Transcriptional Regulation of Genes by Ikaros Tumor Suppressor in Acute Lymphoblastic Leukemia

 , , and
, , and 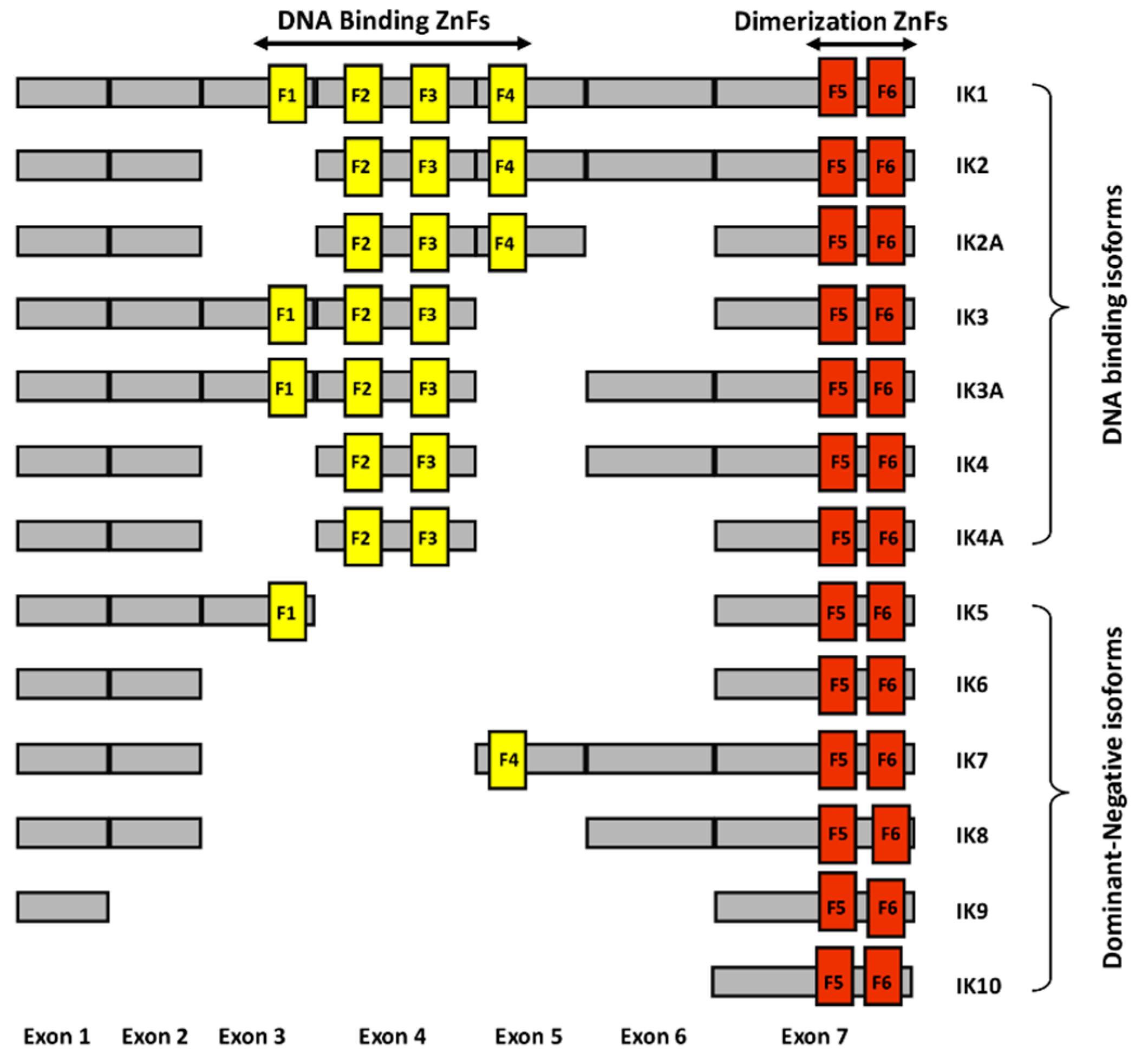{kind=link}
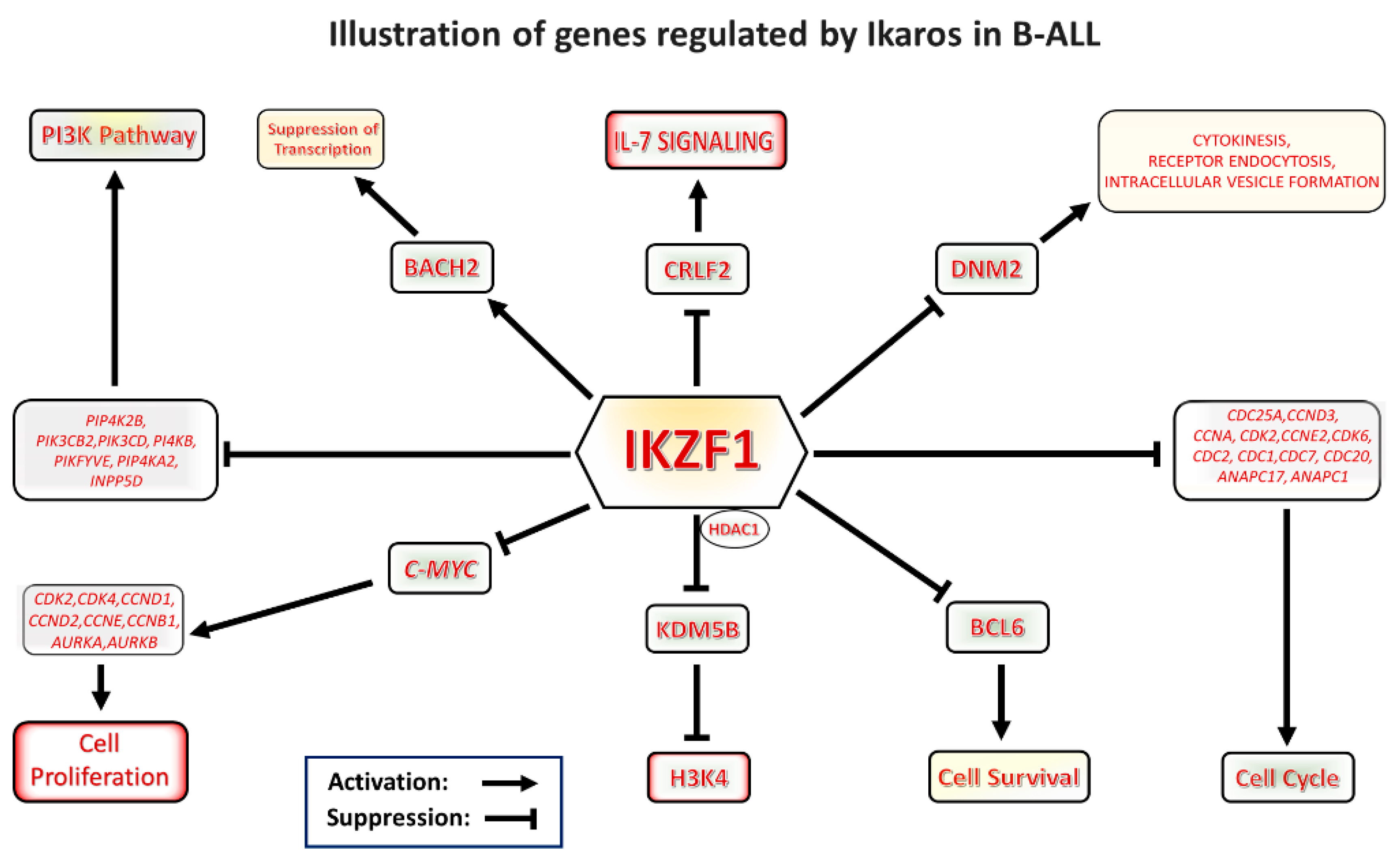{kind=link}
Abstract
:1. Clinical Significance of Ikaros Activity in Leukemia
2. Ikaros as a Transcription Factor and Epigenetic Regulation of Its Target Genes
3. Phosphatidylinositol-3 Kinase (PI3K) Pathway
4. Cell Cycle Pathway
5. Lysine-Specific Demethylase 5B (KDM5B)
6. Plant Homeodomain Finger 2 (PHF2)
7. Cytokine Receptor-like Factor 2 (CRLF2)
8. AT-Rich Interaction Domain 5B (ARID5B)
9. c-MYC and MYC Binding Protein 2 (MYCBP2)
10. B-cell Lymphoma 6 (BCL6) and BTB and CNC Homology 1 Basic Leucine Zipper Transcription Factor 2 (BACH2)
11. Dynamin 2 (DNM2)
12. Interleukin-7 Receptor-α (IL7R) and SH2B Adaptor Protein 3 (SH2B3)
13. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hunger, S.P.; Mullighan, C.G. Acute Lymphoblastic Leukemia in Children. N. Engl. J. Med. 2015, 373, 1541–1552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mullighan, C.G.; Su, X.; Zhang, J.; Radtke, I.; Phillips, L.A.; Miller, C.B.; Ma, J.; Liu, W.; Cheng, C.; Schulman, B.A.; et al. Deletion of IKZF1 and prognosis in acute lymphoblastic leukemia. N. Engl. J. Med. 2009, 360, 470–480. [Google Scholar] [CrossRef] [PubMed]
- Clappier, E.; Grardel, N.; Bakkus, M.; Rapion, J.; De Moerloose, B.; Kastner, P.; Caye, A.; Vivent, J.; Costa, V.; Ferster, A.; et al. IKZF1 deletion is an independent prognostic marker in childhood B-cell precursor acute lymphoblastic leukemia, and distinguishes patients benefiting from pulses during maintenance therapy: Results of the EORTC Children’s Leukemia Group study 58951. Leukemia 2015, 29, 2154. [Google Scholar] [CrossRef] [PubMed]
- Van der Veer, A.; Waanders, E.; Pieters, R.; Willemse, M.E.; Van Reijmersdal, S.V.; Russell, L.J.; Harrison, C.J.; Evans, W.E.; van der Velden, V.H.J.; Hoogerbrugge, P.M.; et al. Independent prognostic value of BCR-ABL1-like signature and IKZF1 deletion, but not high CRLF2 expression, in children with B-cell precursor ALL. Blood 2013, 122, 2622–2629. [Google Scholar] [CrossRef] [PubMed]
- Harris, M.H.; Blonquist, T.M.; Athale, U.H.; Clavell, L.A.; Cole, P.D.; Kelly, K.M.; Laverdiere, C.; Leclerc, J.-M.; Michon, B.; Schorin, M.A.; et al. Ikaros Gene Deletion Significantly Predicts Relapse in Pediatric B-ALL Patients with Low End-Induction Minimal Residual Disease. Blood 2015, 126, 2613. [Google Scholar] [CrossRef]
- Georgopoulos, K.; Bigby, M.; Wang, J.H.; Molnar, A.; Wu, P.; Winandy, S.; Sharpe, A. The Ikaros gene is required for the development of all lymphoid lineages. Cell 1994, 79, 143–156. [Google Scholar] [CrossRef]
- Mullighan, C.G.; Goorha, S.; Radtke, I.; Miller, C.B.; Coustan-Smith, E.; Dalton, J.D.; Girtman, K.; Mathew, S.; Ma, J.; Pounds, S.B.; et al. Genome-wide analysis of genetic alterations in acute lymphoblastic leukaemia. Nature 2007, 446, 758. [Google Scholar] [CrossRef]
- Tokunaga, K.; Yamaguchi, S.; Iwanaga, E.; Nanri, T.; Shimomura, T.; Suzushima, H.; Mitsuya, H.; Asou, N. High frequency of IKZF1 genetic alterations in adult patients with B-cell acute lymphoblastic leukemia. Eur. J. Haematol. 2013, 91, 201–208. [Google Scholar] [CrossRef]
- Mullighan, C.G.; Miller, C.B.; Radtke, I.; Phillips, L.A.; Dalton, J.; Ma, J.; White, D.; Hughes, T.P.; Le Beau, M.M.; Pui, C.H.; et al. BCR-ABL1 lymphoblastic leukaemia is characterized by the deletion of Ikaros. Nature 2008, 453, 110–114. [Google Scholar] [CrossRef]
- Den Boer, M.L.; van Slegtenhorst, M.; De Menezes, R.X.; Cheok, M.H.; Buijs-Gladdines, J.G.; Peters, S.T.; Van Zutven, L.J.; Beverloo, H.B.; Van der Spek, P.J.; Escherich, G.; et al. A subtype of childhood acute lymphoblastic leukaemia with poor treatment outcome: A genome-wide classification study. Lancet Oncol. 2009, 10, 125–134. [Google Scholar] [CrossRef] [Green Version]
- Mullighan, C.G. Genomic profiling of B-progenitor acute lymphoblastic leukemia. Best. Pract. Res. Clin. Haematol. 2011, 24, 489–503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mullighan, C.G. The molecular genetic makeup of acute lymphoblastic leukemia. Hematol. Am Soc Hematol. Educ. Progr. 2012, 2012, 389–396. [Google Scholar] [CrossRef] [Green Version]
- Kuiper, R.P.; Waanders, E.; van der Velden, V.H.J.; van Reijmersdal, S.V.; Venkatachalam, R.; Scheijen, B.; Sonneveld, E.; van Dongen, J.J.M.; Veerman, A.J.P.; van Leeuwen, F.N.; et al. IKZF1 deletions predict relapse in uniformly treated pediatric precursor B-ALL. Leukemia 2010, 24, 1258. [Google Scholar] [CrossRef]
- Yeoh, A.E.J.; Lu, Y.; Chin, W.H.N.; Chiew, E.K.H.; Lim, E.H.; Li, Z.; Kham, S.K.Y.; Chan, Y.H.; Abdullah, W.A.; Lin, H.P.; et al. Intensifying Treatment of Childhood B-Lymphoblastic Leukemia With IKZF1 Deletion Reduces Relapse and Improves Overall Survival: Results of Malaysia-Singapore ALL 2010 Study. J. Clin. Oncol. 2018, 36, 2726–2735. [Google Scholar] [CrossRef]
