Induced Senescence. Mol. Cell. Biol. 2009, 29, 4495–4507. [Google Scholar] [CrossRef] [Green Version]
- Passos, J.F.; Nelson, G.; Wang, C.; Richter, T.; Simillion, C.; Proctor, C.J.; Miwa, S.; Olijslagers, S.; Hallinan, J.; Wipat, A. Feedback between p21 and Reactive Oxygen Production is Necessary for Cell Senescence. Mol. Syst. Biol. 2010, 6, 347. [Google Scholar] [CrossRef]
- Tai, H.; Wang, Z.; Gong, H.; Han, X.; Zhou, J.; Wang, X.; Wei, X.; Ding, Y.; Huang, N.; Qin, J. Autophagy Impairment with Lysosomal and Mitochondrial Dysfunction is an Important Characteristic of Oxidative Stress-Induced Senescence. Autophagy 2017, 13, 99–113. [Google Scholar] [CrossRef] [Green Version]
- Broughton, K.M.; Sussman, M.A. Adult Cardiomyocyte Cell Cycle Detour: Off-Ramp to Quiescent Destinations. Trends Endocrinol. Metab. 2019, 30, 557–567. [Google Scholar] [CrossRef]
- Baker, D.J.; Childs, B.G.; Durik, M.; Wijers, M.E.; Sieben, C.J.; Zhong, J.; Saltness, R.A.; Jeganathan, K.B.; Verzosa, G.C.; Pezeshki, A. Naturally Occurring p16 Ink4a-Positive Cells Shorten Healthy Lifespan. Nature 2016, 530, 184. [Google Scholar] [CrossRef] [Green Version]
- Yosef, R.; Pilpel, N.; Tokarsky-Amiel, R.; Biran, A.; Ovadya, Y.; Cohen, S.; Vadai, E.; Dassa, L.; Shahar, E.; Condiotti, R. Directed Elimination of Senescent Cells by Inhibition of BCL-W and BCL-XL. Nature Commun. 2016, 7, 11190. [Google Scholar] [CrossRef]
- Chang, J.; Wang, Y.; Shao, L.; Laberge, R.; Demaria, M.; Campisi, J.; Janakiraman, K.; Sharpless, N.E.; Ding, S.; Feng, W. Clearance of Senescent Cells by ABT263 Rejuvenates Aged Hematopoietic Stem Cells in Mice. Nat. Med. 2016, 22, 78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Triana-Martinez, F.; Picallos-Rabina, P.; Da Silva-Alvarez, S.; Pietrocola, F.; Llanos, S.; Rodilla, V.; Soprano, E.; Pedrosa, P.; Ferreiros, A.; Barradas, M.; et al. Identification and Characterization of Cardiac Glycosides as Senolytic Compounds. Nat. Commun. 2019, 10, 4731. [Google Scholar] [CrossRef] [PubMed]
- Guerrero, A.; Herranz, N.; Sun, B.; Wagner, V.; Gallage, S.; Guiho, R.; Wolter, K.; Pombo, J.; Irvine, E.E.; Innes, A.J.; et al. Cardiac Glycosides are Broad-Spectrum Senolytics. Nat. Metab. 2019, 1, 1074–1088. [Google Scholar] [CrossRef]
- Zhu, Y.; Tchkonia, T.; Pirtskhalava, T.; Gower, A.C.; Ding, H.; Giorgadze, N.; Palmer, A.K.; Ikeno, Y.; Hubbard, G.B.; Lenburg, M. The Achilles’ Heel of Senescent Cells: From Transcriptome to Senolytic Drugs. Aging Cell 2015, 14, 644–658. [Google Scholar] [CrossRef]
- Schafer, M.J.; White, T.A.; Iijima, K.; Haak, A.J.; Ligresti, G.; Atkinson, E.J.; Oberg, A.L.; Birch, J.; Salmonowicz, H.; Zhu, Y. Cellular Senescence Mediates Fibrotic Pulmonary Disease. Nature Commun. 2017, 8, 14532. [Google Scholar] [CrossRef]
- Lewis-McDougall, F.C.; Ruchaya, P.J.; Domenjo-Vila, E.; Shin Teoh, T.; Prata, L.; Cottle, B.J.; Clark, J.E.; Punjabi, P.P.; Awad, W.; Torella, D. Aged-senescent Cells Contribute to Impaired Heart Regeneration. Aging Cell 2019, 18, e12931. [Google Scholar] [CrossRef] [Green Version]
- Xu, M.; Pirtskhalava, T.; Farr, J.N.; Weigand, B.M.; Palmer, A.K.; Weivoda, M.M.; Inman, C.L.; Ogrodnik, M.B.; Hachfeld, C.M.; Fraser, D.G. Senolytics Improve Physical Function and Increase Lifespan in Old Age. Nat. Med. 2018, 24, 1246. [Google Scholar] [CrossRef]
- Farr, J.N.; Xu, M.; Weivoda, M.M.; Monroe, D.G.; Fraser, D.G.; Onken, J.L.; Negley, B.A.; Sfeir, J.G.; Ogrodnik, M.B.; Hachfeld, C.M. Targeting Cellular Senescence Prevents Age-Related Bone Loss in Mice. Nat. Med. 2017, 23, 1072. [Google Scholar] [CrossRef]
