Remote Ischemic Preconditioning Induces Cardioprotective Autophagy and Signals through the IL-6-Dependent JAK-STAT Pathway

 ,
,  and
and
Abstract
:1. Introduction
2. Results
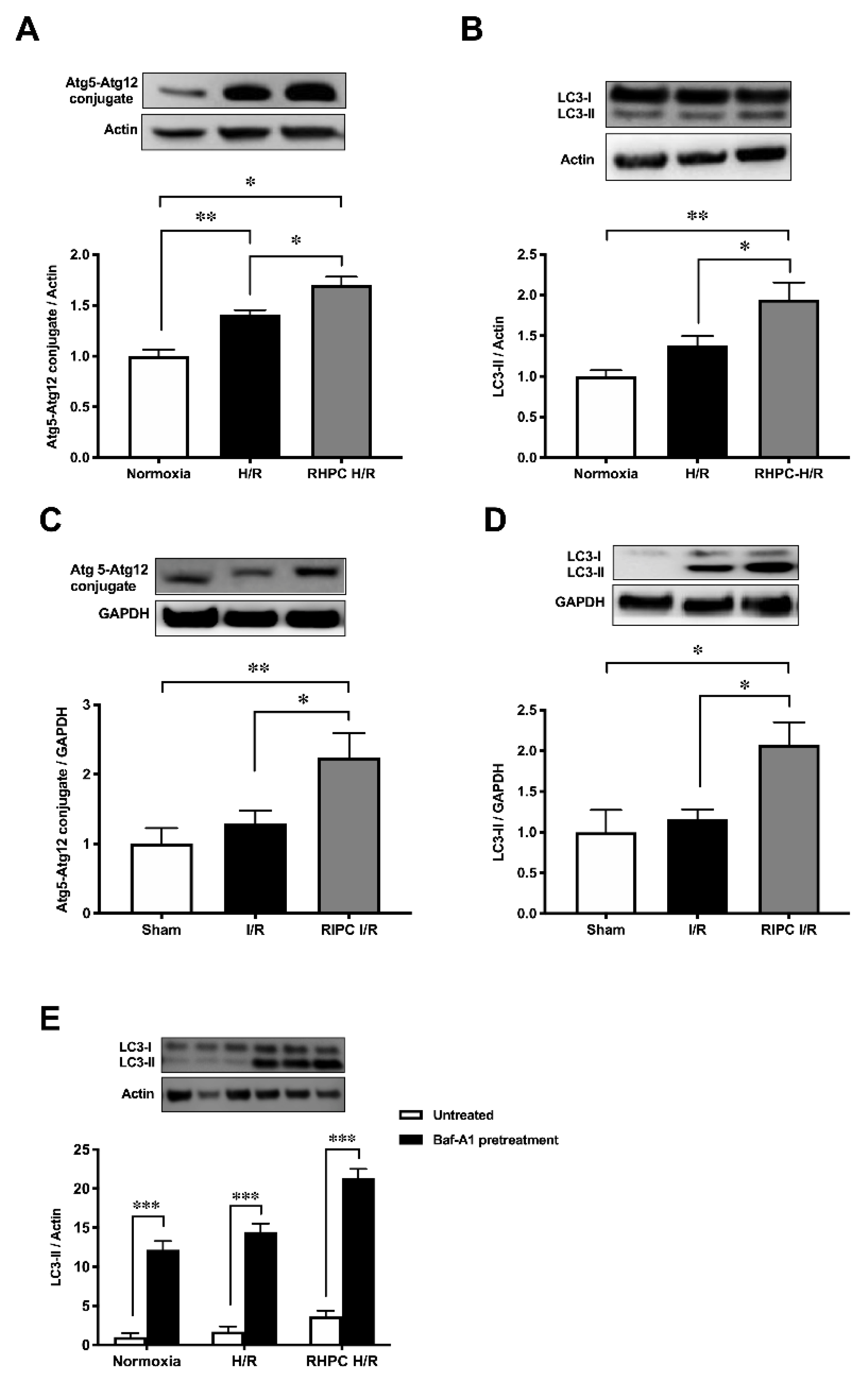
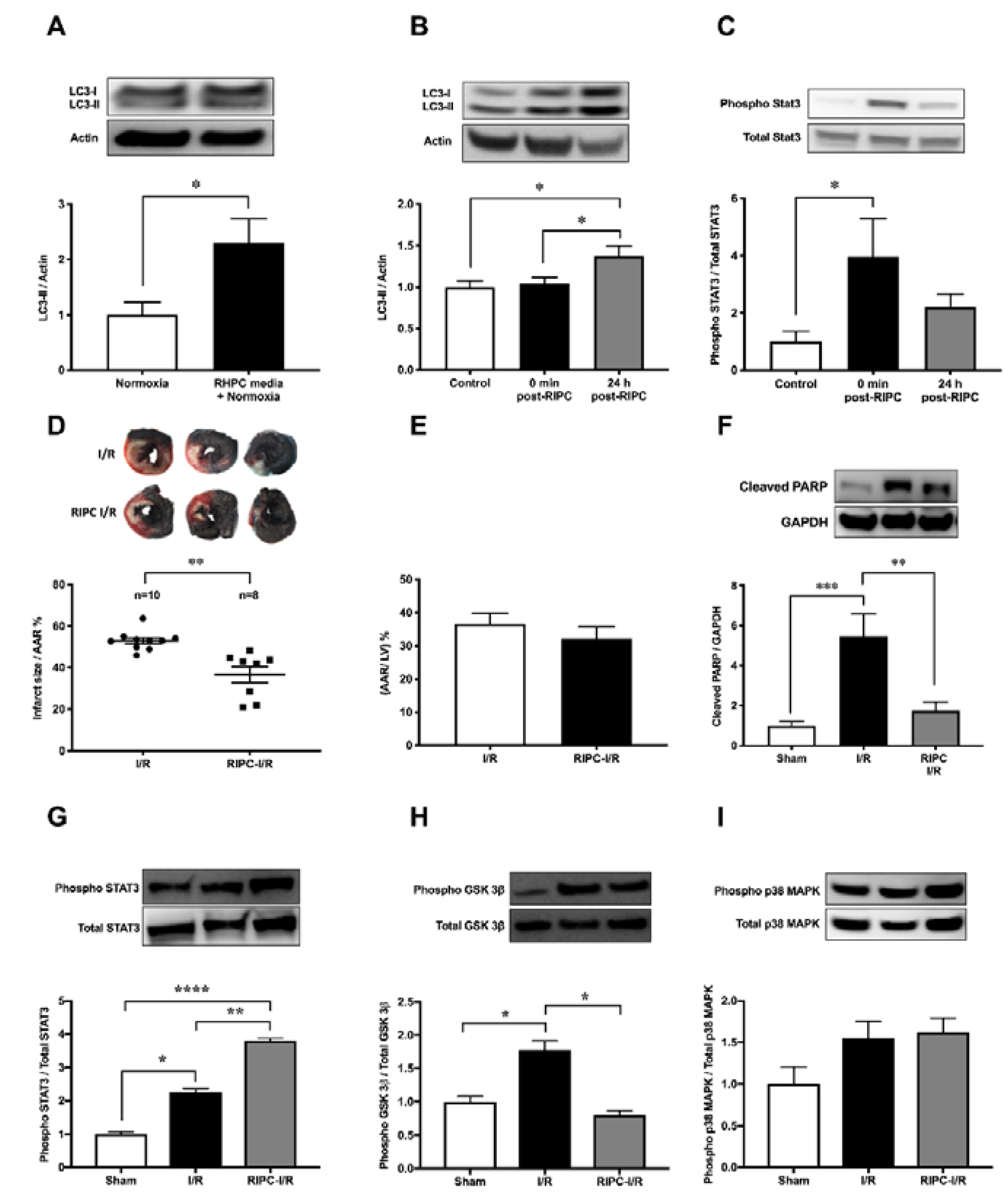
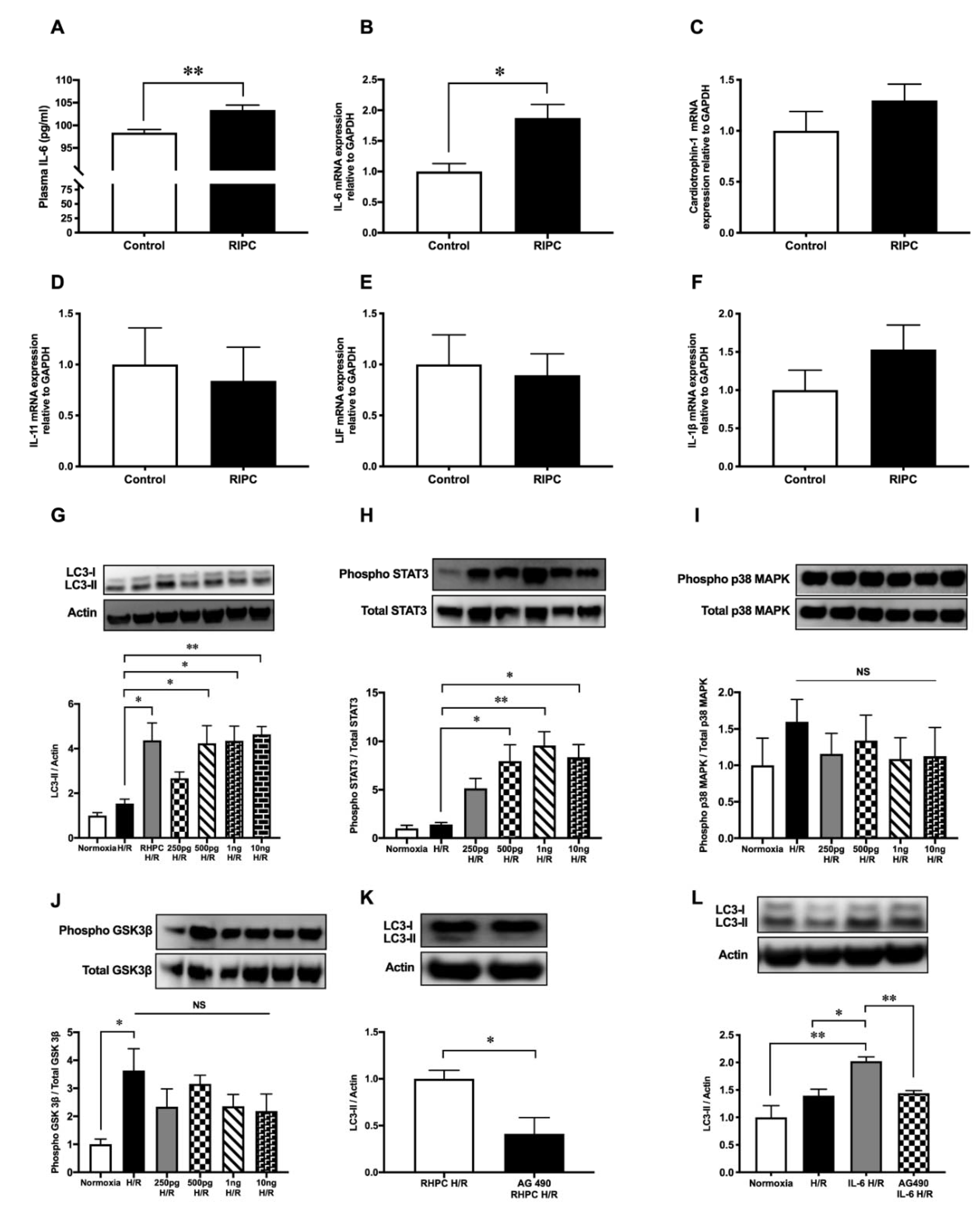
2.1. RIPC Prior to I/R Upregulates Autophagic Activity In Vitro and In Vivo
2.2. Autophagy Functions as a Signaling Mechanism for RIPC and Confers Cardioprotection Against I/R Injury in Rats
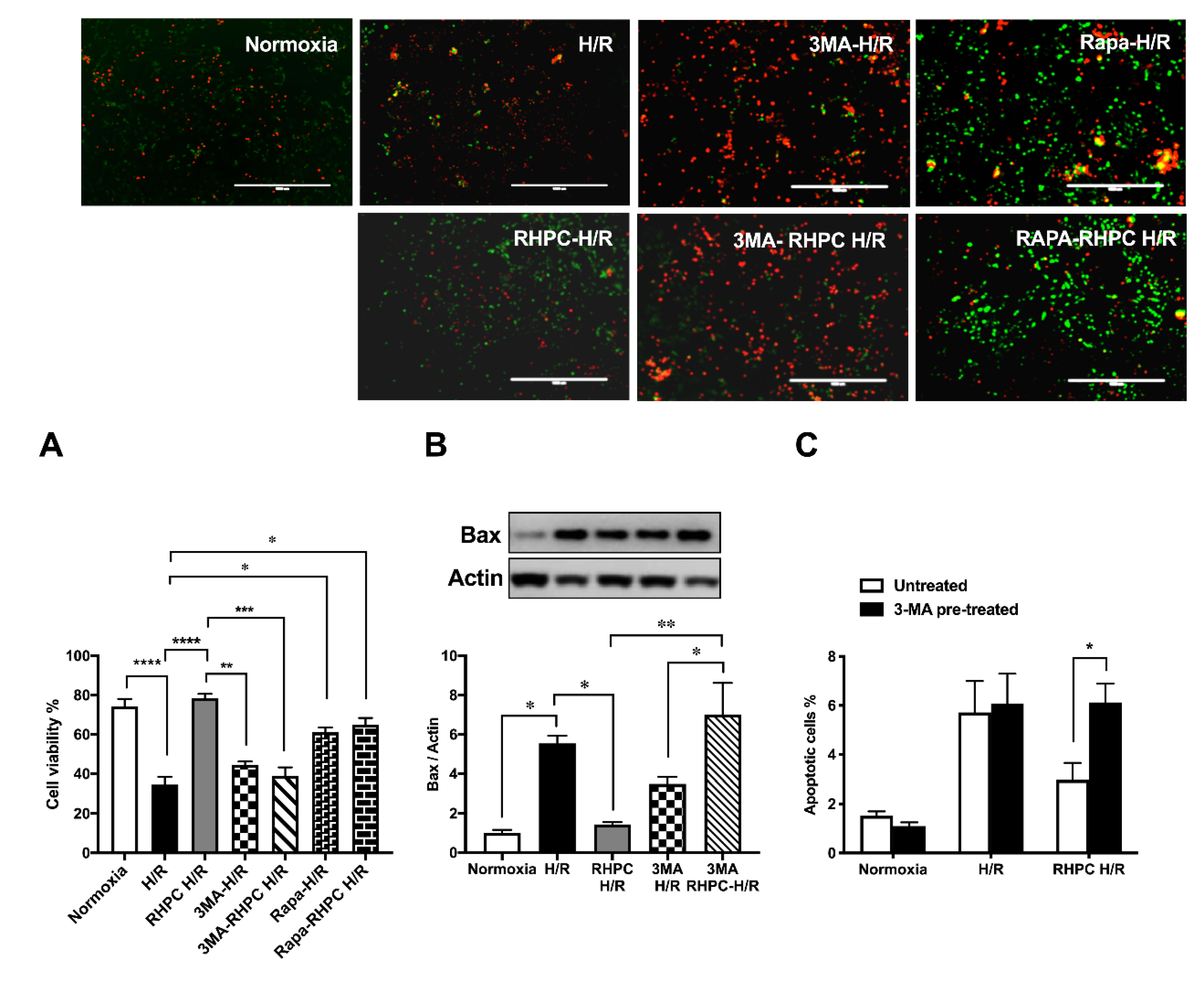
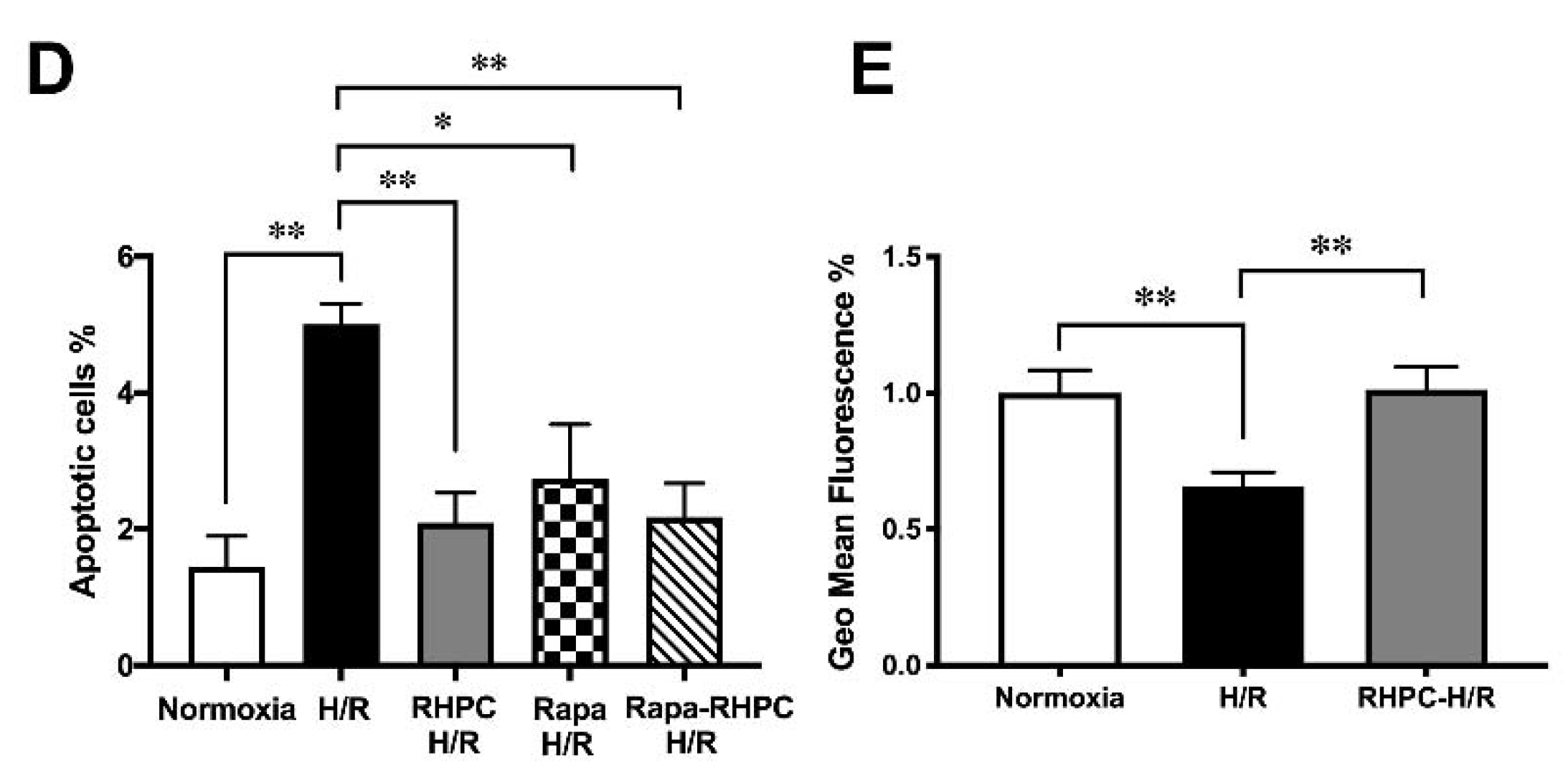
2.3. Autophagy is Essential for RHPC-Induced Cardioprotection In Vitro
2.4. RIPC-Induced Autophagy Regulated by the IL-6-Dependent JAK-STAT Pathway
3. Discussion
4. Study Limitations
5. Materials and Methods
5.1. In Vitro H/R Injury Model
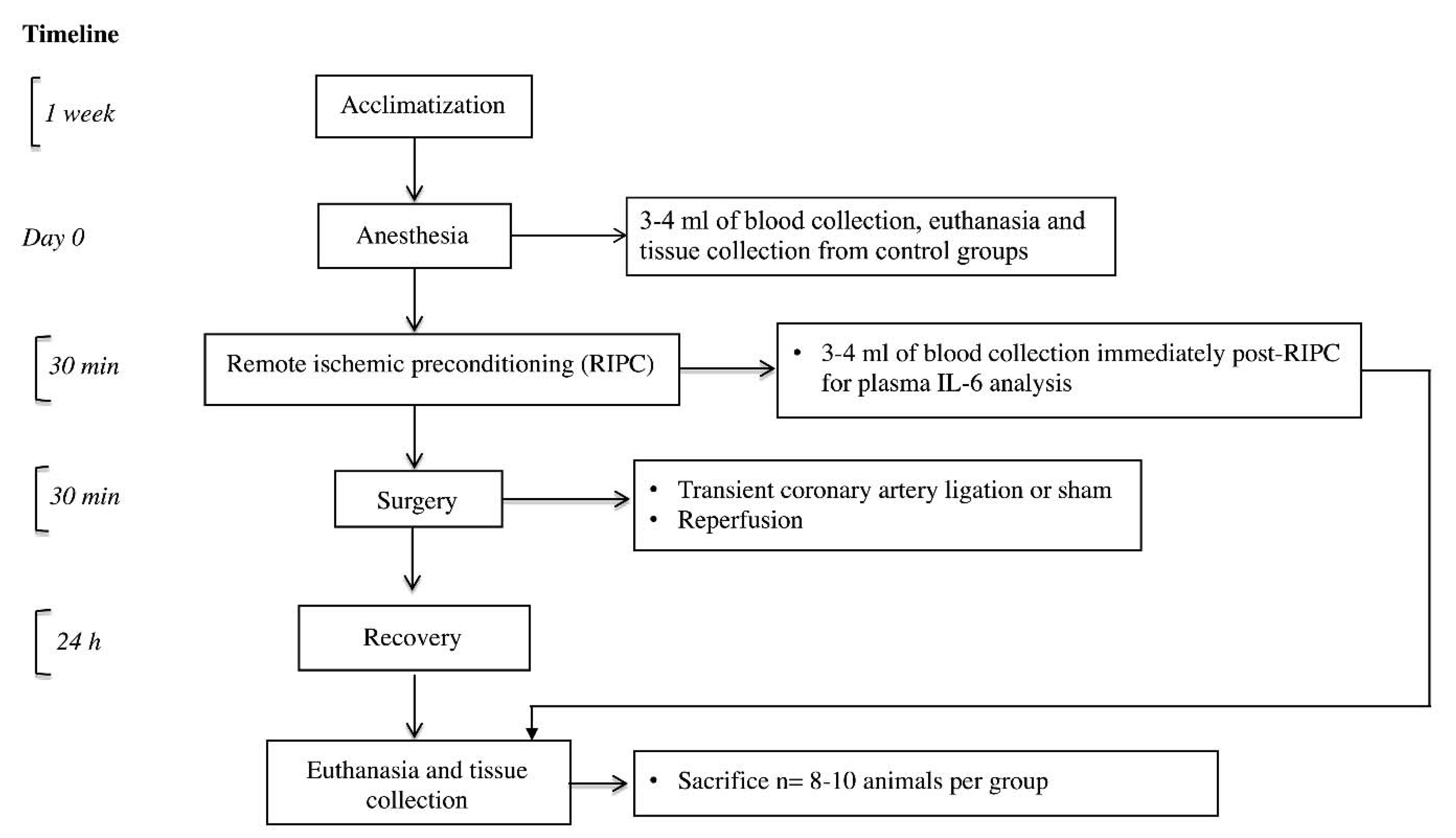
5.2. In Vivo I/R Injury Model
5.3. Measurement of Myocardial Infarct Size and Tissue Collection
5.4. Assessment of Autophagy
5.5. Western Blotting
5.6. Flow Cytometry
5.7. Cell Viability
5.8. Real-Time Polymerase Chain Reaction
5.9. Enzyme-Linked Immunosorbent Assay
5.10. Autophagy Promotion and Inhibition
5.11. Statistical Analysis
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Murry, C.E.; Jennings, R.B.; Reimer, K.A. Preconditioning with ischemia: A delay of lethal cell injury in ischemic myocardium. Circulation 1986, 74, 1124–1136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heusch, G. Molecular basis of cardioprotection: Signal transduction in ischemic pre-, post-, and remote conditioning. Circ. Res. 2015, 116, 674–699. [Google Scholar] [CrossRef] [PubMed]
- Heusch, G. Critical Issues for the Translation of Cardioprotection. Circ. Res. 2017, 120, 1477–1486. [Google Scholar] [CrossRef] [Green Version]
- Kleinbongard, P.; Skyschally, A.; Heusch, G. Cardioprotection by remote ischemic conditioning and its signal transduction. Pflug. Arch. 2017, 469, 159–181. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, A.M.; Ali, G.S.; Tariq, W. Remote ischemic preconditioning is a safe adjuvant technique to myocardial protection but adds no clinical benefit after on-pump coronary artery bypass grafting. Heart Surg Forum 2014, 17, E220–E223. [Google Scholar] [CrossRef]
- Karuppasamy, P.; Chaubey, S.; Dew, T.; Musto, R.; Sherwood, R.; Desai, J.; John, L.; Shah, A.M.; Marber, M.S.; Kunst, G. Remote intermittent ischemia before coronary artery bypass graft surgery: A strategy to reduce injury and inflammation? Basic Res. Cardiol. 2011, 106, 511–519. [Google Scholar] [CrossRef]
- Hong, D.M.; Lee, E.H.; Kim, H.J.; Min, J.J.; Chin, J.H.; Choi, D.K.; Bahk, J.H.; Sim, J.Y.; Choi, I.C.; Jeon, Y. Does remote ischaemic preconditioning with postconditioning improve clinical outcomes of patients undergoing cardiac surgery? Remote Ischaemic Preconditioning with Postconditioning Outcome Trial. Eur. Heart J. 2014, 35, 176–183. [Google Scholar] [CrossRef] [Green Version]
- Lucchinetti, E.; Bestmann, L.; Feng, J.; Freidank, H.; Clanachan, A.S.; Finegan, B.A.; Zaugg, M. Remote ischemic preconditioning applied during isoflurane inhalation provides no benefit to the myocardium of patients undergoing on-pump coronary artery bypass graft surgery: Lack of synergy or evidence of antagonism in cardioprotection? Anesthesiology 2012, 116, 296–310. [Google Scholar] [CrossRef] [Green Version]
- McCrindle, B.W.; Clarizia, N.A.; Khaikin, S.; Holtby, H.M.; Manlhiot, C.; Schwartz, S.M.; Caldarone, C.A.; Coles, J.G.; Van Arsdell, G.S.; Scherer, S.W.; et al. Remote ischemic preconditioning in children undergoing cardiac surgery with cardiopulmonary bypass: A single-center double-blinded randomized trial. J. Am. Heart Assoc. 2014, 3, 000964. [Google Scholar] [CrossRef] [Green Version]
- Botker, H.E.; Lassen, T.R.; Jespersen, N.R. Clinical translation of myocardial conditioning. Am. J. Physiol. Heart Circ. Physiol. 2018, 314, H1225–H1252. [Google Scholar] [CrossRef]
- Munk, K.; Andersen, N.H.; Schmidt, M.R.; Nielsen, S.S.; Terkelsen, C.J.; Sloth, E.; Botker, H.E.; Nielsen, T.T.; Poulsen, S.H. Remote Ischemic Conditioning in Patients with Myocardial Infarction Treated with Primary Angioplasty: Impact on Left Ventricular Function Assessed by Comprehensive Echocardiography and Gated Single-Photon Emission CT. Circ. Cardiovasc. Imaging 2010, 3, 656–662. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, M.; Xin, P.; Li, S.; Tao, J.; Li, Y.; Li, J.; Liu, M.; Li, J.; Zhu, W.; Redington, A.N. Repeated remote ischemic postconditioning protects against adverse left ventricular remodeling and improves survival in a rat model of myocardial infarction. Circ. Res. 2011, 108, 1220–1225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Billah, M.; Ridiandries, A.; Allahwala, U.; Mudaliar, H.; Dona, A.; Hunyor, S.; Khachigian, L.M.; Bhindi, R. Circulating mediators of remote ischemic preconditioning: Search for the missing link between non-lethal ischemia and cardioprotection. Oncotarget 2019, 10, 216–244. [Google Scholar] [CrossRef] [PubMed]