- Liu, Y.; Easton, J.; Shao, Y.; Maciaszek, J.; Wang, Z.; Wilkinson, M.R.; McCastlain, K.; Edmonson, M.; Pounds, S.B.; Shi, L.; et al. The genomic landscape of pediatric and young adult T-lineage acute lymphoblastic leukemia. Nat. Genet 2017, 49, 1211–1218. [Google Scholar] [CrossRef] [Green Version]
- Georgopoulos, K.; Moore, D.D.; Derfler, B. Ikaros, an early lymphoid-specific transcription factor and a putative mediator for T cell commitment. Science 1992, 258, 808–812. [Google Scholar] [CrossRef] [PubMed]
- Molnár, A.; Georgopoulos, K. The Ikaros gene encodes a family of functionally diverse zinc finger DNA-binding proteins. Mol. Cell Biol. 1994, 14, 8292–8303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davis, K.L. Ikaros: Master of hematopoiesis, agent of leukemia. Ther. Adv. Hematol. 2011, 2, 359–368. [Google Scholar] [CrossRef] [Green Version]
- Sun, L.; Liu, A.; Georgopoulos, K. Zinc finger-mediated protein interactions modulate Ikaros activity, a molecular control of lymphocyte development. EMBO J. 1996, 15, 5358–5369. [Google Scholar] [CrossRef]
- Kim, J.; Sif, S.; Jones, B.; Jackson, A.; Koipally, J.; Heller, E.; Winandy, S.; Viel, A.; Sawyer, A.; Ikeda, T.; et al. Ikaros DNA-binding proteins direct formation of chromatin remodeling complexes in lymphocytes. Immunity 1999, 10, 345–355. [Google Scholar] [CrossRef] [Green Version]
- Song, C.; Pan, X.; Ge, Z.; Gowda, C.; Ding, Y.; Li, H.; Li, Z.; Yochum, G.; Muschen, M.; Li, Q.; et al. Epigenetic regulation of gene expression by Ikaros, HDAC1 and Casein Kinase II in leukemia. Leuk. Off. J. Leuk. Soc. Am. Leuk. Res. Fund UK 2016, 30, 1436–1440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, Y.; Zhang, B.; Payne, J.L.; Song, C.; Ge, Z.; Gowda, C.; Iyer, S.; Dhanyamraju, P.K.; Dorsam, G.; Reeves, M.E.; et al. Ikaros tumor suppressor function includes induction of active enhancers and super-enhancers along with pioneering activity. Leukemia 2019, 33, 2720–2731. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stratikopoulos, E.E.; Parsons, R.E. Molecular Pathways: Targeting the PI3K Pathway in Cancer-BET Inhibitors to the Rescue. Clin. Cancer Res. 2016, 22, 2605–2610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, C.; Gowda, C.; Pan, X.; Ding, Y.; Tong, Y.; Tan, B.H.; Wang, H.; Muthusami, S.; Ge, Z.; Sachdev, M.; et al. Targeting casein kinase II restores Ikaros tumor suppressor activity and demonstrates therapeutic efficacy in high-risk leukemia. Blood 2015, 126, 1813–1822. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aleem, E.; Arceci, R.J. Targeting cell cycle regulators in hematologic malignancies. Front. Cell Dev. Biol. 2015, 3, 16. [Google Scholar] [CrossRef] [Green Version]
- Huang, D.; Qiu, Y.; Li, G.; Liu, C.; She, L.; Zhang, D.; Chen, X.; Zhu, G.; Zhang, X.; Tian, Y.; et al. KDM5B overexpression predicts a poor prognosis in patients with squamous cell carcinoma of the head and neck. J. Cancer 2018, 9, 198–204. [Google Scholar] [CrossRef] [Green Version]
- Bamodu, O.A.; Huang, W.C.; Lee, W.H.; Wu, A.; Wang, L.S.; Hsiao, M.; Yeh, C.T.; Chao, T.Y. Aberrant KDM5B expression promotes aggressive breast cancer through MALAT1 overexpression and downregulation of hsa-miR-448. BMC Cancer 2016, 16, 160. [Google Scholar] [CrossRef] [Green Version]
- Han, M.; Xu, W.; Cheng, P.; Jin, H.; Wang, X. Histone demethylase lysine demethylase 5B in development and cancer. Oncotarget 2017, 8, 8980–8991. [Google Scholar] [CrossRef] [Green Version]
- Zheng, Y.C.; Chang, J.; Wang, L.C.; Ren, H.M.; Pang, J.R.; Liu, H.M. Lysine demethylase 5B (KDM5B): A potential anti-cancer drug target. Eur. J. Med. Chem. 2019, 161, 131–140. [Google Scholar] [CrossRef]
- Guerra-Calderas, L.; González-Barrios, R.; Herrera, L.A.; Cantú de León, D.; Soto-Reyes, E. The role of the histone demethylase KDM4A in cancer. Cancer Genet. 2015, 208, 215–224. [Google Scholar] [CrossRef]
- Xhabija, B.; Kidder, B.L. KDM5B is a master regulator of the H3K4-methylome in stem cells, development and cancer. Semin. Cancer Biol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.P.; Hascall, V.C.; Zhang, F.; Linhardt, R.J.; Abbadi, A.; Wang, A. The Responses of Hyperglycemic Dividing Mesangial Cells to Heparin Are Mediated by the Non-reducing Terminal Trisaccharide. J. Biol. Chem. 2015, 290, 29045–29050. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wen, H.; Li, J.; Song, T.; Lu, M.; Kan, P.Y.; Lee, M.G.; Sha, B.; Shi, X. Recognition of histone H3K4 trimethylation by the plant homeodomain of PHF2 modulates histone demethylation. J. Biol. Chem. 2010, 285, 9322–9326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pattabiraman, D.R.; Bierie, B.; Kober, K.I.; Thiru, P.; Krall, J.A.; Zill, C.; Reinhardt, F.; Tam, W.L.; Weinberg, R.A. Activation of PKA leads to mesenchymal-to-epithelial transition and loss of tumor-initiating ability. Science 2016, 351, aad3680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ge, Z.; Gu, Y.; Han, Q.; Sloane, J.; Ge, Q.; Gao, G.; Ma, J.; Song, H.; Hu, J.; Chen, B.; et al. Plant homeodomain finger protein 2 as a novel IKAROS target in acute lymphoblastic leukemia. Epigenomics 2018, 10, 59–69. [Google Scholar] [CrossRef] [PubMed]
