Spotlight on the Ballet of Proteins: The Structural Dynamic Properties of Proteins Illuminated by Solution NMR


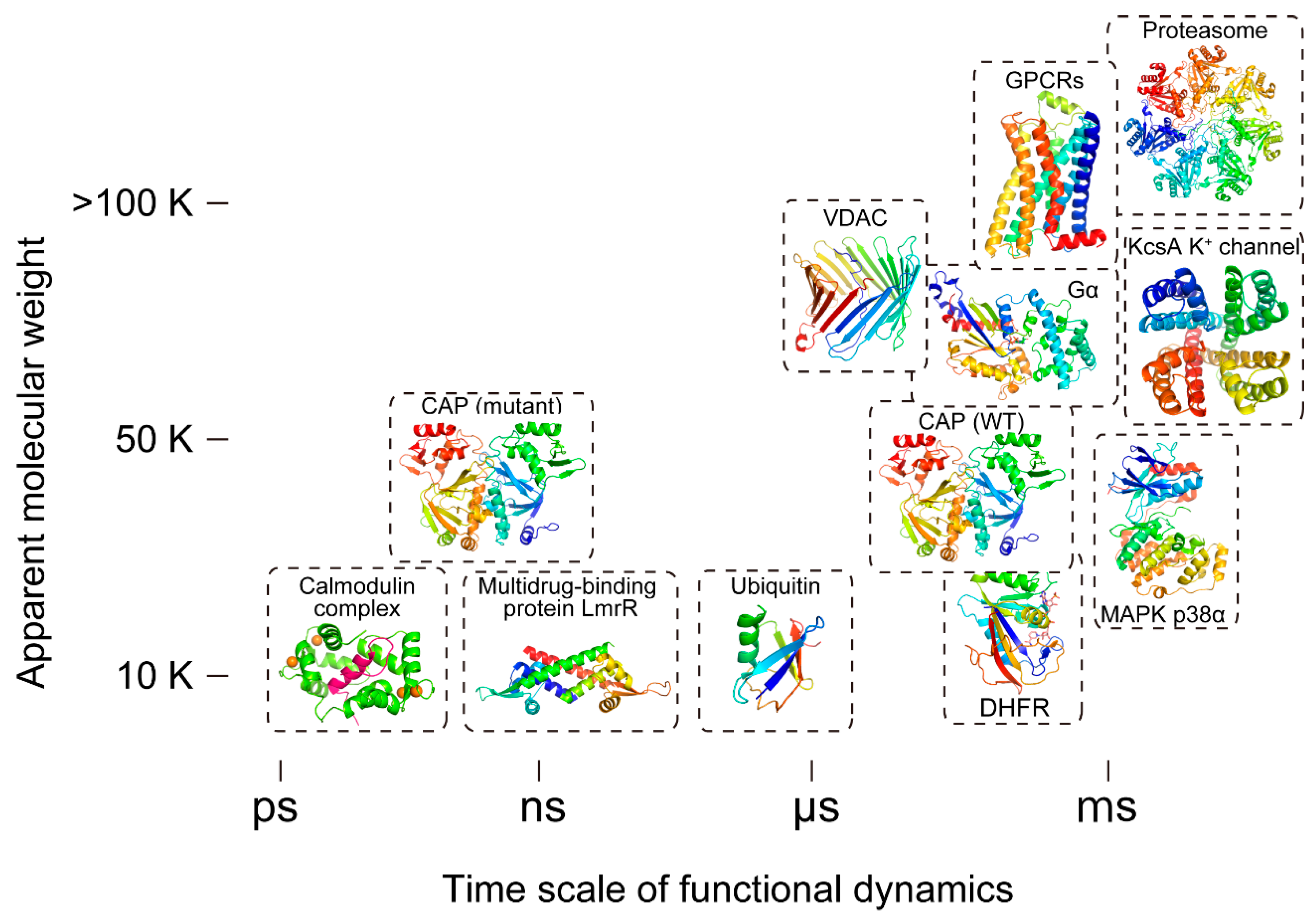{kind=link}
{kind=link}
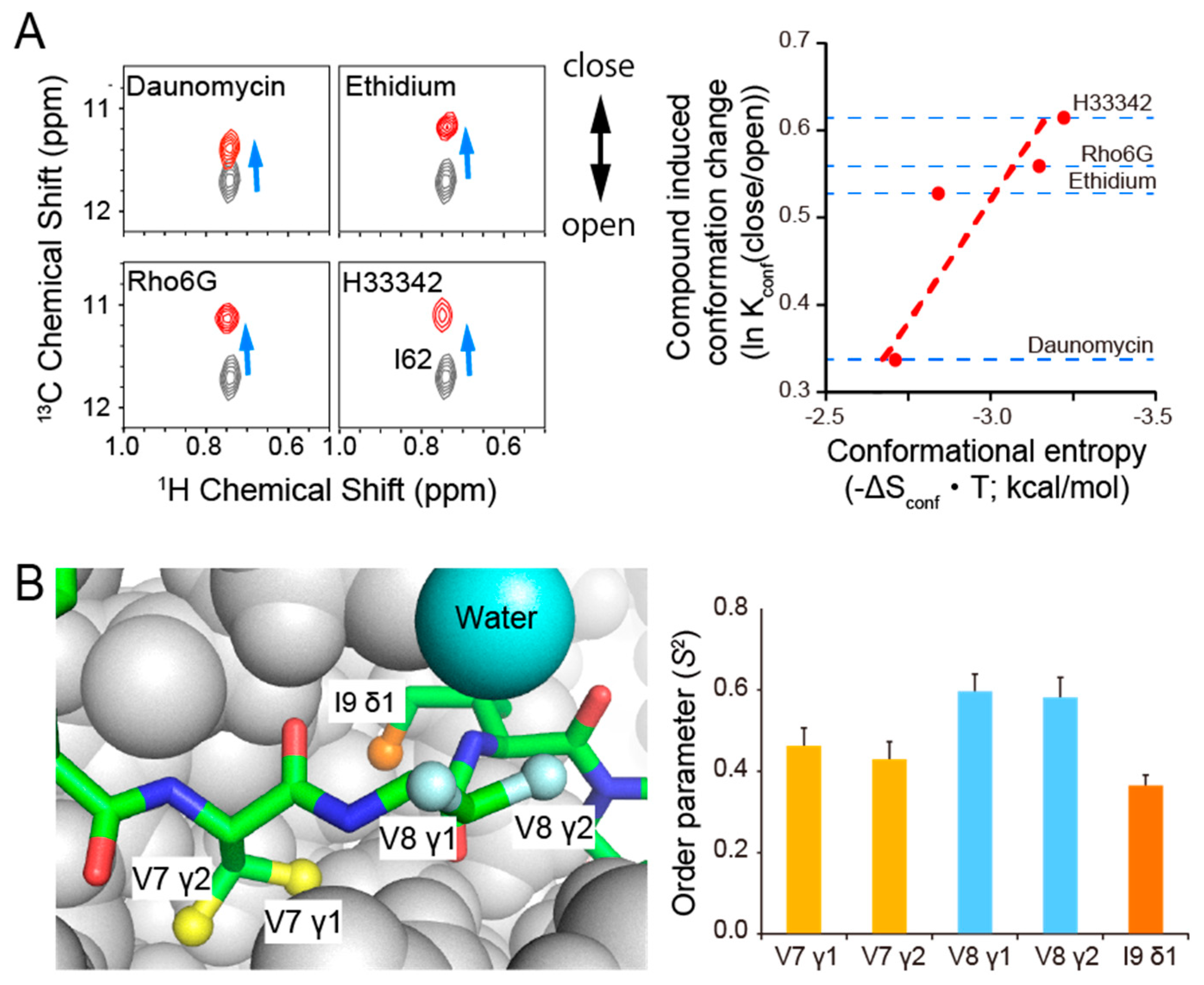{kind=link}
Abstract
:1. Introduction
2. NMR Unveils Protein Dynamics that Dictate Functions in Key Biological Systems.
2.1. G-Protein-Coupled Receptors (GPCRs)
2.2. Ion Channels and Other Membrane Proteins
2.3. Small GTPases
2.4. Kinases
2.5. Enzymes and Other Proteins
2.6. Intrinsically Disordered Proteins (IDPs)
3. Importance of Dynamics in Protein Interactions and Drug Development
3.1. Conformational Entropy: Role of Fast Dynamics in Recognition
3.2. Utilization of Dynamics Information for Drug Design
4. Future Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Wuthrich, K.; Wagner, G. NMR investigations of the dynamics of the aromatic amino acid residues in the basic pancreatic trypsin inhibitor. FEBS Lett. 1975, 50, 265–268. [Google Scholar] [CrossRef] [Green Version]
- Hilger, D.; Masureel, M.; Kobilka, B.K. Structure and dynamics of GPCR signaling complexes. Nat. Struct. Mol. Biol. 2018, 25, 4–12. [Google Scholar] [CrossRef] [PubMed]
- Wootten, D.; Christopoulos, A.; Marti-Solano, M.; Babu, M.M.; Sexton, P.M. Mechanisms of signalling and biased agonism in G protein-coupled receptors. Nat. Rev. Mol. Cell. Biol. 2018, 19, 638–653. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.E.; Melcher, K.; Xu, H.E. Understanding the GPCR biased signaling through G protein and arrestin complex structures. Curr. Opin. Struct. Biol. 2017, 45, 150–159. [Google Scholar] [CrossRef] [PubMed]
- Shimada, I.; Ueda, T.; Kofuku, Y.; Eddy, M.T.; Wüthrich, K. GPCR drug discovery: Integrating solution NMR data with crystal and cryo-EM structures. Nat. Rev. Drug Discov. 2019, 18, 59–82. [Google Scholar] [CrossRef] [PubMed]
- Kofuku, Y.; Ueda, T.; Okude, J.; Shiraishi, Y.; Kondo, K.; Maeda, M.; Tsujishita, H.; Shimada, I. Efficacy of the β₂-adrenergic receptor is determined by conformational equilibrium in the transmembrane region. Nat. Commun. 2012, 3, 1045. [Google Scholar] [CrossRef] [Green Version]
- Ye, L.; Van Eps, N.; Zimmer, M.; Ernst, O.P.; Scott Prosser, R. Activation of the A2A adenosine G-protein-coupled receptor by conformational selection. Nature 2016, 533, 265. [Google Scholar] [CrossRef]
- Manglik, A.; Kim, T.H.; Masureel, M.; Altenbach, C.; Yang, Z.; Hilger, D.; Lerch, M.T.; Kobilka, T.S.; Thian, F.S.; Hubbell, W.L.; et al. Structural Insights into the Dynamic Process of β2-Adrenergic Receptor Signaling. Cell 2015, 161, 1101–1111. [Google Scholar] [CrossRef] [Green Version]