- Gaspar, A.; Lourenco, A.P.; Pereira, M.A.; Azevedo, P.; Roncon-Albuquerque, R., Jr.; Marques, J.; Leite-Moreira, A.F. Randomized controlled trial of remote ischaemic conditioning in ST-elevation myocardial infarction as adjuvant to primary angioplasty (RIC-STEMI). Basic Res. Cardiol. 2018, 113, 14. [Google Scholar] [CrossRef] [PubMed]
- Botker, H.E.; Kharbanda, R.; Schmidt, M.R.; Bottcher, M.; Kaltoft, A.K.; Terkelsen, C.J.; Munk, K.; Andersen, N.H.; Hansen, T.M.; Trautner, S.; et al. Remote ischaemic conditioning before hospital admission, as a complement to angioplasty, and effect on myocardial salvage in patients with acute myocardial infarction: A randomised trial. Lancet 2010, 375, 727–734. [Google Scholar] [CrossRef]
- Hausenloy, D.J.; Kharbanda, R.K.; Moller, U.K.; Ramlall, M.; Aaroe, J.; Butler, R.; Bulluck, H.; Clayton, T.; Dana, A.; Dodd, M.; et al. Effect of remote ischaemic conditioning on clinical outcomes in patients with acute myocardial infarction (CONDI-2/ERIC-PPCI): A single-blind randomised controlled trial. Lancet 2019, 394, 1415–1424. [Google Scholar] [CrossRef] [Green Version]
- Kleinbongard, P.; Botker, H.E.; Ovize, M.; Hausenloy, D.J.; Heusch, G. Co-morbidities and co-medications as confounders of cardioprotection—Does it matter in the clinical setting? Br. J. Pharmacol. 2019. [Google Scholar] [CrossRef] [Green Version]
- Mizushima, N.; Levine, B.; Cuervo, A.M.; Klionsky, D.J. Autophagy fights disease through cellular self-digestion. Nature 2008, 451, 1069–1075. [Google Scholar] [CrossRef] [Green Version]
- Xie, M.; Morales, C.R.; Lavandero, S.; Hill, J.A. Tuning flux: Autophagy as a target of heart disease therapy. Curr. Opin. Cardiol. 2011, 26, 216–222. [Google Scholar] [CrossRef] [Green Version]
- Valentim, L.; Laurence, K.M.; Townsend, P.A.; Carroll, C.J.; Soond, S.; Scarabelli, T.M.; Knight, R.A.; Latchman, D.S.; Stephanou, A. Urocortin inhibits Beclin1-mediated autophagic cell death in cardiac myocytes exposed to ischaemia/reperfusion injury. J. Mol. Cell Cardiol. 2006, 40, 846–852. [Google Scholar] [CrossRef]
- Decker, R.S.; Poole, A.R.; Crie, J.S.; Dingle, J.T.; Wildenthal, K. Lysosomal alterations in hypoxic and reoxygenated hearts. II. Immunohistochemical and biochemical changes in cathepsin D. Am. J. Pathol. 1980, 98, 445–456. [Google Scholar]
- Decker, R.S.; Wildenthal, K. Lysosomal alterations in hypoxic and reoxygenated hearts. I. Ultrastructural and cytochemical changes. Am. J. Pathol. 1980, 98, 425–444. [Google Scholar]
- Gurusamy, N.; Lekli, I.; Gorbunov, N.V.; Gherghiceanu, M.; Popescu, L.M.; Das, D.K. Cardioprotection by adaptation to ischaemia augments autophagy in association with BAG-1 protein. J. Cell. Mol. Med. 2009, 13, 373–387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, C.; Yitzhaki, S.; Perry, C.N.; Liu, W.; Giricz, Z.; Mentzer, R.M., Jr.; Gottlieb, R.A. Autophagy induced by ischemic preconditioning is essential for cardioprotection. J. Cardiovasc. Transl. Res. 2010, 3, 365–373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, C.; Andres, A.M.; Ratliff, E.P.; Hernandez, G.; Lee, P.; Gottlieb, R.A. Preconditioning involves selective mitophagy mediated by Parkin and p62/SQSTM1. PLoS ONE 2011, 6, e20975. [Google Scholar] [CrossRef] [PubMed]
- Wagner, C.; Tillack, D.; Simonis, G.; Strasser, R.H.; Weinbrenner, C. Ischemic post-conditioning reduces infarct size of the in vivo rat heart: Role of PI3-K, mTOR, GSK-3beta, and apoptosis. Mol. Cell. Biochem. 2010, 339, 135–147. [Google Scholar] [CrossRef]
- Wang, Y.; Shen, J.; Xiong, X.; Xu, Y.; Zhang, H.; Huang, C.; Tian, Y.; Jiao, C.; Wang, X.; Li, X. Remote ischemic preconditioning protects against liver ischemia-reperfusion injury via heme oxygenase-1-induced autophagy. PLoS ONE 2014, 9, e98834. [Google Scholar] [CrossRef]
- Su, J.; Zhang, T.; Wang, K.; Zhu, T.; Li, X. Autophagy activation contributes to the neuroprotection of remote ischemic perconditioning against focal cerebral ischemia in rats. Neurochem. Res. 2014, 39, 2068–2077. [Google Scholar] [CrossRef]
- Rohailla, S.; Clarizia, N.; Sourour, M.; Sourour, W.; Gelber, N.; Wei, C.; Li, J.; Redington, A.N. Acute, delayed and chronic remote ischemic conditioning is associated with downregulation of mTOR and enhanced autophagy signaling. PLoS ONE 2014, 9, e111291. [Google Scholar] [CrossRef] [Green Version]
- You, L.; Wang, Z.; Li, H.; Shou, J.; Jing, Z.; Xie, J.; Sui, X.; Pan, H.; Han, W. The role of STAT3 in autophagy. Autophagy 2015, 11, 729–739. [Google Scholar] [CrossRef] [Green Version]