- Ito, T.; Wang, Y.H.; Duramad, O.; Hori, T.; Delespesse, G.J.; Watanabe, N.; Qin, F.X.; Yao, Z.; Cao, W.; Liu, Y.J. TSLP-activated dendritic cells induce an inflammatory T helper type 2 cell response through OX40 ligand. J. Exp. Med. 2005, 202, 1213–1223. [Google Scholar] [CrossRef] [Green Version]
- Ying, S.; O’Connor, B.; Ratoff, J.; Meng, Q.; Mallett, K.; Cousins, D.; Robinson, D.; Zhang, G.; Zhao, J.; Lee, T.H.; et al. Thymic stromal lymphopoietin expression is increased in asthmatic airways and correlates with expression of Th2-attracting chemokines and disease severity. J. Immunol. 2005, 174, 8183–8190. [Google Scholar] [CrossRef]
- Zhou, B.; Comeau, M.R.; De Smedt, T.; Liggitt, H.D.; Dahl, M.E.; Lewis, D.B.; Gyarmati, D.; Aye, T.; Campbell, D.J.; Ziegler, S.F. Thymic stromal lymphopoietin as a key initiator of allergic airway inflammation in mice. Nat. Immunol. 2005, 6, 1047–1053. [Google Scholar] [CrossRef]
- Siracusa, M.C.; Saenz, S.A.; Hill, D.A.; Kim, B.S.; Headley, M.B.; Doering, T.A.; Wherry, E.J.; Jessup, H.K.; Siegel, L.A.; Kambayashi, T.; et al. TSLP promotes interleukin-3-independent basophil haematopoiesis and type 2 inflammation. Nature 2011, 477, 229–233. [Google Scholar] [CrossRef]
- Chen, I.M.; Harvey, R.C.; Mullighan, C.G.; Gastier-Foster, J.; Wharton, W.; Kang, H.; Borowitz, M.J.; Camitta, B.M.; Carroll, A.J.; Devidas, M.; et al. Outcome modeling with CRLF2, IKZF1, JAK, and minimal residual disease in pediatric acute lymphoblastic leukemia: A Children’s Oncology Group study. Blood 2012, 119, 3512–3522. [Google Scholar] [CrossRef]
- Yamashita, Y.; Shimada, A.; Yamada, T.; Yamaji, K.; Hori, T.; Tsurusawa, M.; Watanabe, A.; Kikuta, A.; Asami, K.; Saito, A.M.; et al. IKZF1 and CRLF2 gene alterations correlate with poor prognosis in Japanese BCR-ABL1-negative high-risk B-cell precursor acute lymphoblastic leukemia. Pediatr. Blood Cancer 2013, 60, 1587–1592. [Google Scholar] [CrossRef] [PubMed]
- Hunger, S.P.; Mullighan, C.G. Redefining ALL classification: Toward detecting high-risk ALL and implementing precision medicine. Blood 2015, 125, 3977–3987. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Francis, O.L.; Milford, T.A.; Martinez, S.R.; Baez, I.; Coats, J.S.; Mayagoitia, K.; Concepcion, K.R.; Ginelli, E.; Beldiman, C.; Benitez, A.; et al. A novel xenograft model to study the role of TSLP-induced CRLF2 signals in normal and malignant human B lymphopoiesis. Haematologica 2016, 101, 417–426. [Google Scholar] [CrossRef] [Green Version]
- Findley, H.W., Jr.; Cooper, M.D.; Kim, T.H.; Alvarado, C.; Ragab, A.H. Two new acute lymphoblastic leukemia cell lines with early B-cell phenotypes. Blood 1982, 60, 1305–1309. [Google Scholar] [CrossRef] [Green Version]
- Ge, Z.; Guo, X.; Li, J.; Hartman, M.; Kawasawa, Y.I.; Dovat, S.; Song, C. Clinical significance of high c-MYC and low MYCBP2 expression and their association with Ikaros dysfunction in adult acute lymphoblastic leukemia. Oncotarget 2015, 6, 42300–42311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, C.; Li, Z.; Erbe, A.K.; Savic, A.; Dovat, S. Regulation of Ikaros function by casein kinase 2 and protein phosphatase 1. World J. Biol. Chem. 2011, 2, 126–131. [Google Scholar] [CrossRef]
- Ge, Z.; Gu, Y.; Zhao, G.; Li, J.; Chen, B.; Han, Q.; Guo, X.; Liu, J.; Li, H.; Yu, M.D.; et al. High CRLF2 expression associates with IKZF1 dysfunction in adult acute lymphoblastic leukemia without CRLF2 rearrangement. Oncotarget 2016, 7, 49722–49732. [Google Scholar] [CrossRef] [Green Version]
- Leong, W.Z.; Tan, S.H.; Ngoc, P.C.T.; Amanda, S.; Yam, A.W.Y.; Liau, W.S.; Gong, Z.; Lawton, L.N.; Tenen, D.G.; Sanda, T. ARID5B as a critical downstream target of the TAL1 complex that activates the oncogenic transcriptional program and promotes T-cell leukemogenesis. Genes Dev. 2017, 31, 2343–2360. [Google Scholar] [CrossRef]
- Lin, C.; Song, W.; Bi, X.; Zhao, J.; Huang, Z.; Li, Z.; Zhou, J.; Cai, J.; Zhao, H. Recent advances in the ARID family: Focusing on roles in human cancer. Onco. Targets Ther. 2014, 7, 315–324. [Google Scholar] [CrossRef] [Green Version]
- Wilsker, D.; Patsialou, A.; Dallas, P.B.; Moran, E. ARID proteins: A diverse family of DNA binding proteins implicated in the control of cell growth, differentiation, and development. Cell Growth Differ. 2002, 13, 95–106. [Google Scholar]
- Whitson, R.H.; Huang, T.; Itakura, K. The novel Mrf-2 DNA-binding domain recognizes a five-base core sequence through major and minor-groove contacts. Biochem. Biophys Res. Commun. 1999, 258, 326–331. [Google Scholar] [CrossRef] [PubMed]