- Isogai, S.; Deupi, X.; Opitz, C.; Heydenreich, F.M.; Tsai, C.-J.; Brueckner, F.; Schertler, G.F.X.; Veprintsev, D.B.; Grzesiek, S. Backbone NMR reveals allosteric signal transduction networks in the β1-adrenergic receptor. Nature 2016, 530, 237. [Google Scholar] [CrossRef]
- Eddy, M.T.; Lee, M.-Y.; Gao, Z.-G.; White, K.L.; Didenko, T.; Horst, R.; Audet, M.; Stanczak, P.; McClary, K.M.; Han, G.W.; et al. Allosteric Coupling of Drug Binding and Intracellular Signaling in the A2A Adenosine Receptor. Cell 2018, 172, 68–80.e12. [Google Scholar] [CrossRef] [Green Version]
- Solt, A.S.; Bostock, M.J.; Shrestha, B.; Kumar, P.; Warne, T.; Tate, C.G.; Nietlispach, D. Insight into partial agonism by observing multiple equilibria for ligand-bound and Gs-mimetic nanobody-bound beta1-adrenergic receptor. Nat. Commun. 2017, 8, 1795. [Google Scholar] [CrossRef] [PubMed]
- Casiraghi, M.; Point, E.; Pozza, A.; Moncoq, K.; Baneres, J.L.; Catoire, L.J. NMR analysis of GPCR conformational landscapes and dynamics. Mol. Cell. Endocrinol. 2019, 484, 69–77. [Google Scholar] [CrossRef] [PubMed]
- Abiko, L.; Grahl, A.; Grzesiek, S. High Pressure Shifts the beta(1)-Adrenergic Receptor to the Active Conformation in the Absence of G Protein. J. Am. Chem. Soc. 2019, 141, 16663–16670. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.J.; Horst, R.; Katritch, V.; Stevens, R.C.; Wuthrich, K. Biased signaling pathways in beta2-adrenergic receptor characterized by 19F-NMR. Science 2012, 335, 1106–1110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nygaard, R.; Zou, Y.; Dror, R.O.; Mildorf, T.J.; Arlow, D.H.; Manglik, A.; Pan, A.C.; Liu, C.W.; Fung, J.J.; Bokoch, M.P.; et al. The Dynamic Process of β2-Adrenergic Receptor Activation. Cell 2013, 152, 532–542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shiraishi, Y.; Natsume, M.; Kofuku, Y.; Imai, S.; Nakata, K.; Mizukoshi, T.; Ueda, T.; Iwaï, H.; Shimada, I. Phosphorylation-induced conformation of β2-adrenoceptor related to arrestin recruitment revealed by NMR. Nat. Commun. 2018, 9, 194. [Google Scholar] [CrossRef] [Green Version]
- Okude, J.; Ueda, T.; Kofuku, Y.; Sato, M.; Nobuyama, N.; Kondo, K.; Shiraishi, Y.; Mizumura, T.; Onishi, K.; Natsume, M.; et al. Identification of a Conformational Equilibrium That Determines the Efficacy and Functional Selectivity of the μ-Opioid Receptor. Angew. Chem. Int. Ed. 2015, 54, 15771–15776. [Google Scholar] [CrossRef] [Green Version]