- Justice, J.N.; Nambiar, A.M.; Tchkonia, T.; LeBrasseur, N.K.; Pascual, R.; Hashmi, S.K.; Prata, L.; Masternak, M.M.; Kritchevsky, S.B.; Musi, N. Senolytics in Idiopathic Pulmonary Fibrosis: Results from a First-in-Human, Open-Label, Pilot Study. EBioMedicine 2019, 40, 554–563. [Google Scholar] [CrossRef] [Green Version]
- Hickson, L.J.; Prata, L.G.L.; Bobart, S.A.; Evans, T.K.; Giorgadze, N.; Hashmi, S.K.; Herrmann, S.M.; Jensen, M.D.; Jia, Q.; Jordan, K.L. Senolytics Decrease Senescent Cells in Humans: Preliminary Report from a Clinical Trial of Dasatinib Plus Quercetin in Individuals with Diabetic Kidney Disease. EBioMedicine 2019, 47, 446–456. [Google Scholar] [CrossRef] [Green Version]
- Xie, J.; Chen, Y.; Hu, C.; Pan, Q.; Wang, B.; Li, X.; Geng, J.; Xu, B. Premature Senescence of Cardiac Fibroblasts and Atrial Fibrosis in Patients with Atrial Fibrillation. Oncotarget 2017, 8, 57981–57990. [Google Scholar] [CrossRef] [PubMed]
- Meyer, K.; Hodwin, B.; Ramanujam, D.; Engelhardt, S.; Sarikas, A. Essential Role for Premature Senescence of Myofibroblasts in Myocardial Fibrosis. J. Am. Coll. Cardiol. 2016, 67, 2018–2028. [Google Scholar] [CrossRef] [PubMed]
- Cui, S.; Xue, L.; Yang, F.; Dai, S.; Han, Z.; Liu, K.; Liu, B.; Yuan, Q.; Cui, Z.; Zhang, Y. Postinfarction Hearts are Protected by Premature Senescent Cardiomyocytes Via GATA 4-Dependent CCN 1 Secretion. J. Am. Heart Assoc. 2018, 7, e009111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- EUGenMed Cardiovascular Clinical Study Group; Regitz-Zagrosek, V.; Oertelt-Prigione, S.; Prescott, E.; Franconi, F.; Gerdts, E.; Foryst-Ludwig, A.; Maas, A.H.; Kautzky-Willer, A.; Knappe-Wegner, D.; et al. Gender in Cardiovascular Diseases: Impact on Clinical Manifestations, Management, and Outcomes. Eur. Heart J. 2016, 37, 24–34. [Google Scholar] [PubMed] [Green Version]
- Benjamin, E.J.; Muntner, P.; Alonso, A.; Bittencourt, M.S.; Callaway, C.W.; Carson, A.P.; Chamberlain, A.M.; Chang, A.R.; Cheng, S.; Das, S.R.; et al. Heart Disease and Stroke Statistics-2019 Update: A Report from the American Heart Association. Circulation 2019, 139, e56–e528. [Google Scholar] [CrossRef]
- Ho, J.E.; Gona, P.; Pencina, M.J.; Tu, J.V.; Austin, P.C.; Vasan, R.S.; Kannel, W.B.; D’Agostino, R.B.; Lee, D.S.; Levy, D. Discriminating Clinical Features of Heart Failure with Preserved Vs. Reduced Ejection Fraction in the Community. Eur. Heart J. 2012, 33, 1734–1741. [Google Scholar] [CrossRef] [Green Version]
- Gori, M.; Lam, C.S.; Gupta, D.K.; Santos, A.B.; Cheng, S.; Shah, A.M.; Claggett, B.; Zile, M.R.; Kraigher-Krainer, E.; Pieske, B.; et al. Sex-Specific Cardiovascular Structure and Function in Heart Failure with Preserved Ejection Fraction. Eur. J. Heart Fail. 2014, 16, 535–542. [Google Scholar] [CrossRef]
- Zhao, Z.; Wang, H.; Jessup, J.A.; Lindsey, S.H.; Chappell, M.C.; Groban, L. Role of Estrogen in Diastolic Dysfunction. Am. J. Physiol. Heart Circ. Physiol. 2014, 306, H628–H640. [Google Scholar] [CrossRef] [Green Version]
- Myerburg, R.J.; Junttila, M.J. Sudden Cardiac Death Caused by Coronary Heart Disease. Circulation 2012, 125, 1043–1052. [Google Scholar] [CrossRef] [Green Version]
- Haukilahti, M.A.E.; Holmstrom, L.; Vahatalo, J.; Kentta, T.; Tikkanen, J.; Pakanen, L.; Kortelainen, M.L.; Perkiomaki, J.; Huikuri, H.; Myerburg, R.J.; et al. Sudden Cardiac Death in Women. Circulation 2019, 139, 1012–1021. [Google Scholar] [CrossRef]