- Lahoud, M.H.; Ristevski, S.; Venter, D.J.; Jermiin, L.S.; Bertoncello, I.; Zavarsek, S.; Hasthorpe, S.; Drago, J.; de Kretser, D.; Hertzog, P.J.; et al. Gene targeting of Desrt, a novel ARID class DNA-binding protein, causes growth retardation and abnormal development of reproductive organs. Genome Res. 2001, 11, 1327–1334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baba, A.; Ohtake, F.; Okuno, Y.; Yokota, K.; Okada, M.; Imai, Y.; Ni, M.; Meyer, C.A.; Igarashi, K.; Kanno, J.; et al. PKA-dependent regulation of the histone lysine demethylase complex PHF2-ARID5B. Nat. Cell Biol. 2011, 13, 668–675. [Google Scholar] [CrossRef] [PubMed]
- Hata, K.; Takashima, R.; Amano, K.; Ono, K.; Nakanishi, M.; Yoshida, M.; Wakabayashi, M.; Matsuda, A.; Maeda, Y.; Suzuki, Y.; et al. Arid5b facilitates chondrogenesis by recruiting the histone demethylase Phf2 to Sox9-regulated genes. Nat. Commun. 2013, 4, 2850. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joshi, P.; Greco, T.M.; Guise, A.J.; Luo, Y.; Yu, F.; Nesvizhskii, A.I.; Cristea, I.M. The functional interactome landscape of the human histone deacetylase family. Mol. Syst. Biol. 2013, 9, 672. [Google Scholar] [CrossRef] [Green Version]
- Papaemmanuil, E.; Hosking, F.J.; Vijayakrishnan, J.; Price, A.; Olver, B.; Sheridan, E.; Kinsey, S.E.; Lightfoot, T.; Roman, E.; Irving, J.A.; et al. Loci on 7p12.2, 10q21.2 and 14q11.2 are associated with risk of childhood acute lymphoblastic leukemia. Nat. Genet. 2009, 41, 1006–1010. [Google Scholar] [CrossRef] [Green Version]
- Trevino, L.R.; Yang, W.; French, D.; Hunger, S.P.; Carroll, W.L.; Devidas, M.; Willman, C.; Neale, G.; Downing, J.; Raimondi, S.C.; et al. Germline genomic variants associated with childhood acute lymphoblastic leukemia. Nat. Genet. 2009, 41, 1001–1005. [Google Scholar] [CrossRef] [Green Version]
- Barrena, S.; Almeida, J.; Yunta, M.; Lopez, A.; Fernandez-Mosteirin, N.; Giralt, M.; Romero, M.; Perdiguer, L.; Delgado, M.; Orfao, A.; et al. Aberrant expression of tetraspanin molecules in B-cell chronic lymphoproliferative disorders and its correlation with normal B-cell maturation. Leukemia 2005, 19, 1376–1383. [Google Scholar] [CrossRef] [Green Version]
- Rudant, J.; Orsi, L.; Bonaventure, A.; Goujon-Bellec, S.; Baruchel, A.; Petit, A.; Bertrand, Y.; Nelken, B.; Pasquet, M.; Michel, G.; et al. ARID5B, IKZF1 and non-genetic factors in the etiology of childhood acute lymphoblastic leukemia: The ESCALE study. PLoS ONE 2015, 10, e0121348. [Google Scholar] [CrossRef] [Green Version]
- Evans, T.J.; Milne, E.; Anderson, D.; de Klerk, N.H.; Jamieson, S.E.; Talseth-Palmer, B.A.; Bowden, N.A.; Holliday, E.G.; Rudant, J.; Orsi, L.; et al. Confirmation of childhood acute lymphoblastic leukemia variants, ARID5B and IKZF1, and interaction with parental environmental exposures. PLoS ONE 2014, 9, e110255. [Google Scholar] [CrossRef] [Green Version]
- Rudant, J.; Orsi, L.; Bonaventure, A.; Goujon-Bellec, S.; Corda, E.; Baruchel, A.; Bertrand, Y.; Nelken, B.; Robert, A.; Michel, G.; et al. Are ARID5B and IKZF1 polymorphisms also associated with childhood acute myeloblastic leukemia: The ESCALE study (SFCE)? Leukemia 2013, 27, 746–748. [Google Scholar] [CrossRef] [PubMed]
- Linabery, A.M.; Blommer, C.N.; Spector, L.G.; Davies, S.M.; Robison, L.L.; Ross, J.A. ARID5B and IKZF1 variants, selected demographic factors, and childhood acute lymphoblastic leukemia: A report from the Children’s Oncology Group. Leuk. Res. 2013, 37, 936–942. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peyrouze, P.; Guihard, S.; Grardel, N.; Berthon, C.; Pottier, N.; Pigneux, A.; Cahn, J.Y.; Bene, M.C.; Lheritier, V.; Delabesse, E.; et al. Genetic polymorphisms in ARID5B, CEBPE, IKZF1 and CDKN2A in relation with risk of acute lymphoblastic leukaemia in adults: A Group for Research on Adult Acute Lymphoblastic Leukaemia (GRAALL) study. Br. J. Haematol. 2012, 159, 599–602. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Chen, J.; Li, J.; Deng, J.; Rui, Y.; Lu, Q.; Wang, M.; Tong, N.; Zhang, Z.; Fang, Y. Association of three polymorphisms in ARID5B, IKZF1 and CEBPE with the risk of childhood acute lymphoblastic leukemia in a Chinese population. Gene 2013, 524, 203–207. [Google Scholar] [CrossRef] [PubMed]
- Bhandari, P.; Ahmad, F.; Mandava, S.; Das, B.R. Association of Genetic Variants in ARID5B, IKZF1 and CEBPE with Risk of Childhood de novo B-Lineage Acute Lymphoblastic Leukemia in India. Asian Pac. J. Cancer Prev. 2016, 17, 3989–3995. [Google Scholar]
- Gharbi, H.; Ben Hassine, I.; Soltani, I.; Safra, I.; Ouerhani, S.; Bel Haj Othmen, H.; Teber, M.; Farah, A.; Amouri, H.; Toumi, N.H.; et al. Association of genetic variation in IKZF1, ARID5B, CDKN2A, and CEBPE with the risk of acute lymphoblastic leukemia in Tunisian children and their contribution to racial differences in leukemia incidence. Pediatr. Hematol. Oncol. 2016, 33, 157–167. [Google Scholar] [CrossRef]
- Hsu, L.I.; Chokkalingam, A.P.; Briggs, F.B.; Walsh, K.; Crouse, V.; Fu, C.; Metayer, C.; Wiemels, J.L.; Barcellos, L.F.; Buffler, P.A. Association of genetic variation in IKZF1, ARID5B, and CEBPE and surrogates for early-life infections with the risk of acute lymphoblastic leukemia in Hispanic children. Cancer Causes. Control. 2015, 26, 609–619. [Google Scholar] [CrossRef] [Green Version]