- Eddy, M.T.; Didenko, T.; Stevens, R.C.; Wüthrich, K. β2-Adrenergic Receptor Conformational Response to Fusion Protein in the Third Intracellular Loop. Structure 2016, 24, 2190–2197. [Google Scholar] [CrossRef] [Green Version]
- Kofuku, Y.; Ueda, T.; Okude, J.; Shiraishi, Y.; Kondo, K.; Mizumura, T.; Suzuki, S.; Shimada, I. Functional dynamics of deuterated β2 -adrenergic receptor in lipid bilayers revealed by NMR spectroscopy. Angew. Chem. Int. Ed. Engl. 2014, 53, 13376–13379. [Google Scholar] [CrossRef]
- Minato, Y.; Suzuki, S.; Hara, T.; Kofuku, Y.; Kasuya, G.; Fujiwara, Y.; Igarashi, S.; Suzuki, E.; Nureki, O.; Hattori, M.; et al. Conductance of P2X4 purinergic receptor is determined by conformational equilibrium in the transmembrane region. Proc. Natl. Acad. Sci. USA 2016, 113, 4741–4746. [Google Scholar] [CrossRef] [Green Version]
- Clark, L.D.; Dikiy, I.; Chapman, K.; Rödström, K.E.J.; Aramini, J.; LeVine, M.V.; Khelashvili, G.; Rasmussen, S.G.F.; Gardner, K.H.; Rosenbaum, D.M. Ligand modulation of sidechain dynamics in a wild-type human GPCR. eLife 2017, 6, e28505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, L.; Neale, C.; Sljoka, A.; Lyda, B.; Pichugin, D.; Tsuchimura, N.; Larda, S.T.; Pomes, R.; Garcia, A.E.; Ernst, O.P.; et al. Mechanistic insights into allosteric regulation of the A2A adenosine G protein-coupled receptor by physiological cations. Nat. Commun. 2018, 9, 1372. [Google Scholar] [CrossRef] [PubMed]
- Goricanec, D.; Stehle, R.; Egloff, P.; Grigoriu, S.; Pluckthun, A.; Wagner, G.; Hagn, F. Conformational dynamics of a G-protein alpha subunit is tightly regulated by nucleotide binding. Proc. Natl. Acad. Sci. USA 2016, 113, E3629–E3638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toyama, Y.; Kano, H.; Mase, Y.; Yokogawa, M.; Osawa, M.; Shimada, I. Dynamic regulation of GDP binding to G proteins revealed by magnetic field-dependent NMR relaxation analyses. Nat. Commun. 2017, 8, 14523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takeuchi, K.; Takahashi, H.; Kawano, S.; Shimada, I. Identification and Characterization of the Slowly Exchanging pH-dependent Conformational Rearrangement in KcsA. J. Biol. Chem. 2007, 282, 15179–15186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cuello, L.G.; Romero, J.G.; Cortes, D.M.; Perozo, E. pH-Dependent Gating in the Streptomyces lividans K+ Channel. Biochemistry 1998, 37, 3229–3236. [Google Scholar] [CrossRef]