- Jonchere, B.; Belanger, A.; Guette, C.; Barre, B.; Coqueret, O.C. STAT3 as a new autophagy regulator. Jak-Stat 2013, 2, e24353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mudaliar, H.; Rayner, B.; Billah, M.; Kapoor, N.; Lay, W.; Dona, A.; Bhindi, R. Remote ischemic preconditioning attenuates EGR-1 expression following myocardial ischemia reperfusion injury through activation of the JAK-STAT pathway. Int. J. Cardiol. 2017, 228, 729–741. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, T.; Narazaki, M.; Kishimoto, T. IL-6 in inflammation, immunity, and disease. Cold Spring Harb. Perspect. Biol. 2014, 6, a016295. [Google Scholar] [CrossRef] [PubMed]
- Fontes, J.A.; Rose, N.R.; Cihakova, D. The varying faces of IL-6: From cardiac protection to cardiac failure. Cytokine 2015, 74, 62–68. [Google Scholar] [CrossRef] [Green Version]
- Kanda, T.; McManus, J.E.; Nagai, R.; Imai, S.; Suzuki, T.; Yang, D.; McManus, B.M.; Kobayashi, I. Modification of viral myocarditis in mice by interleukin-6. Circ. Res. 1996, 78, 848–856. [Google Scholar] [CrossRef]
- McGinnis, G.R.; Ballmann, C.; Peters, B.; Nanayakkara, G.; Roberts, M.; Amin, R.; Quindry, J.C. Interleukin-6 mediates exercise preconditioning against myocardial ischemia reperfusion injury. Am. J. Physiol. Heart Circ. Physiol. 2015, 308, H1423–H1433. [Google Scholar] [CrossRef] [Green Version]
- Qin, B.; Zhou, Z.; He, J.; Yan, C.; Ding, S. IL-6 Inhibits Starvation-induced Autophagy via the STAT3/Bcl-2 Signaling Pathway. Sci. Rep. 2015, 5, 15701. [Google Scholar] [CrossRef] [Green Version]
- Chang, P.C.; Wang, T.Y.; Chang, Y.T.; Chu, C.Y.; Lee, C.L.; Hsu, H.W.; Zhou, T.A.; Wu, Z.; Kim, R.H.; Desai, S.J.; et al. Autophagy pathway is required for IL-6 induced neuroendocrine differentiation and chemoresistance of prostate cancer LNCaP cells. PLoS ONE 2014, 9, e88556. [Google Scholar] [CrossRef] [Green Version]
- Linnemann, A.K.; Blumer, J.; Marasco, M.R.; Battiola, T.J.; Umhoefer, H.M.; Han, J.Y.; Lamming, D.W.; Davis, D.B. Interleukin 6 protects pancreatic beta cells from apoptosis by stimulation of autophagy. FASEB J. 2017, 31, 4140–4152. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; van Boxel-Dezaire, A.H.; Cheon, H.; Yang, J.; Stark, G.R. STAT3 activation in response to IL-6 is prolonged by the binding of IL-6 receptor to EGF receptor. Proc. Natl. Acad. Sci. USA 2013, 110, 16975–16980. [Google Scholar] [CrossRef] [Green Version]
- Hilfiker-Kleiner, D.; Hilfiker, A.; Fuchs, M.; Kaminski, K.; Schaefer, A.; Schieffer, B.; Hillmer, A.; Schmiedl, A.; Ding, Z.; Podewski, E.; et al. Signal transducer and activator of transcription 3 is required for myocardial capillary growth, control of interstitial matrix deposition, and heart protection from ischemic injury. Circ. Res. 2004, 95, 187–195. [Google Scholar] [CrossRef] [PubMed]
- Dawn, B.; Xuan, Y.T.; Guo, Y.; Rezazadeh, A.; Stein, A.B.; Hunt, G.; Wu, W.J.; Tan, W.; Bolli, R. IL-6 plays an obligatory role in late preconditioning via JAK-STAT signaling and upregulation of iNOS and COX-2. Cardiovasc. Res. 2004, 64, 61–71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klionsky, D.J.; Abdelmohsen, K.; Abe, A.; Abedin, M.J.; Abeliovich, H.; Acevedo Arozena, A.; Adachi, H.; Adams, C.M.; Adams, P.D.; Adeli, K.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy 2016, 12, 1–222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rubinsztein, D.C.; Cuervo, A.M.; Ravikumar, B.; Sarkar, S.; Korolchuk, V.; Kaushik, S.; Klionsky, D.J. In search of an “autophagomometer”. Autophagy 2009, 5, 585–589. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, S.; Ravikumar, B.; Rubinsztein, D.C. Autophagic clearance of aggregate-prone proteins associated with neurodegeneration. Methods Enzym. 2009, 453, 83–110. [Google Scholar]
- Kroemer, G.; Dallaporta, B.; Resche-Rigon, M. The mitochondrial death/life regulator in apoptosis and necrosis. Annu. Rev. Physiol. 1998, 60, 619–642. [Google Scholar] [CrossRef]
- Crompton, M. The mitochondrial permeability transition pore and its role in cell death. Biochem. J. 1999, 341 Pt 2, 233–249. [Google Scholar] [CrossRef]
- Kroemer, G.; Reed, J.C. Mitochondrial control of cell death. Nat. Med. 2000, 6, 513–519. [Google Scholar] [CrossRef]
- Kramer, D.K.; Bouzakri, K.; Holmqvist, O.; Al-Khalili, L.; Krook, A. Effect of serum replacement with plysate on cell growth and metabolismin primary cultures of human skeletal muscle. Cytotechnology 2005, 48, 89–95. [Google Scholar] [CrossRef] [Green Version]
- Mannello, F.; Tonti, G.A. Concise review: No breakthroughs for human mesenchymal and embryonic stem cell culture: Conditioned medium, feeder layer, or feeder-free; medium with fetal calf serum, human serum, or enriched plasma; serum-free, serum replacement nonconditioned medium, or ad hoc formula? All that glitters is not gold! Stem Cells 2007, 25, 1603–1609. [Google Scholar]