- Burmeister, T.; Bartels, G.; Groger, D.; Trautmann, H.; Schwartz, S.; Lenz, K.; Tietze-Burger, C.; Viardot, A.; Wasch, R.; Horst, H.A.; et al. Germline variants in IKZF1, ARID5B, and CEBPE as risk factors for adult-onset acute lymphoblastic leukemia: An analysis from the GMALL study group. Haematologica 2014, 99, e23–e25. [Google Scholar] [CrossRef]
- Bartram, T.; Burkhardt, B.; Wossmann, W.; Seidemann, K.; Zimmermann, M.; Cario, G.; Lisfeld, J.; Ellinghaus, E.; Franke, A.; Houlston, R.S.; et al. Childhood acute lymphoblastic leukemia-associated risk-loci IKZF1, ARID5B and CEBPE and risk of pediatric non-Hodgkin lymphoma: A report from the Berlin-Frankfurt-Munster Study Group. Leuk. Lymphoma 2015, 56, 814–816. [Google Scholar] [CrossRef]
- Lin, C.Y.; Li, M.J.; Chang, J.G.; Liu, S.C.; Weng, T.; Wu, K.H.; Yang, S.F.; Huang, F.K.; Lo, W.Y.; Peng, C.T. High-resolution melting analyses for genetic variants in ARID5B and IKZF1 with childhood acute lymphoblastic leukemia susceptibility loci in Taiwan. Blood Cells Mol. Dis. 2014, 52, 140–145. [Google Scholar] [CrossRef]
- Pastorczak, A.; Gorniak, P.; Sherborne, A.; Hosking, F.; Trelinska, J.; Lejman, M.; Szczepanski, T.; Borowiec, M.; Fendler, W.; Kowalczyk, J.; et al. Role of 657del5 NBN mutation and 7p12.2 (IKZF1), 9p21 (CDKN2A), 10q21.2 (ARID5B) and 14q11.2 (CEBPE) variation and risk of childhood ALL in the Polish population. Leuk. Res. 2011, 35, 1534–1536. [Google Scholar] [CrossRef] [PubMed]
- Ge, Z.; Han, Q.; Gu, Y.; Ge, Q.; Ma, J.; Sloane, J.; Gao, G.; Payne, K.J.; Szekely, L.; Song, C.; et al. Aberrant ARID5B expression and its association with Ikaros dysfunction in acute lymphoblastic leukemia. Oncogenesis 2018, 7, 84. [Google Scholar] [CrossRef] [PubMed]
- Guo, Q.; Xie, J.; Dang, C.V.; Liu, E.T.; Bishop, J.M. Identification of a large Myc-binding protein that contains RCC1-like repeats. Proc. Natl. Acad. Sci. USA 1998, 95, 9172–9177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ehnert, C.; Tegeder, I.; Pierre, S.; Birod, K.; Nguyen, H.V.; Schmidtko, A.; Geisslinger, G.; Scholich, K. Protein associated with Myc (PAM) is involved in spinal nociceptive processing. J. Neurochem. 2004, 88, 948–957. [Google Scholar] [CrossRef] [PubMed]
- Maeurer, C.; Holland, S.; Pierre, S.; Potstada, W.; Scholich, K. Sphingosine-1-phosphate induced mTOR-activation is mediated by the E3-ubiquitin ligase PAM. Cell Signal. 2009, 21, 293–300. [Google Scholar] [CrossRef] [PubMed]
- Murthy, V.; Han, S.; Beauchamp, R.L.; Smith, N.; Haddad, L.A.; Ito, N.; Ramesh, V. Pam and its ortholog highwire interact with and may negatively regulate the TSC1.TSC2 complex. J. Biol. Chem. 2004, 279, 1351–1358. [Google Scholar] [CrossRef] [Green Version]
- Pierre, S.C.; Häusler, J.; Birod, K.; Geisslinger, G.; Scholich, K. PAM mediates sustained inhibition of cAMP signaling by sphingosine-1-phosphate. EMBO J. 2004, 23, 3031–3040. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bredrup, C.; Johansson, S.; Bindoff, L.A.; Sztromwasser, P.; Kråkenes, J.; Mellgren, A.E.; Brurås, K.R.; Lind, O.; Boman, H.; Knappskog, P.M.; et al. High myopia-excavated optic disc anomaly associated with a frameshift mutation in the MYC-binding protein 2 gene (MYCBP2). Am. J. Ophthalmol. 2015, 159, 973–979. [Google Scholar] [CrossRef]
- Ye, B.H.; Chaganti, S.; Chang, C.C.; Niu, H.; Corradini, P.; Chaganti, R.S.; Dalla-Favera, R. Chromosomal translocations cause deregulated BCL6 expression by promoter substitution in B cell lymphoma. EMBO J. 1995, 14, 6209–6217. [Google Scholar] [CrossRef]
- Dent, A.L.; Vasanwala, F.H.; Toney, L.M. Regulation of gene expression by the proto-oncogene BCL-6. Crit. Rev. Oncol. Hematol. 2002, 41, 1–9. [Google Scholar] [CrossRef]
- Ye, B.H.; Cattoretti, G.; Shen, Q.; Zhang, J.; Hawe, N.; de Waard, R.; Leung, C.; Nouri-Shirazi, M.; Orazi, A.; Chaganti, R.S.; et al. The BCL-6 proto-oncogene controls germinal-centre formation and Th2-type inflammation. Nat. Genet 1997, 16, 161–170. [Google Scholar] [CrossRef] [PubMed]
- Mascle, X.; Albagli, O.; Lemercier, C. Point mutations in BCL6 DNA-binding domain reveal distinct roles for the six zinc fingers. Biochem. Biophys Res. Commun. 2003, 300, 391–396. [Google Scholar] [CrossRef] [Green Version]
- Hurtz, C.; Hatzi, K.; Cerchietti, L.; Braig, M.; Park, E.; Kim, Y.M.; Herzog, S.; Ramezani-Rad, P.; Jumaa, H.; Müller, M.C.; et al. BCL6-mediated repression of p53 is critical for leukemia stem cell survival in chronic myeloid leukemia. J. Exp. Med. 2011, 208, 2163–2174. [Google Scholar] [CrossRef] [PubMed]
- Baron, B.W.; Anastasi, J.; Thirman, M.J.; Furukawa, Y.; Fears, S.; Kim, D.C.; Simone, F.; Birkenbach, M.; Montag, A.; Sadhu, A.; et al. The human programmed cell death-2 (PDCD2) gene is a target of BCL6 repression: Implications for a role of BCL6 in the down-regulation of apoptosis. Proc. Natl. Acad. Sci. USA 2002, 99, 2860–2865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, H.B.; Lv, W.F.; Wang, Y.X.; Li, Y.Y.; Guo, W. BCL6 promotes the methotrexate-resistance by upregulating ZEB1 expression in children with acute B lymphocytic leukemia. Eur. Rev. Med. Pharmacol. Sci. 2018, 22, 5240–5247. [Google Scholar] [CrossRef]