- Tokunaga, Y.; Takeuchi, K.; Takahashi, H.; Shimada, I. Allosteric enhancement of MAP kinase p38 alpha’s activity and substrate selectivity by docking interactions. Nat. Struct. Mol. Biol. 2014, 21, 704–711. [Google Scholar] [CrossRef]
- Kim, D.M.; Dikiy, I.; Upadhyay, V.; Posson, D.J.; Eliezer, D.; Nimigean, C.M. Conformational heterogeneity in closed and open states of the KcsA potassium channel in lipid bicelles. J. Gen. Physiol. 2016, 148, 119–132. [Google Scholar] [CrossRef] [Green Version]
- Imai, S.; Osawa, M.; Takeuchi, K.; Shimada, I. Structural basis underlying the dual gate properties of KcsA. Proc. Natl. Acad. Sci. USA 2010, 107, 6216–6221. [Google Scholar] [CrossRef] [Green Version]
- Baker, K.A.; Tzitzilonis, C.; Kwiatkowski, W.; Choe, S.; Riek, R. Conformational dynamics of the KcsA potassium channel governs gating properties. Nat. Struct. Mol. Biol. 2007, 14, 1089–1095. [Google Scholar] [CrossRef]
- Imai, S.; Osawa, M.; Mita, K.; Toyonaga, S.; Machiyama, A.; Ueda, T.; Takeuchi, K.; Oiki, S.; Shimada, I. Functional Equilibrium of the KcsA Structure Revealed by NMR. J. Biol. Chem. 2012, 287, 39634–39641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morrison, E.A.; DeKoster, G.T.; Dutta, S.; Vafabakhsh, R.; Clarkson, M.W.; Bahl, A.; Kern, D.; Ha, T.; Henzler-Wildman, K.A. Antiparallel EmrE exports drugs by exchanging between asymmetric structures. Nature 2012, 481, 45–50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morrison, E.A.; Henzler-Wildman, K.A. Transported Substrate Determines Exchange Rate in the Multidrug Resistance Transporter EmrE. J. Biol. Chem. 2014, 289, 6825–6836. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brettmann, J.B.; Urusova, D.; Tonelli, M.; Silva, J.R.; Henzler-Wildman, K.A. Role of protein dynamics in ion selectivity and allosteric coupling in the NaK channel. Proc. Natl. Acad. Sci. USA 2015, 112, 15366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robinson, A.J.; Kunji, E.R.S. Mitochondrial carriers in the cytoplasmic state have a common substrate binding site. Proc. Natl. Acad. Sci. USA 2006, 103, 2617. [Google Scholar] [CrossRef] [Green Version]
- Brüschweiler, S.; Yang, Q.; Run, C.; Chou, J.J. Substrate-modulated ADP/ATP-transporter dynamics revealed by NMR relaxation dispersion. Nat. Struct. Amp Mol. Biol. 2015, 22, 636. [Google Scholar] [CrossRef] [Green Version]
- Liang, B.; Tamm, L.K. Structure of outer membrane protein G by solution NMR spectroscopy. Proc. Natl. Acad. Sci. USA 2007, 104, 16140. [Google Scholar] [CrossRef] [Green Version]