- Van der Valk, J.; Brunner, D.; De Smet, K.; Fex Svenningsen, A.; Honegger, P.; Knudsen, L.E.; Lindl, T.; Noraberg, J.; Price, A.; Scarino, M.L.; et al. Optimization of chemically defined cell culture media--replacing fetal bovine serum in mammalian in vitro methods. Toxicol. Vitr. 2010, 24, 1053–1063. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, X.; Baker, H.; Hancock, W.S.; Fawaz, F.; McCaman, M.; Pungor, E., Jr. Proteomic analysis for the assessment of different lots of fetal bovine serum as a raw material for cell culture. Part IV. Application of proteomics to the manufacture of biological drugs. Biotechnol. Prog. 2006, 22, 1294–1300. [Google Scholar] [CrossRef] [PubMed]
- Colzani, M.; Waridel, P.; Laurent, J.; Faes, E.; Ruegg, C.; Quadroni, M. Metabolic labeling and protein linearization technology allow the study of proteins secreted by cultured cells in serum-containing media. J. Proteome Res. 2009, 8, 4779–4788. [Google Scholar] [CrossRef]
- Lambert, K.; Pirt, S.J. Growth of human diploid cells (strain MRC-5) in defined medium; replacement of serum by a fraction of serum ultrafiltrate. J. Cell Sci. 1979, 35, 381–392. [Google Scholar] [PubMed]
- Codeluppi, S.; Gregory, E.N.; Kjell, J.; Wigerblad, G.; Olson, L.; Svensson, C.I. Influence of rat substrain and growth conditions on the characteristics of primary cultures of adult rat spinal cord astrocytes. J. Neurosci. Methods 2011, 197, 118–127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pontarin, G.; Ferraro, P.; Rampazzo, C.; Kollberg, G.; Holme, E.; Reichard, P.; Bianchi, V. Deoxyribonucleotide metabolism in cycling and resting human fibroblasts with a missense mutation in p53R2, a subunit of ribonucleotide reductase. J. Biol. Chem. 2011, 286, 11132–11140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Rechem, C.; Boulay, G.; Pinte, S.; Stankovic-Valentin, N.; Guerardel, C.; Leprince, D. Differential regulation of HIC1 target genes by CtBP and NuRD, via an acetylation/SUMOylation switch, in quiescent versus proliferating cells. Mol. Cell Biol. 2010, 30, 4045–4059. [Google Scholar] [CrossRef] [Green Version]
- Chao, W.; Shen, Y.; Zhu, X.; Zhao, H.; Novikov, M.; Schmidt, U.; Rosenzweig, A. Lipopolysaccharide improves cardiomyocyte survival and function after serum deprivation. J. Biol. Chem. 2005, 280, 21997–22005. [Google Scholar] [CrossRef] [Green Version]
- Borlongan, C.V.; Yamamoto, M.; Takei, N.; Kumazaki, M.; Ungsuparkorn, C.; Hida, H.; Sanberg, P.R.; Nishino, H. Glial cell survival is enhanced during melatonin-induced neuroprotection against cerebral ischemia. FASEB J. 2000, 14, 1307–1317. [Google Scholar]
- Chen, Y.; Wang, H.; Zhang, Y.; Wang, Z.; Liu, S.; Cui, L. Pretreatment of ghrelin protects H9c2 cells against hypoxia/reoxygenation-induced cell death via PI3K/AKT and AMPK pathways. Artif. Cells Nanomed. Biotechnol. 2019, 47, 2179–2187. [Google Scholar] [CrossRef] [Green Version]
- Zhao, J.; Jie, Q.; Li, G.; Li, Y.; Liu, B.; Li, H.; Luo, J.; Qin, X.; Li, Z.; Wei, Y. Rac1 promotes the survival of H9c2 cells during serum deficiency targeting JNK/c-JUN/Cyclin-D1 and AKT2/MCL1 pathways. Int. J. Med. Sci. 2018, 15, 1062–1071. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yellon, D.M.; Hausenloy, D.J. Myocardial reperfusion injury. N. Engl. J. Med. 2007, 357, 1121–1135. [Google Scholar] [CrossRef] [PubMed]
- Rochitte, C.E.; Lima, J.A.; Bluemke, D.A.; Reeder, S.B.; McVeigh, E.R.; Furuta, T.; Becker, L.C.; Melin, J.A. Magnitude and time course of microvascular obstruction and tissue injury after acute myocardial infarction. Circulation 1998, 98, 1006–1014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Z.Q.; Nakamura, M.; Wang, N.P.; Velez, D.A.; Hewan-Lowe, K.O.; Guyton, R.A.; Vinten-Johansen, J. Dynamic progression of contractile and endothelial dysfunction and infarct extension in the late phase of reperfusion. J. Surg Res. 2000, 94, 133–144. [Google Scholar] [CrossRef] [PubMed]
- Qin, Z.H.; Wang, Y.; Kegel, K.B.; Kazantsev, A.; Apostol, B.L.; Thompson, L.M.; Yoder, J.; Aronin, N.; DiFiglia, M. Autophagy regulates the processing of amino terminal huntingtin fragments. Hum. Mol. Genet. 2003, 12, 3231–3244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shintani, T.; Klionsky, D.J. Autophagy in health and disease: A double-edged sword. Science 2004, 306, 990–995. [Google Scholar] [CrossRef] [Green Version]
- Matsui, Y.; Kyoi, S.; Takagi, H.; Hsu, C.P.; Hariharan, N.; Ago, T.; Vatner, S.F.; Sadoshima, J. Molecular mechanisms and physiological significance of autophagy during myocardial ischemia and reperfusion. Autophagy 2008, 4, 409–415. [Google Scholar] [CrossRef] [Green Version]
- Sala-Mercado, J.A.; Wider, J.; Undyala, V.V.; Jahania, S.; Yoo, W.; Mentzer, R.M., Jr.; Gottlieb, R.A.; Przyklenk, K. Profound cardioprotection with chloramphenicol succinate in the swine model of myocardial ischemia-reperfusion injury. Circulation 2010, 122 (Suppl. 11), S179–S184. [Google Scholar] [CrossRef] [Green Version]
- Yan, L.; Vatner, D.E.; Kim, S.J.; Ge, H.; Masurekar, M.; Massover, W.H.; Yang, G.; Matsui, Y.; Sadoshima, J.; Vatner, S.F. Autophagy in chronically ischemic myocardium. Proc. Natl. Acad. Sci. USA 2005, 102, 13807–13812. [Google Scholar] [CrossRef] [Green Version]