- Bink, D.I.; Lozano-Vidal, N.; Boon, R.A. Long Non-Coding RNA in Vascular Disease and Aging. Non-coding RNA 2019, 5, 26. [Google Scholar] [CrossRef] [Green Version]
- Greco, S.; Gaetano, C.; Martelli, F. Long Noncoding Competing Endogenous RNA Networks in Age-Associated Cardiovascular Diseases. Int. J. Mol. Sci. 2019, 20, 3079. [Google Scholar] [CrossRef] [Green Version]
- Puvvula, P.K. LncRNAs Regulatory Networks in Cellular Senescence. Int. J. Mol. Sci. 2019, 20, 2615. [Google Scholar] [CrossRef] [Green Version]
- Lozano-Vidal, N.; Bink, D.I.; Boon, R.A. Long Noncoding RNA in Cardiac Aging and Disease. J. Mol. Cell Biol. 2019, 11, 860–867. [Google Scholar] [CrossRef] [PubMed]
- Hermans-Beijnsberger, S.; van Bilsen, M.; Schroen, B. Long Non-Coding RNAs in the Failing Heart and Vasculature. Non-coding RNA Res. 2018, 3, 118–130. [Google Scholar] [CrossRef] [PubMed]
- Suh, N. MicroRNA Controls of Cellular Senescence. BMB Rep. 2018, 51, 493–499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Lucia, C.; Komici, K.; Borghetti, G.; Femminella, G.D.; Bencivenga, L.; Cannavo, A.; Corbi, G.; Ferrara, N.; Houser, S.R.; Koch, W.J. microRNA in Cardiovascular Aging and Age-Related Cardiovascular Diseases. Front. Med. 2017, 4, 74. [Google Scholar] [CrossRef] [Green Version]
- Bai, X.Y.; Ma, Y.; Ding, R.; Fu, B.; Shi, S.; Chen, X.M. miR-335 and miR-34a Promote Renal Senescence by Suppressing Mitochondrial Antioxidative Enzymes. J. Am. Soc. Nephrol. 2011, 22, 1252–1261. [Google Scholar] [CrossRef] [Green Version]
- Ito, T.; Yagi, S.; Yamakuchi, M. MicroRNA-34a Regulation of Endothelial Senescence. Biochem. Biophys. Res. Commun. 2010, 398, 735–740. [Google Scholar] [CrossRef]
- Tazawa, H.; Tsuchiya, N.; Izumiya, M.; Nakagama, H. Tumor-Suppressive miR-34a Induces Senescence-Like Growth Arrest through Modulation of the E2F Pathway in Human Colon Cancer Cells. Proc. Natl. Acad. Sci. USA 2007, 104, 15472–15477. [Google Scholar] [CrossRef] [Green Version]
- Boon, R.A.; Iekushi, K.; Lechner, S.; Seeger, T.; Fischer, A.; Heydt, S.; Kaluza, D.; Tréguer, K.; Carmona, G.; Bonauer, A. MicroRNA-34a Regulates Cardiac Ageing and Function. Nature 2013, 495, 107. [Google Scholar] [CrossRef]
- Verjans, R.; Derks, W.J.; Korn, K.; Sönnichsen, B.; van Leeuwen, R.E.; Schroen, B.; van Bilsen, M.; Heymans, S. Functional Screening Identifies MicroRNAs as Multi-Cellular Regulators of Heart Failure. Sci. Rep. 2019, 9, 6055. [Google Scholar] [CrossRef]
- Nagpal, V.; Rai, R.; Place, A.T.; Murphy, S.B.; Verma, S.K.; Ghosh, A.K.; Vaughan, D.E. MiR-125b is Critical for Fibroblast-to-Myofibroblast Transition and Cardiac Fibrosis. Circulation 2016, 133, 291–301. [Google Scholar] [CrossRef] [Green Version]
- Thum, T.; Gross, C.; Fiedler, J.; Fischer, T.; Kissler, S.; Bussen, M.; Galuppo, P.; Just, S.; Rottbauer, W.; Frantz, S. MicroRNA-21 Contributes to Myocardial Disease by Stimulating MAP Kinase Signalling in Fibroblasts. Nature 2008, 456, 980. [Google Scholar] [CrossRef]
- Van Almen, G.C.; Verhesen, W.; van Leeuwen, R.E.; van de Vrie, M.; Eurlings, C.; Schellings, M.W.; Swinnen, M.; Cleutjens, J.P.; van Zandvoort, M.A.; Heymans, S. MicroRNA-18 and microRNA-19 Regulate CTGF and TSP-1 Expression in Age-related Heart Failure. Aging Cell 2011, 10, 769–779. [Google Scholar] [CrossRef] [Green Version]
- Van Rooij, E.; Sutherland, L.B.; Thatcher, J.E.; DiMaio, J.M.; Naseem, R.H.; Marshall, W.S.; Hill, J.A.; Olson, E.N. Dysregulation of microRNAs After Myocardial Infarction Reveals a Role of miR-29 in Cardiac Fibrosis. Proc. Natl. Acad. Sci. USA 2008, 105, 13027–13032. [Google Scholar] [CrossRef] [Green Version]