- Zhuang, T.; Chisholm, C.; Chen, M.; Tamm, L.K. NMR-Based Conformational Ensembles Explain pH-Gated Opening and Closing of OmpG Channel. J. Am. Chem. Soc. 2013, 135, 15101–15113. [Google Scholar] [CrossRef] [Green Version]
- Arora, A.; Abildgaard, F.; Bushweller, J.H.; Tamm, L.K. Structure of outer membrane protein A transmembrane domain by NMR spectroscopy. Nat. Struct. Biol. 2001, 8, 334. [Google Scholar] [CrossRef]
- Horst, R.; Stanczak, P.; Wüthrich, K. NMR Polypeptide Backbone Conformation of the E. coli Outer Membrane Protein, W. Structure 2014, 22, 1204–1209. [Google Scholar] [CrossRef] [Green Version]
- Hwang, P.M.; Choy, W.-Y.; Lo, E.I.; Chen, L.; Forman-Kay, J.D.; Raetz, C.R.H.; Privé, G.G.; Bishop, R.E.; Kay, L.E. Solution structure and dynamics of the outer membrane enzyme PagP by NMR. Proc. Natl. Acad. Sci. USA 2002, 99, 13560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernández, C.; Hilty, C.; Wider, G.; Güntert, P.; Wüthrich, K. NMR Structure of the Integral Membrane Protein OmpX. J. Mol. Biol. 2004, 336, 1211–1221. [Google Scholar] [CrossRef] [PubMed]
- Hiller, S.; Garces, R.G.; Malia, T.J.; Orekhov, V.Y.; Colombini, M.; Wagner, G. Solution Structure of the Integral Human Membrane Protein VDAC-1 in Detergent Micelles. Science 2008, 321, 1206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raschle, T.; Hiller, S.; Yu, T.-Y.; Rice, A.J.; Walz, T.; Wagner, G. Structural and Functional Characterization of the Integral Membrane Protein VDAC-1 in Lipid Bilayer Nanodiscs. J. Am. Chem. Soc. 2009, 131, 17777–17779. [Google Scholar] [CrossRef] [Green Version]
- Villinger, S.; Briones, R.; Giller, K.; Zachariae, U.; Lange, A.; de Groot, B.L.; Griesinger, C.; Becker, S.; Zweckstetter, M. Functional dynamics in the voltage-dependent anion channel. Proc. Natl. Acad. Sci. USA 2010, 107, 22546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bayrhuber, M.; Meins, T.; Habeck, M.; Becker, S.; Giller, K.; Villinger, S.; Vonrhein, C.; Griesinger, C.; Zweckstetter, M.; Zeth, K. Structure of the human voltage-dependent anion channel. Proc. Natl. Acad. Sci. USA 2008, 105, 15370. [Google Scholar] [CrossRef] [Green Version]
- Berardi, M.J.; Shih, W.M.; Harrison, S.C.; Chou, J.J. Mitochondrial uncoupling protein 2 structure determined by NMR molecular fragment searching. Nature 2011, 476, 109. [Google Scholar] [CrossRef] [Green Version]
- Oxenoid, K.; Dong, Y.; Cao, C.; Cui, T.; Sancak, Y.; Markhard, A.L.; Grabarek, Z.; Kong, L.; Liu, Z.; Ouyang, B.; et al. Architecture of the mitochondrial calcium uniporter. Nature 2016, 533, 269. [Google Scholar] [CrossRef] [Green Version]