- Gedik, N.; Thielmann, M.; Kottenberg, E.; Peters, J.; Jakob, H.; Heusch, G.; Kleinbongard, P. No evidence for activated autophagy in left ventricular myocardium at early reperfusion with protection by remote ischemic preconditioning in patients undergoing coronary artery bypass grafting. PLoS ONE 2014, 9, e96567. [Google Scholar] [CrossRef]
- Zhu, J.; Lu, T.; Yue, S.; Shen, X.; Gao, F.; Busuttil, R.W.; Kupiec-Weglinski, J.W.; Xia, Q.; Zhai, Y. Rapamycin protection of livers from ischemia and reperfusion injury is dependent on both autophagy induction and mammalian target of rapamycin complex 2-Akt activation. Transplantation 2015, 99, 48–55. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.L.; Zhang, J.; Cui, L.Y.; Yang, S. Autophagy activation attenuates renal ischemia-reperfusion injury in rats. Exp. Biol. Med. 2015, 240, 1590–1598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdul-Ghani, S.; Heesom, K.J.; Angelini, G.D.; Suleiman, M.S. Cardiac phosphoproteomics during remote ischemic preconditioning: A role for the sarcomeric Z-disk proteins. Biomed. Res. Int. 2014, 2014, 767812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gedik, N.; Kottenberg, E.; Thielmann, M.; Frey, U.H.; Jakob, H.; Peters, J.; Heusch, G.; Kleinbongard, P. Potential humoral mediators of remote ischemic preconditioning in patients undergoing surgical coronary revascularization. Sci. Rep. 2017, 7, 12660. [Google Scholar] [CrossRef] [Green Version]
- Szczepanek, K.; Chen, Q.; Derecka, M.; Salloum, F.N.; Zhang, Q.; Szelag, M.; Cichy, J.; Kukreja, R.C.; Dulak, J.; Lesnefsky, E.J.; et al. Mitochondrial-targeted Signal transducer and activator of transcription 3 (STAT3) protects against ischemia-induced changes in the electron transport chain and the generation of reactive oxygen species. J. Biol. Chem. 2011, 286, 29610–29620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yeh, C.C.; Li, H.; Malhotra, D.; Turcato, S.; Nicholas, S.; Tu, R.; Zhu, B.Q.; Cha, J.; Swigart, P.M.; Myagmar, B.E.; et al. Distinctive ERK and p38 signaling in remote and infarcted myocardium during post-MI remodeling in the mouse. J. Cell Biochem. 2010, 109, 1185–1191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yin, T.; Sandhu, G.; Wolfgang, C.D.; Burrier, A.; Webb, R.L.; Rigel, D.F.; Hai, T.; Whelan, J. Tissue-specific pattern of stress kinase activation in ischemic/reperfused heart and kidney. J. Biol. Chem. 1997, 272, 19943–19950. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Xuan, W.; Yan, R.; Tropak, M.B.; Jean-St-Michel, E.; Liang, W.; Gladstone, R.; Backx, P.H.; Kharbanda, R.K.; Redington, A.N. Remote preconditioning provides potent cardioprotection via PI3K/Akt activation and is associated with nuclear accumulation of beta-catenin. Clin. Sci. 2011, 120, 451–462. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.S.; Loughran, P.A.; Billiar, T.R. Carbon monoxide decreases the level of iNOS protein and active dimer in IL-1beta-stimulated hepatocytes. Nitric. Oxide. 2008, 18, 256–265. [Google Scholar] [CrossRef] [Green Version]
- Zhai, P.; Sadoshima, J. Glycogen synthase kinase-3beta controls autophagy during myocardial ischemia and reperfusion. Autophagy 2012, 8, 138–139. [Google Scholar] [CrossRef] [Green Version]
- Zhai, P.; Sciarretta, S.; Galeotti, J.; Volpe, M.; Sadoshima, J. Differential roles of GSK-3beta during myocardial ischemia and ischemia/reperfusion. Circ. Res. 2011, 109, 502–511. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.; Hu, S.; Yang, S.; Chen, M.; Zhang, P.; Liu, J.; Abbott, G.W. Remote Liver Ischemic Preconditioning Protects against Sudden Cardiac Death via an ERK/GSK-3beta-Dependent Mechanism. PLoS ONE 2016, 11, e0165123. [Google Scholar] [CrossRef] [PubMed]
- Runwal, G.; Stamatakou, E.; Siddiqi, F.H.; Puri, C.; Zhu, Y.; Rubinsztein, D.C. LC3-positive structures are prominent in autophagy-deficient cells. Sci. Rep. 2019, 9, 10147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bjorkoy, G.; Lamark, T.; Pankiv, S.; Overvatn, A.; Brech, A.; Johansen, T. Monitoring autophagic degradation of p62/SQSTM1. Methods Enzym. 2009, 452, 181–197. [Google Scholar]
- Heusch, G. Remote Ischemic Conditioning in Cardiovascular Surgery. J. Cardiovasc. Pharmacol. 2017, 22, 297–301. [Google Scholar] [CrossRef]
- Zangrillo, A.; Musu, M.; Greco, T.; Di Prima, A.L.; Matteazzi, A.; Testa, V.; Nardelli, P.; Febres, D.; Monaco, F.; Calabro, M.G.; et al. Additive Effect on Survival of Anaesthetic Cardiac Protection and Remote Ischemic Preconditioning in Cardiac Surgery: A Bayesian Network Meta-Analysis of Randomized Trials. PLoS ONE 2015, 10, e0134264. [Google Scholar] [CrossRef]
- Zhou, C.; Liu, Y.; Yao, Y.; Zhou, S.; Fang, N.; Wang, W.; Li, L. beta-blockers and volatile anesthetics may attenuate cardioprotection by remote preconditioning in adult cardiac surgery: A meta-analysis of 15 randomized trials. J. Cardiothorac. Vasc. Anesth. 2013, 27, 305–311. [Google Scholar] [CrossRef]