- Yuan, J.; Liu, H.; Gao, W.; Zhang, L.; Ye, Y.; Yuan, L.; Ding, Z.; Wu, J.; Kang, L.; Zhang, X.; et al. MicroRNA-378 Suppresses Myocardial Fibrosis through a Paracrine Mechanism at the Early Stage of Cardiac Hypertrophy Following Mechanical Stress. Theranostics 2018, 8, 2565–2582. [Google Scholar] [CrossRef]
- Small, E.M.; Frost, R.J.; Olson, E.N. MicroRNAs Add a New Dimension to Cardiovascular Disease. Circulation 2010, 121, 1022–1032. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Wang, H.; Li, Y.; Zhang, C.; Zhou, C.; Wang, L.; Xia, Y.; Du, J.; Li, H. MicroRNA Let-7i Negatively Regulates Cardiac Inflammation and Fibrosis. Hypertension 2015, 66, 776–785. [Google Scholar] [CrossRef]
- Sawaki, D.; Czibik, G.; Pini, M.; Ternacle, J.; Suffee, N.; Mercedes, R.; Marcelin, G.; Surenaud, M.; Marcos, E.; Gual, P. Visceral Adipose Tissue Drives Cardiac Aging through Modulation of Fibroblast Senescence by Osteopontin Production. Circulation 2018, 138, 809–822. [Google Scholar] [CrossRef]
- Lin, J.; Lopez, E.F.; Jin, Y.; Van Remmen, H.; Bauch, T.; Han, H.; Lindsey, M.L. Age-Related Cardiac Muscle Sarcopenia: Combining Experimental and Mathematical Modeling to Identify Mechanisms. Exp. Gerontol. 2008, 43, 296–306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, N.; Youle, R.J.; Finkel, T. The Mitochondrial Basis of Aging. Mol. Cell 2016, 61, 654–666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chia, C.W.; Egan, J.M.; Ferrucci, L. Age-Related Changes in Glucose Metabolism, Hyperglycemia, and Cardiovascular Risk. Circ. Res. 2018, 123, 886–904. [Google Scholar] [CrossRef] [PubMed]
- Lopaschuk, G.D.; Collins-Nakai, R.L.; Itoi, T. Developmental Changes in Energy Substrate use by the Heart. Cardiovasc. Res. 1992, 26, 1172–1180. [Google Scholar] [CrossRef]
- Razeghi, P.; Young, M.E.; Alcorn, J.L.; Moravec, C.S.; Frazier, O.; Taegtmeyer, H. Metabolic Gene Expression in Fetal and Failing Human Heart. Circulation 2001, 104, 2923–2931. [Google Scholar] [CrossRef] [Green Version]
- El Azzouzi, H.; Leptidis, S.; Dirkx, E.; Hoeks, J.; van Bree, B.; Brand, K.; McClellan, E.A.; Poels, E.; Sluimer, J.C.; Van den Hoogenhof, M.M.; et al. The Hypoxia-Inducible microRNA Cluster miR-199a∼ 214 Targets Myocardial PPARδ and Impairs Mitochondrial Fatty Acid Oxidation. Cell Metab. 2013, 18, 341–354. [Google Scholar] [CrossRef] [Green Version]
- Doenst, T.; Nguyen, T.D.; Abel, E.D. Cardiac Metabolism in Heart Failure: Implications Beyond ATP Production. Circ. Res. 2013, 113, 709–724. [Google Scholar] [CrossRef] [Green Version]
- Trifunovic, A.; Wredenberg, A.; Falkenberg, M.; Spelbrink, J.N.; Rovio, A.T.; Bruder, C.E.; Bohlooly-Y, M.; Gidlöf, S.; Oldfors, A.; Wibom, R. Premature Ageing in Mice Expressing Defective Mitochondrial DNA Polymerase. Nature 2004, 429, 417. [Google Scholar] [CrossRef]
- Pinto, M.; Pickrell, A.M.; Wang, X.; Bacman, S.R.; Yu, A.; Hida, A.; Dillon, L.M.; Morton, P.D.; Malek, T.R.; Williams, S.L. Transient Mitochondrial DNA Double Strand Breaks in Mice Cause Accelerated Aging Phenotypes in a ROS-Dependent but p53/p21-Independent Manner. Cell Death Differ. 2017, 24, 288. [Google Scholar] [CrossRef] [Green Version]
- Wiley, C.D.; Velarde, M.C.; Lecot, P.; Liu, S.; Sarnoski, E.A.; Freund, A.; Shirakawa, K.; Lim, H.W.; Davis, S.S.; Ramanathan, A. Mitochondrial Dysfunction Induces Senescence with a Distinct Secretory Phenotype. Cell Metab. 2016, 23, 303–314. [Google Scholar] [CrossRef] [Green Version]