- OuYang, B.; Xie, S.; Berardi, M.J.; Zhao, X.; Dev, J.; Yu, W.; Sun, B.; Chou, J.J. Unusual architecture of the p7 channel from hepatitis C virus. Nature 2013, 498, 521. [Google Scholar] [CrossRef]
- Van Horn, W.D.; Kim, H.-J.; Ellis, C.D.; Hadziselimovic, A.; Sulistijo, E.S.; Karra, M.D.; Tian, C.; Sönnichsen, F.D.; Sanders, C.R. Solution Nuclear Magnetic Resonance Structure of Membrane-Integral Diacylglycerol Kinase. Science 2009, 324, 1726. [Google Scholar] [CrossRef] [Green Version]
- Zhou, S.; Pettersson, P.; Huang, J.; Sjöholm, J.; Sjöstrand, D.; Pomès, R.; Högbom, M.; Brzezinski, P.; Mäler, L.; Ädelroth, P. Solution NMR structure of yeast Rcf1, a protein involved in respiratory supercomplex formation. Proc. Natl. Acad. Sci. USA 2018, 115, 3048. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Y.; Cierpicki, T.; Jimenez, R.H.F.; Lukasik, S.M.; Ellena, J.F.; Cafiso, D.S.; Kadokura, H.; Beckwith, J.; Bushweller, J.H. NMR Solution Structure of the Integral Membrane Enzyme DsbB: Functional Insights into DsbB-Catalyzed Disulfide Bond Formation. Mol. Cell 2008, 31, 896–908. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reckel, S.; Gottstein, D.; Stehle, J.; Löhr, F.; Verhoefen, M.K.; Takeda, M.; Silvers, R.; Kainosho, M.; Glaubitz, C.; Wachtveitl, J.; et al. Solution NMR Structure of Proteorhodopsin. Angew. Chem. Int. Ed. 2011, 50, 11942–11946. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mazhab-Jafari, M.T.; Marshall, C.B.; Smith, M.J.; Gasmi-Seabrook, G.M.C.; Stathopulos, P.B.; Inagaki, F.; Kay, L.E.; Neel, B.G.; Ikura, M. Oncogenic and RASopathy-associated K-RAS mutations relieve membrane-dependent occlusion of the effector-binding site. Proc. Natl. Acad. Sci. USA 2015, 112, 6625. [Google Scholar] [CrossRef] [Green Version]
- Fang, Z.; Marshall, C.B.; Nishikawa, T.; Gossert, A.D.; Jansen, J.M.; Jahnke, W.; Ikura, M. Inhibition of K-RAS4B by a Unique Mechanism of Action: Stabilizing Membrane-Dependent Occlusion of the Effector-Binding Site. Cell Chem. Biol. 2018, 25, 1327–1336.e4. [Google Scholar] [CrossRef] [Green Version]
- Bossemeyer, D. Protein kinases—Structure and function. FEBS Lett. 1995, 369, 57–61. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Cole, P.A. Catalytic mechanisms and regulation of protein kinases. Methods Enzymol. 2014, 548, 1–21. [Google Scholar] [PubMed] [Green Version]
- Shen, K.; Hines, A.C.; Schwarzer, D.; Pickin, K.A.; Cole, P.A. Protein kinase structure and function analysis with chemical tools. Biochim. Biophys. Acta 2005, 1754, 65–78. [Google Scholar] [CrossRef] [PubMed]