- Noh, H.S.; Shin, I.W.; Ha, J.H.; Hah, Y.S.; Baek, S.M.; Kim, D.R. Propofol protects the autophagic cell death induced by the ischemia/reperfusion injury in rats. Mol. Cells 2010, 30, 455–460. [Google Scholar] [CrossRef]
- Qiao, S.; Xie, H.; Wang, C.; Wu, X.; Liu, H.; Liu, C. Delayed anesthetic preconditioning protects against myocardial infarction via activation of nuclear factor-kappaB and upregulation of autophagy. J. Anesth. 2013, 27, 251–260. [Google Scholar] [CrossRef]
- Kersten, J.R.; Schmeling, T.J.; Pagel, P.S.; Gross, G.J.; Warltier, D.C. Isoflurane mimics ischemic preconditioning via activation of K (ATP) channels: Reduction of myocardial infarct size with an acute memory phase. Anesthesiology 1997, 87, 361–370. [Google Scholar] [CrossRef]
- Behmenburg, F.; van Caster, P.; Bunte, S.; Brandenburger, T.; Heinen, A.; Hollmann, M.W.; Huhn, R. Impact of Anesthetic Regimen on Remote Ischemic Preconditioning in the Rat Heart in vivo. Anesth. Analg. 2018, 126, 1377–1380. [Google Scholar] [CrossRef] [PubMed]
- Bunte, S.; Behmenburg, F.; Eckelskemper, F.; Mohr, F.; Stroethoff, M.; Raupach, A.; Heinen, A.; Hollmann, M.W.; Huhn, R. Cardioprotection by Humoral Factors Released After Remote Ischemic Preconditioning Depends on Anesthetic Regimen. Crit. Care Med. 2019, 47, e250–e255. [Google Scholar] [CrossRef] [PubMed]
- Berger, M.M.; Huhn, R.; Oei, G.T.; Heinen, A.; Winzer, A.; Bauer, I.; Preckel, B.; Weber, N.C.; Schlack, W.; Hollmann, M.W. Hypoxia induces late preconditioning in the rat heart in vivo. Anesthesiology 2010, 113, 1351–1360. [Google Scholar] [CrossRef] [PubMed]
- Sheng, R.; Zhang, T.T.; Felice, V.D.; Qin, T.; Qin, Z.H.; Smith, C.D.; Sapp, E.; Difiglia, M.; Waeber, C. Preconditioning stimuli induce autophagy via sphingosine kinase 2 in mouse cortical neurons. J. Biol. Chem. 2014, 289, 20845–20857. [Google Scholar] [CrossRef] [Green Version]
- Heusch, G.; Musiolik, J.; Gedik, N.; Skyschally, A. Mitochondrial STAT3 activation and cardioprotection by ischemic postconditioning in pigs with regional myocardial ischemia/reperfusion. Circ. Res. 2011, 109, 1302–1308. [Google Scholar] [CrossRef] [Green Version]
- Heusch, G.; Musiolik, J.; Kottenberg, E.; Peters, J.; Jakob, H.; Thielmann, M. STAT5 activation and cardioprotection by remote ischemic preconditioning in humans: Short communication. Circ. Res. 2012, 110, 111–115. [Google Scholar] [CrossRef]
- Pepe, S.; Liaw, N.Y.; Hepponstall, M.; Sheeran, F.L.; Yong, M.S.; d’Udekem, Y.; Cheung, M.M.; Konstantinov, I.E. Effect of remote ischemic preconditioning on phosphorylated protein signaling in children undergoing tetralogy of Fallot repair: A randomized controlled trial. J. Am. Heart Assoc. 2013, 2, e000095. [Google Scholar] [CrossRef] [Green Version]
- Wu, Q.; Wang, T.; Chen, S.; Zhou, Q.; Li, H.; Hu, N.; Feng, Y.; Dong, N.; Yao, S.; Xia, Z. Cardiac protective effects of remote ischaemic preconditioning in children undergoing tetralogy of fallot repair surgery: A randomized controlled trial. Eur. Heart J. 2018, 39, 1028–1037. [Google Scholar] [CrossRef]
- Liang, Y.; Li, Y.P.; He, F.; Liu, X.Q.; Zhang, J.Y. Long-term, regular remote ischemic preconditioning improves endothelial function in patients with coronary heart disease. Braz. J. Med. Biol Res. 2015, 48, 568–576. [Google Scholar] [CrossRef]
- Hausenloy, D.J.; Yellon, D.M. Myocardial ischemia-reperfusion injury: A neglected therapeutic target. J. Clin. Investig. 2013, 123, 92–100. [Google Scholar] [CrossRef]
- Shimizu, M.; Konstantinov, I.E.; Kharbanda, R.K.; Cheung, M.H.; Redington, A.N. Effects of intermittent lower limb ischaemia on coronary blood flow and coronary resistance in pigs. Acta Physiol. 2007, 190, 103–109. [Google Scholar] [CrossRef] [PubMed]
- Kono, Y.; Fukuda, S.; Hanatani, A.; Nakanishi, K.; Otsuka, K.; Taguchi, H.; Shimada, K. Remote ischemic conditioning improves coronary microcirculation in healthy subjects and patients with heart failure. Drug Des. Dev. 2014, 8, 1175–1181. [Google Scholar]
- Iwai-Kanai, E.; Yuan, H.; Huang, C.; Sayen, M.R.; Perry-Garza, C.N.; Kim, L.; Gottlieb, R.A. A method to measure cardiac autophagic flux in vivo. Autophagy 2008, 4, 322–329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, H.; Tannous, P.; Johnstone, J.L.; Kong, Y.; Shelton, J.M.; Richardson, J.A.; Le, V.; Levine, B.; Rothermel, B.A.; Hill, J.A. Cardiac autophagy is a maladaptive response to hemodynamic stress. J. Clin. Investig. 2007, 117, 1782–1793. [Google Scholar] [CrossRef] [PubMed]
- Abas, L.; Bogoyevitch, M.A.; Guppy, M. Mitochondrial ATP production is necessary for activation of the extracellular-signal-regulated kinases during ischaemia/reperfusion in rat myocyte-derived H9c2 cells. Biochem. J. 2000, 349 Pt 1, 119–126. [Google Scholar] [CrossRef]
- Kuznetsov, A.V.; Javadov, S.; Sickinger, S.; Frotschnig, S.; Grimm, M. H9c2 and HL-1 cells demonstrate distinct features of energy metabolism, mitochondrial function and sensitivity to hypoxia-reoxygenation. Biochim. Biophys. Acta 2015, 1853, 276–284. [Google Scholar] [CrossRef] [Green Version]