- Hoshino, A.; Mita, Y.; Okawa, Y.; Ariyoshi, M.; Iwai-Kanai, E.; Ueyama, T.; Ikeda, K.; Ogata, T.; Matoba, S. Cytosolic p53 Inhibits Parkin-Mediated Mitophagy and Promotes Mitochondrial Dysfunction in the Mouse Heart. Nature Commun. 2013, 4, 2308. [Google Scholar] [CrossRef] [Green Version]
- Nomura, S.; Satoh, M.; Fujita, T.; Higo, T.; Sumida, T.; Ko, T.; Yamaguchi, T.; Tobita, T.; Naito, A.T.; Ito, M. Cardiomyocyte Gene Programs Encoding Morphological and Functional Signatures in Cardiac Hypertrophy and Failure. Nature Commun. 2018, 9, 4435. [Google Scholar] [CrossRef] [PubMed]
- Mouchiroud, L.; Houtkooper, R.H.; Moullan, N.; Katsyuba, E.; Ryu, D.; Cantó, C.; Mottis, A.; Jo, Y.; Viswanathan, M.; Schoonjans, K. The NAD /Sirtuin Pathway Modulates Longevity through Activation of Mitochondrial UPR and FOXO Signaling. Cell 2013, 154, 430–441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lagouge, M.; Argmann, C.; Gerhart-Hines, Z.; Meziane, H.; Lerin, C.; Daussin, F.; Messadeq, N.; Milne, J.; Lambert, P.; Elliott, P. Resveratrol Improves Mitochondrial Function and Protects Against Metabolic Disease by Activating SIRT1 and PGC-1α. Cell 2006, 127, 1109–1122. [Google Scholar] [CrossRef]
- Youle, R.J.; Narendra, D.P. Mechanisms of Mitophagy. Nat. Rev. Mol. Cell Biol. 2011, 12, 9. [Google Scholar] [CrossRef]
- Sebastian, D.; Palacin, M.; Zorzano, A. Mitochondrial Dynamics: Coupling Mitochondrial Fitness with Healthy Aging. Trends Mol. Med. 2017, 23, 201–215. [Google Scholar] [CrossRef]
- Bonora, M.; Wieckowski, M.R.; Sinclair, D.A.; Kroemer, G.; Pinton, P.; Galluzzi, L. Targeting Mitochondria for Cardiovascular Disorders: Therapeutic Potential and Obstacles. Nat. Rev. Cardiol. 2019, 16, 33–55. [Google Scholar] [CrossRef]
- Weir, H.J.; Yao, P.; Huynh, F.K.; Escoubas, C.C.; Goncalves, R.L.; Burkewitz, K.; Laboy, R.; Hirschey, M.D.; Mair, W.B. Dietary Restriction and AMPK Increase Lifespan Via Mitochondrial Network and Peroxisome Remodeling. Cell Metab. 2017, 26, 884–896. [Google Scholar] [CrossRef] [Green Version]
- Song, M.; Franco, A.; Fleischer, J.A.; Zhang, L.; Dorn II, G.W. Abrogating Mitochondrial Dynamics in Mouse Hearts Accelerates Mitochondrial Senescence. Cell Metab. 2017, 26, 872–883. [Google Scholar] [CrossRef] [Green Version]
- Shirakabe, A.; Zhai, P.; Ikeda, Y.; Saito, T.; Maejima, Y.; Hsu, C.; Nomura, M.; Egashira, K.; Levine, B.; Sadoshima, J. Drp1-Dependent Mitochondrial Autophagy Plays a Protective Role Against Pressure Overload–induced Mitochondrial Dysfunction and Heart Failure. Circulation 2016, 133, 1249–1263. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.; Yao, X.; Zhang, Q.; Zhu, M.; Liu, Z.; Ci, B.; Xie, Y.; Carlson, D.; Rothermel, B.A.; Sun, Y. Beclin-1-Dependent Autophagy Protects the Heart during Sepsis. Circulation 2018, 138, 2247–2262. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Bie, Z.; Sun, C. Long Noncoding RNA AK088388 Regulates Autophagy through miR-30a to Affect Cardiomyocyte Injury. J. Cell. Biochem. 2019, 120, 10155–10163. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Zhang, X.; Zhuang, H.; Chen, H.G.; Chen, Y.; Tian, W.; Wu, W.; Li, Y.; Wang, S.; Zhang, L.; et al. MicroRNA-137 is a Novel Hypoxia-Responsive microRNA that Inhibits Mitophagy Via Regulation of Two Mitophagy Receptors FUNDC1 and NIX. J. Biol. Chem. 2014, 289, 10691–10701. [Google Scholar] [CrossRef] [Green Version]