- Duy, C.; Hurtz, C.; Shojaee, S.; Cerchietti, L.; Geng, H.; Swaminathan, S.; Klemm, L.; Kweon, S.M.; Nahar, R.; Braig, M.; et al. BCL6 enables Ph+ acute lymphoblastic leukaemia cells to survive BCR-ABL1 kinase inhibition. Nature 2011, 473, 384–388. [Google Scholar] [CrossRef]
- Muto, A.; Tashiro, S.; Nakajima, O.; Hoshino, H.; Takahashi, S.; Sakoda, E.; Ikebe, D.; Yamamoto, M.; Igarashi, K. The transcriptional programme of antibody class switching involves the repressor Bach2. Nature 2004, 429, 566–571. [Google Scholar] [CrossRef] [Green Version]
- Ye, B.H.; Mai, Y. A Bach2 link between pre-B cell receptor checkpoint and pre-B cell ALL. Cancer Cell 2013, 24, 282–284. [Google Scholar] [CrossRef] [Green Version]
- Nutt, S.L.; Taubenheim, N.; Hasbold, J.; Corcoran, L.M.; Hodgkin, P.D. The genetic network controlling plasma cell differentiation. Semin. Immunol. 2011, 23, 341–349. [Google Scholar] [CrossRef]
- Dave, S.S. The polyphony of BACH2. Blood 2014, 123, 950. [Google Scholar] [CrossRef] [Green Version]
- Muto, A.; Hoshino, H.; Madisen, L.; Yanai, N.; Obinata, M.; Karasuyama, H.; Hayashi, N.; Nakauchi, H.; Yamamoto, M.; Groudine, M.; et al. Identification of Bach2 as a B-cell-specific partner for small maf proteins that negatively regulate the immunoglobulin heavy chain gene 3′ enhancer. EMBO J. 1998, 17, 5734–5743. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swaminathan, S.; Huang, C.; Geng, H.; Chen, Z.; Harvey, R.; Kang, H.; Ng, C.; Titz, B.; Hurtz, C.; Sadiyah, M.F.; et al. BACH2 mediates negative selection and p53-dependent tumor suppression at the pre-B cell receptor checkpoint. Nat. Med. 2013, 19, 1014–1022. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Casolari, D.A.; Makri, M.; Yoshida, C.; Muto, A.; Igarashi, K.; Melo, J.V. Transcriptional suppression of BACH2 by the Bcr-Abl oncoprotein is mediated by PAX5. Leukemia 2013, 27, 409–415. [Google Scholar] [CrossRef] [PubMed]
- Merup, M.; Moreno, T.C.; Heyman, M.; Rönnberg, K.; Grandér, D.; Detlofsson, R.; Rasool, O.; Liu, Y.; Söderhäll, S.; Juliusson, G.; et al. 6q deletions in acute lymphoblastic leukemia and non-Hodgkin’s lymphomas. Blood 1998, 91, 3397–3400. [Google Scholar] [CrossRef] [PubMed]
- Takakuwa, T.; Luo, W.J.; Ham, M.F.; Sakane-Ishikawa, F.; Wada, N.; Aozasa, K. Integration of Epstein-Barr virus into chromosome 6q15 of Burkitt lymphoma cell line (Raji) induces loss of BACH2 expression. Am. J. Pathol. 2004, 164, 967–974. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Sørensen, A.B.; Wang, B.; Wabl, M.; Nielsen, A.L.; Pedersen, F.S. Identification of novel Bach2 transcripts and protein isoforms through tagging analysis of retroviral integrations in B-cell lymphomas. BMC Mol. Biol. 2009, 10, 2. [Google Scholar] [CrossRef] [Green Version]
- Rasmussen, M.H.; Ballarín-González, B.; Liu, J.; Lassen, L.B.; Füchtbauer, A.; Füchtbauer, E.M.; Nielsen, A.L.; Pedersen, F.S. Antisense transcription in gammaretroviruses as a mechanism of insertional activation of host genes. J. Virol. 2010, 84, 3780–3788. [Google Scholar] [CrossRef] [Green Version]
- Vassiliou, G.S.; Cooper, J.L.; Rad, R.; Li, J.; Rice, S.; Uren, A.; Rad, L.; Ellis, P.; Andrews, R.; Banerjee, R.; et al. Mutant nucleophosmin and cooperating pathways drive leukemia initiation and progression in mice. Nat. Genet. 2011, 43, 470–475. [Google Scholar] [CrossRef]
- Swaminathan, S.; Duy, C.; Müschen, M. BACH2-BCL6 balance regulates selection at the pre-B cell receptor checkpoint. Trends Immunol. 2014, 35, 131–137. [Google Scholar] [CrossRef] [Green Version]
- Ge, Z.; Zhou, X.; Gu, Y.; Han, Q.; Li, J.; Chen, B.; Ge, Q.; Dovat, E.; Payne, J.L.; Sun, T.; et al. Ikaros regulation of the BCL6/BACH2 axis and its clinical relevance in acute lymphoblastic leukemia. Oncotarget 2017, 8, 8022–8034. [Google Scholar] [CrossRef] [Green Version]
- Shi, D.; Xie, D.; Zhang, H.; Zhao, H.; Huang, J.; Li, C.; Liu, Y.; Lv, F.; The, E.; Yuan, T.; et al. Reduction in dynamin-2 is implicated in ischaemic cardiac arrhythmias. J. Cell Mol. Med. 2014, 18, 1992–1999. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Nolan, M.; Yamada, H.; Watanabe, M.; Nasu, Y.; Takei, K.; Takeda, T. Dynamin2 GTPase contributes to invadopodia formation in invasive bladder cancer cells. Biochem. Biophys Res. Commun. 2016, 480, 409–414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Xu, L.; Ye, J.; Li, X.; Zhang, D.; Liang, D.; Xu, X.; Qi, M.; Li, C.; Zhang, H.; et al. Aberrant dynamin 2-dependent Na(+)/H(+) exchanger-1 trafficking contributes to cardiomyocyte apoptosis. J. Cell Mol. Med. 2013, 17, 1119–1127. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Zhang, D.S.; Ye, J.C.; Li, C.M.; Qi, M.; Liang, D.D.; Xu, X.R.; Xu, L.; Liu, Y.; Zhang, H.; et al. Dynamin-2 mediates heart failure by modulating Ca2+ -dependent cardiomyocyte apoptosis. Int. J. Cardiol. 2013, 168, 2109–2119. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Ding, L.; Holmfeldt, L.; Wu, G.; Heatley, S.L.; Payne-Turner, D.; Easton, J.; Chen, X.; Wang, J.; Rusch, M.; et al. The genetic basis of early T-cell precursor acute lymphoblastic leukaemia. Nature 2012, 481, 157–163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neumann, M.; Heesch, S.; Schlee, C.; Schwartz, S.; Gökbuget, N.; Hoelzer, D.; Konstandin, N.P.; Ksienzyk, B.; Vosberg, S.; Graf, A.; et al. Whole-exome sequencing in adult ETP-ALL reveals a high rate of DNMT3A mutations. Blood 2013, 121, 4749–4752. [Google Scholar] [CrossRef]
- Ge, Z.; Li, M.; Zhao, G.; Xiao, L.; Gu, Y.; Zhou, X.; Yu, M.D.; Li, J.; Dovat, S.; Song, C. Novel dynamin 2 mutations in adult T-cell acute lymphoblastic leukemia. Oncol. Lett. 2016, 12, 2746–2751. [Google Scholar] [CrossRef] [Green Version]
- Ge, Z.; Gu, Y.; Han, Q.; Zhao, G.; Li, M.; Li, J.; Chen, B.; Sun, T.; Dovat, S.; Gale, R.P.; et al. Targeting High Dynamin-2 (DNM2) Expression by Restoring Ikaros Function in Acute Lymphoblastic Leukemia. Sci Rep 2016, 6, 38004. [Google Scholar] [CrossRef] [Green Version]
- Neumann, M.; Vosberg, S.; Schlee, C.; Heesch, S.; Schwartz, S.; Gökbuget, N.; Hoelzer, D.; Graf, A.; Krebs, S.; Bartram, I.; et al. Mutational spectrum of adult T-ALL. Oncotarget 2015, 6, 2754–2766. [Google Scholar] [CrossRef] [Green Version]
- Le Borgne, R.; Bardin, A.; Schweisguth, F. The roles of receptor and ligand endocytosis in regulating Notch signaling. Development 2005, 132, 1751–1762. [Google Scholar] [CrossRef] [Green Version]
- Auer, F.; Ingenhag, D.; Pinkert, S.; Kracker, S.; Hacein-Bey-Abina, S.; Cavazzana, M.; Gombert, M.; Martin-Lorenzo, A.; Lin, M.H.; Vicente-Dueñas, C.; et al. Activation-induced cytidine deaminase prevents pro-B cell acute lymphoblastic leukemia by functioning as a negative regulator in Rag1 deficient pro-B cells. Oncotarget 2017, 8, 75797–75807. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tremblay, C.S.; Brown, F.C.; Collett, M.; Saw, J.; Chiu, S.K.; Sonderegger, S.E.; Lucas, S.E.; Alserihi, R.; Chau, N.; Toribio, M.L.; et al. Loss-of-function mutations of Dynamin 2 promote T-ALL by enhancing IL-7 signalling. Leukemia 2016, 30, 1993–2001. [Google Scholar] [CrossRef] [PubMed]
- Noronha, E.P.; Marques, L.V.C.; Andrade, F.G.; Sardou-Cezar, I.; Dos Santos-Bueno, F.V.; Zampier, C.D.P.; Terra-Granado, E.; Pombo-de-Oliveira, M.S. T-lymphoid/myeloid mixed phenotype acute leukemia and early T-cell precursor lymphoblastic leukemia similarities with. Cancer Manag. Res. 2019, 11, 3933–3943. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bazdar, D.A.; Kalinowska, M.; Panigrahi, S.; Sieg, S.F. Recycled IL-7 Can Be Delivered to Neighboring T Cells. J. Immunol. 2015, 194, 4698–4704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ko, R.H.; Ji, L.; Barnette, P.; Bostrom, B.; Hutchinson, R.; Raetz, E.; Seibel, N.L.; Twist, C.J.; Eckroth, E.; Sposto, R.; et al. Outcome of patients treated for relapsed or refractory acute lymphoblastic leukemia: A Therapeutic Advances in Childhood Leukemia Consortium study. J. Clin. Oncol. 2010, 28, 648–654. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raetz, E.A.; Bhatla, T. Where do we stand in the treatment of relapsed acute lymphoblastic leukemia? Hematol. Am. Soc. Hematol. Educ. Program 2012, 2012, 129–136. [Google Scholar] [CrossRef] [Green Version]
- Ziegler, S.E.; Morella, K.K.; Anderson, D.; Kumaki, N.; Leonard, W.J.; Cosman, D.; Baumann, H. Reconstitution of a functional interleukin (IL)-7 receptor demonstrates that the IL-2 receptor gamma chain is required for IL-7 signal transduction. Eur. J. Immunol. 1995, 25, 399–404. [Google Scholar] [CrossRef]
- Roberts, K.G.; Yang, Y.L.; Payne-Turner, D.; Lin, W.; Files, J.K.; Dickerson, K.; Gu, Z.; Taunton, J.; Janke, L.J.; Chen, T.; et al. Oncogenic role and therapeutic targeting of ABL-class and JAK-STAT activating kinase alterations in Ph-like ALL. Blood Adv. 2017, 1, 1657–1671. [Google Scholar] [CrossRef]
- Kondo, M.; Takeshita, T.; Higuchi, M.; Nakamura, M.; Sudo, T.; Nishikawa, S.; Sugamura, K. Functional participation of the IL-2 receptor gamma chain in IL-7 receptor complexes. Science 1994, 263, 1453–1454. [Google Scholar] [CrossRef]
- Noguchi, M.; Nakamura, Y.; Russell, S.M.; Ziegler, S.F.; Tsang, M.; Cao, X.; Leonard, W.J. Interleukin-2 receptor gamma chain: A functional component of the interleukin-7 receptor. Science 1993, 262, 1877–1880. [Google Scholar] [CrossRef] [Green Version]
- Peschon, J.J.; Morrissey, P.J.; Grabstein, K.H.; Ramsdell, F.J.; Maraskovsky, E.; Gliniak, B.C.; Park, L.S.; Ziegler, S.F.; Williams, D.E.; Ware, C.B.; et al. Early lymphocyte expansion is severely impaired in interleukin 7 receptor-deficient mice. J. Exp. Med. 1994, 180, 1955–1960. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oliveira, M.L.; Akkapeddi, P.; Ribeiro, D.; Melão, A.; Barata, J.T. IL-7R-mediated signaling in T-cell acute lymphoblastic leukemia: An update. Adv. Biol. Regul. 2019, 71, 88–96. [Google Scholar] [CrossRef] [PubMed]