- Kimes, B.W.; Brandt, B.L. Properties of a clonal muscle cell line from rat heart. Exp. Cell Res. 1976, 98, 367–381. [Google Scholar] [CrossRef]
- Mejia-Alvarez, R.; Tomaselli, G.F.; Marban, E. Simultaneous expression of cardiac and skeletal muscle isoforms of the L-type Ca2+ channel in a rat heart muscle cell line. J. Physiol. 1994, 478 Pt 2, 315–329. [Google Scholar] [CrossRef]
- Botker, H.E.; Hausenloy, D.; Andreadou, I.; Antonucci, S.; Boengler, K.; Davidson, S.M.; Deshwal, S.; Devaux, Y.; Di Lisa, F.; Di Sante, M.; et al. Practical guidelines for rigor and reproducibility in preclinical and clinical studies on cardioprotection. Basic Res. Cardiol. 2018, 113, 39. [Google Scholar] [CrossRef] [Green Version]
- Zheng, Q.; Wang, X. Autophagy and the ubiquitin-proteasome system in cardiac dysfunction. Panminerva Med. 2010, 52, 9–25. [Google Scholar]
- Wang, X.; Robbins, J. Heart failure and protein quality control. Circ. Res. 2006, 99, 1315–1328. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.; Tian, Y.; Xu, H.; Pan, B.; Terpstra, E.M.; Wu, P.; Wang, H.; Li, F.; Liu, J.; Wang, X. Inadequate ubiquitination-proteasome coupling contributes to myocardial ischemia-reperfusion injury. J. Clin. Investig. 2018, 128, 5294–5306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Divald, A.; Kivity, S.; Wang, P.; Hochhauser, E.; Roberts, B.; Teichberg, S.; Gomes, A.V.; Powell, S.R. Myocardial ischemic preconditioning preserves postischemic function of the 26S proteasome through diminished oxidative damage to 19S regulatory particle subunits. Circ. Res. 2010, 106, 1829–1838. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Billah, M.; Ridiandries, A.; Rayner, B.S.; Allahwala, U.K.; Dona, A.; Khachigian, L.M.; Bhindi, R. Egr-1 functions as a master switch regulator of remote ischemic preconditioning-induced cardioprotection. Basic Res. Cardiol. 2019, 115, 3. [Google Scholar] [CrossRef]
- Li, C.; Jackson, R.M. Reactive species mechanisms of cellular hypoxia-reoxygenation injury. Am. J. Physiol. Cell Physiol. 2002, 282, C227–C241. [Google Scholar] [CrossRef] [Green Version]
- Webster, K.A.; Discher, D.J.; Kaiser, S.; Hernandez, O.; Sato, B.; Bishopric, N.H. Hypoxia-activated apoptosis of cardiac myocytes requires reoxygenation or a pH shift and is independent of p53. J. Clin. Investig. 1999, 104, 239–252. [Google Scholar]
- Shang, L.; Ananthakrishnan, R.; Li, Q.; Quadri, N.; Abdillahi, M.; Zhu, Z.; Qu, W.; Rosario, R.; Toure, F.; Yan, S.F.; et al. RAGE modulates hypoxia/reoxygenation injury in adult murine cardiomyocytes via JNK and GSK-3beta signaling pathways. PLoS ONE 2010, 5, e10092. [Google Scholar] [CrossRef] [Green Version]
- Calvillo, L.; Vanoli, E.; Andreoli, E.; Besana, A.; Omodeo, E.; Gnecchi, M.; Zerbi, P.; Vago, G.; Busca, G.; Schwartz, P.J. Vagal stimulation, through its nicotinic action, limits infarct size and the inflammatory response to myocardial ischemia and reperfusion. J. Cardiovasc. Pharmacol. 2011, 58, 500–507. [Google Scholar] [CrossRef] [Green Version]
- Guo, J.; Zhu, J.; Ma, L.; Shi, H.; Hu, J.; Zhang, S.; Hou, L.; Xu, F.; An, Y.; Yu, H.; et al. Chronic Kidney Disease Exacerbates Myocardial Ischemia Reperfusion Injury: Role of Endoplasmic Reticulum Stress-Mediated Apoptosis. Shock 2018, 49, 712–720. [Google Scholar] [CrossRef]
- Bohl, S.; Medway, D.J.; Schulz-Menger, J.; Schneider, J.E.; Neubauer, S.; Lygate, C.A. Refined approach for quantification of in vivo ischemia-reperfusion injury in the mouse heart. Am. J. Physiol. Heart Circ. Physiol. 2009, 297, H2054–H2058. [Google Scholar] [CrossRef] [PubMed]
- Deng, C.; Sun, Z.; Tong, G.; Yi, W.; Ma, L.; Zhao, B.; Cheng, L.; Zhang, J.; Cao, F.; Yi, D. alpha-Lipoic acid reduces infarct size and preserves cardiac function in rat myocardial ischemia/reperfusion injury through activation of PI3K/Akt/Nrf2 pathway. PLoS ONE 2013, 8, e58371. [Google Scholar]
- Price, A.N.; Cheung, K.K.; Lim, S.Y.; Yellon, D.M.; Hausenloy, D.J.; Lythgoe, M.F. Rapid assessment of myocardial infarct size in rodents using multi-slice inversion recovery late gadolinium enhancement CMR at 9.4T. J. Cardiovasc. Magn. Reson. 2011, 13, 44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Jia, P.; Huang, Z.; Liu, S.; Miao, J.; Guo, Y.; Wu, N.; Jia, D. Lycopene protects against myocardial ischemia-reperfusion injury by inhibiting mitochondrial permeability transition pore opening. Drug Des. Dev. 2019, 13, 2331–2342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, D.; Huang, J.; Zhang, Z.; Gao, H.; Li, J.; Shen, M.; Cao, F.; Wang, H. Luteolin limits infarct size and improves cardiac function after myocardium ischemia/reperfusion injury in diabetic rats. PLoS ONE 2012, 7, e33491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsuda, T.; Gao, E.; Evangelisti, L.; Markova, D.; Ma, X.; Chu, M.L. Post-ischemic myocardial fibrosis occurs independent of hemodynamic changes. Cardiovasc. Res. 2003, 59, 926–933. [Google Scholar] [CrossRef] [Green Version]