- Zhao, T.; Qiu, Z.; Gao, Y. MiR-137-3p Exacerbates the Ischemia-Reperfusion Injured Cardiomyocyte Apoptosis by Targeting KLF15. Naunyn Schmiedebergs Arch. Pharmacol. 2019, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Prosdocimo, D.A.; Anand, P.; Liao, X.; Zhu, H.; Shelkay, S.; Artero-Calderon, P.; Zhang, L.; Kirsh, J.; Moore, D.; Rosca, M.G.; et al. Kruppel-Like Factor 15 is a Critical Regulator of Cardiac Lipid Metabolism. J. Biol. Chem. 2014, 289, 5914–5924. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haldar, S.M.; Jeyaraj, D.; Anand, P.; Zhu, H.; Lu, Y.; Prosdocimo, D.A.; Eapen, B.; Kawanami, D.; Okutsu, M.; Brotto, L.; et al. Kruppel-Like Factor 15 Regulates Skeletal Muscle Lipid Flux and Exercise Adaptation. Proc. Natl. Acad. Sci. USA 2012, 109, 6739–6744. [Google Scholar] [CrossRef] [Green Version]
- Rowe, G.C.; Jiang, A.; Arany, Z. PGC-1 Coactivators in Cardiac Development and Disease. Circ. Res. 2010, 107, 825–838. [Google Scholar] [CrossRef] [Green Version]
- Lehman, J.J.; Barger, P.M.; Kovacs, A.; Saffitz, J.E.; Medeiros, D.M.; Kelly, D.P. Peroxisome Proliferator-Activated Receptor Gamma Coactivator-1 Promotes Cardiac Mitochondrial Biogenesis. J. Clin. Invest. 2000, 106, 847–856. [Google Scholar] [CrossRef] [Green Version]
- Arany, Z.; He, H.; Lin, J.; Hoyer, K.; Handschin, C.; Toka, O.; Ahmad, F.; Matsui, T.; Chin, S.; Wu, P. Transcriptional Coactivator PGC-1α Controls the Energy State and Contractile Function of Cardiac Muscle. Cell Metab. 2005, 1, 259–271. [Google Scholar] [CrossRef] [Green Version]
- Arany, Z.; Foo, S.; Ma, Y.; Ruas, J.L.; Bommi-Reddy, A.; Girnun, G.; Cooper, M.; Laznik, D.; Chinsomboon, J.; Rangwala, S.M. HIF-Independent Regulation of VEGF and Angiogenesis by the Transcriptional Coactivator PGC-1α. Nature 2008, 451, 1008. [Google Scholar] [CrossRef] [Green Version]
- Liang, J.; Liu, C.; Qiao, A.; Cui, Y.; Zhang, H.; Cui, A.; Zhang, S.; Yang, Y.; Xiao, X.; Chen, Y. MicroRNA-29a-C Decrease Fasting Blood Glucose Levels by Negatively Regulating Hepatic Gluconeogenesis. J. Hepatol. 2013, 58, 535–542. [Google Scholar] [CrossRef] [PubMed]
- Caravia, X.M.; Fanjul, V.; Oliver, E.; Roiz-Valle, D.; Morán-Álvarez, A.; Desdín-Micó, G.; Mittelbrunn, M.; Cabo, R.; Vega, J.A.; Rodríguez, F. The microRNA-29/PGC1α Regulatory Axis is Critical for Metabolic Control of Cardiac Function. PLoS Biol. 2018, 16, e2006247. [Google Scholar] [CrossRef]
- Long, J.; Badal, S.S.; Ye, Z.; Wang, Y.; Ayanga, B.A.; Galvan, D.L.; Green, N.H.; Chang, B.H.; Overbeek, P.A.; Danesh, F.R. Long Noncoding RNA Tug1 Regulates Mitochondrial Bioenergetics in Diabetic Nephropathy. J. Clin. Invest. 2016, 126, 4205–4218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bai, Z.; Chai, X.; Yoon, M.J.; Kim, H.; LO, K.A.; Zhang, Z.; Xu, D.; Siang, D.T.C.; Walet, A.C.E.; Xu, S. Dynamic Transcriptome Changes during Adipose Tissue Energy Expenditure Reveal Critical Roles for Long Noncoding RNA Regulators. PLoS Biol. 2017, 15, e2002176. [Google Scholar] [CrossRef] [PubMed]
- Kleiner, S.; Nguyen-Tran, V.; Bare, O.; Huang, X.; Spiegelman, B.; Wu, Z. PPAR{Delta} Agonism Activates Fatty Acid Oxidation Via PGC-1{Alpha} but does Not Increase Mitochondrial Gene Expression and Function. J. Biol. Chem. 2009, 284, 18624–18633. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sen, P.; Shah, P.P.; Nativio, R.; Berger, S.L. Epigenetic Mechanisms of Longevity and Aging. Cell 2016, 166, 822–839. [Google Scholar] [CrossRef] [Green Version]
- Das, A.; Huang, G.X.; Bonkowski, M.S.; Longchamp, A.; Li, C.; Schultz, M.B.; Kim, L.; Osborne, B.; Joshi, S.; Lu, Y. Impairment of an Endothelial NAD -H2S Signaling Network is a Reversible Cause of Vascular Aging. Cell 2018, 173, 74–89. [Google Scholar] [CrossRef] [Green Version]