- Gianfelici, V.; Messina, M.; Paoloni, F.; Peragine, N.; Lauretti, A.; Fedullo, A.L.; Di Giacomo, F.; Vignetti, M.; Vitale, A.; Guarini, A.; et al. IL7R overexpression in adult acute lymphoblastic leukemia is associated to JAK/STAT pathway mutations and identifies patients who could benefit from targeted therapies. Leuk. Lymphoma 2019, 60, 829–832. [Google Scholar] [CrossRef] [PubMed]
- Shochat, C.; Tal, N.; Gryshkova, V.; Birger, Y.; Bandapalli, O.R.; Cazzaniga, G.; Gershman, N.; Kulozik, A.E.; Biondi, A.; Mansour, M.R.; et al. Novel activating mutations lacking cysteine in type I cytokine receptors in acute lymphoblastic leukemia. Blood 2014, 124, 106–110. [Google Scholar] [CrossRef] [Green Version]
- Zenatti, P.P.; Ribeiro, D.; Li, W.; Zuurbier, L.; Silva, M.C.; Paganin, M.; Tritapoe, J.; Hixon, J.A.; Silveira, A.B.; Cardoso, B.A.; et al. Oncogenic IL7R gain-of-function mutations in childhood T-cell acute lymphoblastic leukemia. Nat. Genet. 2011, 43, 932–939. [Google Scholar] [CrossRef]
- Perez-Garcia, A.; Ambesi-Impiombato, A.; Hadler, M.; Rigo, I.; LeDuc, C.A.; Kelly, K.; Jalas, C.; Paietta, E.; Racevskis, J.; Rowe, J.M.; et al. Genetic loss of SH2B3 in acute lymphoblastic leukemia. Blood 2013, 122, 2425–2432. [Google Scholar] [CrossRef] [Green Version]
- Cheng, Y.; Chikwava, K.; Wu, C.; Zhang, H.; Bhagat, A.; Pei, D.; Choi, J.K.; Tong, W. LNK/SH2B3 regulates IL-7 receptor signaling in normal and malignant B-progenitors. J. Clin. Investig. 2016, 126, 1267–1281. [Google Scholar] [CrossRef] [Green Version]
- Willman, C.L. SH2B3: A new leukemia predisposition gene. Blood 2013, 122, 2293–2295. [Google Scholar] [CrossRef] [Green Version]
- Lin, D.C.; Yin, T.; Koren-Michowitz, M.; Ding, L.W.; Gueller, S.; Gery, S.; Tabayashi, T.; Bergholz, U.; Kazi, J.U.; Rönnstrand, L.; et al. Adaptor protein Lnk binds to and inhibits normal and leukemic FLT3. Blood 2012, 120, 3310–3317. [Google Scholar] [CrossRef] [Green Version]
- Baran-Marszak, F.; Magdoud, H.; Desterke, C.; Alvarado, A.; Roger, C.; Harel, S.; Mazoyer, E.; Cassinat, B.; Chevret, S.; Tonetti, C.; et al. Expression level and differential JAK2-V617F-binding of the adaptor protein Lnk regulates JAK2-mediated signals in myeloproliferative neoplasms. Blood 2010, 116, 5961–5971. [Google Scholar] [CrossRef]
- Jang, W.; Park, J.; Kwon, A.; Choi, H.; Kim, J.; Lee, G.D.; Han, E.; Jekarl, D.W.; Chae, H.; Han, K.; et al. CDKN2B downregulation and other genetic characteristics in T-acute lymphoblastic leukemia. Exp. Mol. Med. 2019, 51, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alexander, T.B.; Gu, Z.; Iacobucci, I.; Dickerson, K.; Choi, J.K.; Xu, B.; Payne-Turner, D.; Yoshihara, H.; Loh, M.L.; Horan, J.; et al. The genetic basis and cell of origin of mixed phenotype acute leukaemia. Nature 2018, 562, 373–379. [Google Scholar] [CrossRef] [PubMed]
- Maslah, N.; Cassinat, B.; Verger, E.; Kiladjian, J.J.; Velazquez, L. The role of LNK/SH2B3 genetic alterations in myeloproliferative neoplasms and other hematological disorders. Leukemia 2017, 31, 1661–1670. [Google Scholar] [CrossRef] [PubMed]
- Ge, Z.; Gu, Y.; Xiao, L.; Han, Q.; Li, J.; Chen, B.; Yu, J.; Kawasawa, Y.I.; Payne, K.J.; Dovat, S.; et al. Co-existence of IL7R high and SH2B3 low expression distinguishes a novel high-risk acute lymphoblastic leukemia with Ikaros dysfunction. Oncotarget 2016, 7, 46014–46027. [Google Scholar] [CrossRef] [Green Version]


© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dhanyamraju, P.K.; Iyer, S.; Smink, G.; Bamme, Y.; Bhadauria, P.; Payne, J.L.; Dovat, E.; Klink, M.; Ding, Y. Transcriptional Regulation of Genes by Ikaros Tumor Suppressor in Acute Lymphoblastic Leukemia. Int. J. Mol. Sci. 2020, 21, 1377. https://doi.org/10.3390/ijms21041377
Dhanyamraju PK, Iyer S, Smink G, Bamme Y, Bhadauria P, Payne JL, Dovat E, Klink M, Ding Y. Transcriptional Regulation of Genes by Ikaros Tumor Suppressor in Acute Lymphoblastic Leukemia. International Journal of Molecular Sciences. 2020; 21(4):1377. https://doi.org/10.3390/ijms21041377
Chicago/Turabian StyleDhanyamraju, Pavan Kumar, Soumya Iyer, Gayle Smink, Yevgeniya Bamme, Preeti Bhadauria, Jonathon L Payne, Elanora Dovat, Morgann Klink, and Yali Ding. 2020. "Transcriptional Regulation of Genes by Ikaros Tumor Suppressor in Acute Lymphoblastic Leukemia" International Journal of Molecular Sciences 21, no. 4: 1377. https://doi.org/10.3390/ijms21041377
APA StyleDhanyamraju, P. K., Iyer, S., Smink, G., Bamme, Y., Bhadauria, P., Payne, J. L., Dovat, E., Klink, M., & Ding, Y. (2020). Transcriptional Regulation of Genes by Ikaros Tumor Suppressor in Acute Lymphoblastic Leukemia. International Journal of Molecular Sciences, 21(4), 1377. https://doi.org/10.3390/ijms21041377