- Eckle, T.; Grenz, A.; Kohler, D.; Redel, A.; Falk, M.; Rolauffs, B.; Osswald, H.; Kehl, F.; Eltzschig, H.K. Systematic evaluation of a novel model for cardiac ischemic preconditioning in mice. Am. J. Physiol. Heart Circ. Physiol. 2006, 291, H2533–H2540. [Google Scholar] [CrossRef] [Green Version]
- Tanase, H.; Yamori, Y.; Hansen, C.T.; Lovenberg, W. Heart size in inbred strains of rats. Part 1. Genetic determination of the development of cardiovascular enlargement in rats. Hypertension 1982, 4, 864–872. [Google Scholar] [CrossRef] [Green Version]
- Cao, C.M.; Zhang, Y.; Weisleder, N.; Ferrante, C.; Wang, X.; Lv, F.; Zhang, Y.; Song, R.; Hwang, M.; Jin, L.; et al. MG53 constitutes a primary determinant of cardiac ischemic preconditioning. Circulation 2010, 121, 2565–2574. [Google Scholar] [CrossRef] [Green Version]
- Shen, Y.T.; Depre, C.; Yan, L.; Park, J.Y.; Tian, B.; Jain, K.; Chen, L.; Zhang, Y.; Kudej, R.K.; Zhao, X.; et al. Repetitive ischemia by coronary stenosis induces a novel window of ischemic preconditioning. Circulation 2008, 118, 1961–1969. [Google Scholar] [CrossRef] [Green Version]
- Kabeya, Y.; Mizushima, N.; Ueno, T.; Yamamoto, A.; Kirisako, T.; Noda, T.; Kominami, E.; Ohsumi, Y.; Yoshimori, T. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000, 19, 5720–5728. [Google Scholar] [CrossRef]
- Goldman, A.; Harper, S.; Speicher, D.W. Detection of Proteins on Blot Membranes. Curr. Protoc. Protein Sci. 2016, 86, 10 8 1–10 8 11. [Google Scholar] [CrossRef]
- Tremblay, F.; Brule, S.; Hee Um, S.; Li, Y.; Masuda, K.; Roden, M.; Sun, X.J.; Krebs, M.; Polakiewicz, R.D.; Thomas, G.; et al. Identification of IRS-1 Ser-1101 as a target of S6K1 in nutrient- and obesity-induced insulin resistance. Proc. Natl. Acad. Sci. USA 2007, 104, 14056–14061. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yung, H.W.; Charnock-Jones, D.S.; Burton, G.J. Regulation of AKT phosphorylation at Ser473 and Thr308 by endoplasmic reticulum stress modulates substrate specificity in a severity dependent manner. PLoS ONE 2011, 6, e17894. [Google Scholar] [CrossRef] [PubMed]
- Aldridge, G.M.; Podrebarac, D.M.; Greenough, W.T.; Weiler, I.J. The use of total protein stains as loading controls: An alternative to high-abundance single-protein controls in semi-quantitative immunoblotting. J. Neurosci. Methods 2008, 172, 250–254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eaton, S.L.; Roche, S.L.; Llavero Hurtado, M.; Oldknow, K.J.; Farquharson, C.; Gillingwater, T.H.; Wishart, T.M. Total protein analysis as a reliable loading control for quantitative fluorescent Western blotting. PLoS ONE 2013, 8, e72457. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Primer Sequence | Annealing Temperature |
|---|---|---|
| Cardiotrophin-1 | Forward 5′-AGCCTTCCTGATTTCTCAGGCT-3′ Reverse 5′-AATT TCTGTGTTGGCGCAGTGTGG-3′ | 60 °C |
| LIF | Forward 5′-TGTGGTAGCAGCCGAGTCATAAT-3′ Reverse 5′-CGTGGCTTGTGTGTTCGGTTTCAT-3′ | 60 °C |
| IL-6 | Forward 5′-GCTCGTCGTCGACAACGGCCTC-3′ Reverse 5′-CAAACATGATCTGGGTCATCTTCTC-3′ | 60 °C |
| IL-11 | Forward 5′-CGTGAAGCTGTGTTGTCCTG-3′ Reverse 5′-GCTCCTAGGACTGTCTTCTTC-3′ | 60 °C |
| IL-1β | Forward 5′-CTGTGACTCGTGGGATGATG-3′ Reverse 5′-GGGATTTTGTCGTTGCTTGT-3′ | 60 °C |
| GAPDH | Forward 5′-ATGGGAAGCTGGTCATCAAC-3′ Reverse 5′ GTGGTTCACACCCATCACAA-3′ | 60 °C |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Billah, M.; Ridiandries, A.; Allahwala, U.K.; Mudaliar, H.; Dona, A.; Hunyor, S.; Khachigian, L.M.; Bhindi, R. Remote Ischemic Preconditioning Induces Cardioprotective Autophagy and Signals through the IL-6-Dependent JAK-STAT Pathway. Int. J. Mol. Sci. 2020, 21, 1692. https://doi.org/10.3390/ijms21051692
Billah M, Ridiandries A, Allahwala UK, Mudaliar H, Dona A, Hunyor S, Khachigian LM, Bhindi R. Remote Ischemic Preconditioning Induces Cardioprotective Autophagy and Signals through the IL-6-Dependent JAK-STAT Pathway. International Journal of Molecular Sciences. 2020; 21(5):1692. https://doi.org/10.3390/ijms21051692
Chicago/Turabian StyleBillah, Muntasir, Anisyah Ridiandries, Usaid K Allahwala, Harshini Mudaliar, Anthony Dona, Stephen Hunyor, Levon M. Khachigian, and Ravinay Bhindi. 2020. "Remote Ischemic Preconditioning Induces Cardioprotective Autophagy and Signals through the IL-6-Dependent JAK-STAT Pathway" International Journal of Molecular Sciences 21, no. 5: 1692. https://doi.org/10.3390/ijms21051692
APA StyleBillah, M., Ridiandries, A., Allahwala, U. K., Mudaliar, H., Dona, A., Hunyor, S., Khachigian, L. M., & Bhindi, R. (2020). Remote Ischemic Preconditioning Induces Cardioprotective Autophagy and Signals through the IL-6-Dependent JAK-STAT Pathway. International Journal of Molecular Sciences, 21(5), 1692. https://doi.org/10.3390/ijms21051692