- Alcendor, R.R.; Gao, S.; Zhai, P.; Zablocki, D.; Holle, E.; Yu, X.; Tian, B.; Wagner, T.; Vatner, S.F.; Sadoshima, J. Sirt1 Regulates Aging and Resistance to Oxidative Stress in the Heart. Circ. Res. 2007, 100, 1512–1521. [Google Scholar] [CrossRef]
- Yamakuchi, M.; Ferlito, M.; Lowenstein, C.J. miR-34a Repression of SIRT1 Regulates Apoptosis. Proc. Natl. Acad. Sci. USA 2008, 105, 13421–13426. [Google Scholar] [CrossRef] [Green Version]
- Badi, I.; Burba, I.; Ruggeri, C.; Zeni, F.; Bertolotti, M.; Scopece, A.; Pompilio, G.; Raucci, A. MicroRNA-34a Induces Vascular Smooth Muscle Cells Senescence by SIRT1 Downregulation and Promotes the Expression of Age-Associated Pro-Inflammatory Secretory Factors. J. Gerontol. Ser. A Biomed. Sci. Med Sci. 2014, 70, 1304–1311. [Google Scholar] [CrossRef]
- Wang, G.; Wang, Y.; Xiong, Y.; Chen, X.; Ma, M.; Cai, R.; Gao, Y.; Sun, Y.; Yang, G.; Pang, W. Sirt1 AS lncRNA Interacts with its mRNA to Inhibit Muscle Formation by Attenuating Function of miR-34a. Sci. Rep. 2016, 6, 21865. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Hu, Y.; Li, X.; Jin, G.; Chen, X.; Chen, G.; Chen, Y.; Huang, S.; Liao, W.; Liao, Y. Sirt1 Antisense Long Noncoding RNA Promotes Cardiomyocyte Proliferation by Enhancing the Stability of Sirt1. J. Am. Heart Assoc. 2018, 7, e009700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, X.; Yang, Y.; Xu, C.; Peng, Z.; Zhang, M.; Lei, L.; Gao, W.; Dong, Y.; Shi, Z.; Sun, X.; et al. Upregulation of miR-181a Impairs Hepatic Glucose and Lipid Homeostasis. Oncotarget 2017, 8, 91362–91378. [Google Scholar] [CrossRef]
- Das, S.; Kohr, M.; Dunkerly-Eyring, B.; Lee, D.I.; Bedja, D.; Kent, O.A.; Leung, A.K.; Henao-Mejia, J.; Flavell, R.A.; Steenbergen, C. Divergent Effects of miR-181 Family Members on Myocardial Function through Protective Cytosolic and Detrimental Mitochondrial microRNA Targets. J. Am. Heart Assoc. 2017, 6, e004694. [Google Scholar] [CrossRef]
- Edelberg, J.M.; Reed, M.J. Aging and Angiogenesis. Front. Biosci. 2003, 8, s1199–s1209. [Google Scholar] [CrossRef]
- Tian, X.; Firsanov, D.; Zhang, Z.; Cheng, Y.; Luo, L.; Tombline, G.; Tan, R.; Simon, M.; Henderson, S.; Steffan, J. SIRT6 is Responsible for More Efficient DNA Double-Strand Break Repair in Long-Lived Species. Cell 2019, 177, 622–638. [Google Scholar] [CrossRef] [Green Version]
- Elhanati, S.; Ben-Hamo, R.; Kanfi, Y.; Varvak, A.; Glazz, R.; Lerrer, B.; Efroni, S.; Cohen, H.Y. Reciprocal Regulation between SIRT6 and miR-122 Controls Liver Metabolism and Predicts Hepatocarcinoma Prognosis. Cell Rep. 2016, 14, 234–242. [Google Scholar] [CrossRef] [Green Version]
- Ieda, M.; Fu, J.; Delgado-Olguin, P.; Vedantham, V.; Hayashi, Y.; Bruneau, B.G.; Srivastava, D. Direct Reprogramming of Fibroblasts into Functional Cardiomyocytes by Defined Factors. Cell 2010, 142, 375–386. [Google Scholar] [CrossRef] [Green Version]
- Song, K.; Nam, Y.; Luo, X.; Qi, X.; Tan, W.; Huang, G.N.; Acharya, A.; Smith, C.L.; Tallquist, M.D.; Neilson, E.G. Heart Repair by Reprogramming Non-Myocytes with Cardiac Transcription Factors. Nature 2012, 485, 599. [Google Scholar] [CrossRef] [Green Version]
- Muraoka, N.; Nara, K.; Tamura, F.; Kojima, H.; Yamakawa, H.; Sadahiro, T.; Miyamoto, K.; Isomi, M.; Haginiwa, S.; Tani, H. Role of Cyclooxygenase-2-Mediated Prostaglandin E2-Prostaglandin E Receptor 4 Signaling in Cardiac Reprogramming. Nat. Commun. 2019, 10, 674. [Google Scholar] [CrossRef]
- Diez-Cuñado, M.; Wei, K.; Bushway, P.J.; Maurya, M.R.; Perera, R.; Subramaniam, S.; Ruiz-Lozano, P.; Mercola, M. miRNAs that Induce Human Cardiomyocyte Proliferation Converge on the Hippo Pathway. Cell Rep. 2018, 23, 2168–2174. [Google Scholar] [CrossRef] [PubMed]
- Torrini, C.; Cubero, R.J.; Dirkx, E.; Braga, L.; Ali, H.; Prosdocimo, G.; Gutierrez, M.I.; Collesi, C.; Licastro, D.; Zentilin, L. Common Regulatory Pathways Mediate Activity of MicroRNAs Inducing Cardiomyocyte Proliferation. Cell Rep. 2019, 27, 2759–2771. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, W.; Feng, Y.; Liang, J.; Yu, H.; Wang, C.; Wang, B.; Wang, M.; Jiang, L.; Meng, W.; Cai, W. Loss of microRNA-128 Promotes Cardiomyocyte Proliferation and Heart Regeneration. Nat. Commun. 2018, 9, 700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, Y.; Yan, Y.; Lundy, D.J.; Lo, A.H.; Wang, Y.; Ruan, S.; Lin, P.; Hsieh, P.C. Reprogramming-derived Gene Cocktail Increases Cardiomyocyte Proliferation for Heart Regeneration. EMBO Mol. Med. 2017, 9, 251–264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ponnusamy, M.; Liu, F.; Zhang, Y.; Li, R.; Zhai, M.; Liu, F.; Zhou, L.; Liu, C.; Yan, K.; Dong, Y. Long Noncoding RNA CPR (Cardiomyocyte Proliferation Regulator) Regulates Cardiomyocyte Proliferation and Cardiac Repair. Circulation 2019, 139, 2668–2684. [Google Scholar] [CrossRef]
- Munoz-Espin, D.; Canamero, M.; Maraver, A.; Gomez-Lopez, G.; Contreras, J.; Murillo-Cuesta, S.; Rodriguez-Baeza, A.; Varela-Nieto, I.; Ruberte, J.; Collado, M.; et al. Programmed Cell Senescence during Mammalian Embryonic Development. Cell 2013, 155, 1104–1118. [Google Scholar] [CrossRef] [Green Version]
- Storer, M.; Mas, A.; Robert-Moreno, A.; Pecoraro, M.; Ortells, M.C.; Di Giacomo, V.; Yosef, R.; Pilpel, N.; Krizhanovsky, V.; Sharpe, J.; et al. Senescence is a Developmental Mechanism that Contributes to Embryonic Growth and Patterning. Cell 2013, 155, 1119–1130. [Google Scholar] [CrossRef] [Green Version]
- Puente, B.N.; Kimura, W.; Muralidhar, S.A.; Moon, J.; Amatruda, J.F.; Phelps, K.L.; Grinsfelder, D.; Rothermel, B.A.; Chen, R.; Garcia, J.A.; et al. The Oxygen-Rich Postnatal Environment Induces Cardiomyocyte Cell-Cycle Arrest through DNA Damage Response. Cell 2014, 157, 565–579. [Google Scholar] [CrossRef] [Green Version]
- Nakada, Y.; Canseco, D.C.; Thet, S.; Abdisalaam, S.; Asaithamby, A.; Santos, C.X.; Shah, A.M.; Zhang, H.; Faber, J.E.; Kinter, M.T.; et al. Hypoxia Induces Heart Regeneration in Adult Mice. Nature 2017, 541, 222–227. [Google Scholar] [CrossRef]
- Van Heesch, S.; Witte, F.; Schneider-Lunitz, V.; Schulz, J.F.; Adami, E.; Faber, A.B.; Kirchner, M.; Maatz, H.; Blachut, S.; Sandmann, C.L.; et al. The Translational Landscape of the Human Heart. Cell 2019, 178, 242–260. [Google Scholar] [CrossRef] [Green Version]

© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lin, R.; Kerkelä, R. Regulatory Mechanisms of Mitochondrial Function and Cardiac Aging. Int. J. Mol. Sci. 2020, 21, 1359. https://doi.org/10.3390/ijms21041359
Lin R, Kerkelä R. Regulatory Mechanisms of Mitochondrial Function and Cardiac Aging. International Journal of Molecular Sciences. 2020; 21(4):1359. https://doi.org/10.3390/ijms21041359
Chicago/Turabian StyleLin, Ruizhu, and Risto Kerkelä. 2020. "Regulatory Mechanisms of Mitochondrial Function and Cardiac Aging" International Journal of Molecular Sciences 21, no. 4: 1359. https://doi.org/10.3390/ijms21041359
APA StyleLin, R., & Kerkelä, R. (2020). Regulatory Mechanisms of Mitochondrial Function and Cardiac Aging. International Journal of Molecular Sciences, 21(4), 1359. https://doi.org/10.3390/ijms21041359