- Huse, M.; Kuriyan, J. The conformational plasticity of protein kinases. Cell 2002, 109, 275–282. [Google Scholar] [CrossRef] [Green Version]
- Endicott, J.A.; Noble, M.E.M.; Johnson, L.N. The Structural Basis for Control of Eukaryotic Protein Kinases. Ann. Rev. Biochem. 2012, 81, 587–613. [Google Scholar] [CrossRef] [PubMed]
- Taylor, S.S.; Knighton, D.R.; Zheng, J.; Ten Eyck, L.F.; Sowadski, J.M. Structural framework for the protein kinase family. Annu. Rev. Cell Biol. 1992, 8, 429–462. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Strand, A.; Robbins, D.; Cobb, M.H.; Goldsmith, E.J. Atomic structure of the MAP kinase ERK2 at 2.3 A resolution. Nature 1994, 367, 704–711. [Google Scholar] [CrossRef] [PubMed]
- Canagarajah, B.; Khokhlatchev, A.; Cobb, M.; Goldsmith, E. Activation mechanism of the MAP kinase ERK2 by dual phosphorylation. Cell 1997, 90, 859–869. [Google Scholar] [CrossRef] [Green Version]
- Xiao, Y.; Lee, T.; Latham, M.P.; Warner, L.R.; Tanimoto, A.; Pardi, A.; Ahn, N.G. Phosphorylation releases constraints to domain motion in ERK2. Proc. Natl. Acad. Sci. USA 2014, 111, 2506. [Google Scholar] [CrossRef] [Green Version]
- Aoto, P.C.; Stanfield, R.L.; Wilson, I.A.; Dyson, H.J.; Wright, P.E. A Dynamic Switch in Inactive p38γ Leads to an Excited State on the Pathway to an Active Kinase. Biochemistry 2019, 58, 5160–5172. [Google Scholar] [CrossRef]
- Henzler-Wildman, K.A.; Lei, M.; Thai, V.; Kerns, S.J.; Karplus, M.; Kern, D. A hierarchy of timescales in protein dynamics is linked to enzyme catalysis. Nature 2007, 450, 913–916. [Google Scholar] [CrossRef]
- Henzler-Wildman, K.A.; Thai, V.; Lei, M.; Ott, M.; Wolf-Watz, M.; Fenn, T.; Pozharski, E.; Wilson, M.A.; Petsko, G.A.; Karplus, M.; et al. Intrinsic motions along an enzymatic reaction trajectory. Nature 2007, 450, 838. [Google Scholar] [CrossRef]
- Masterson, L.R.; Cembran, A.; Shi, L.; Veglia, G. Allostery and binding cooperativity of the catalytic subunit of protein kinase A by NMR spectroscopy and molecular dynamics simulations. Adv. Protein Chem. Struct. Biol. 2012, 87, 363–389. [Google Scholar]
- Saleh, T.; Rossi, P.; Kalodimos, C.G. Atomic view of the energy landscape in the allosteric regulation of Abl kinase. Nat. Struct. Mol. Biol. 2017, 24, 893–901. [Google Scholar] [CrossRef]
- Jahnke, W.; Grotzfeld, R.M.; Pelle, X.; Strauss, A.; Fendrich, G.; Cowan-Jacob, S.W.; Cotesta, S.; Fabbro, D.; Furet, P.; Mestan, J.; et al. Binding or bending: Distinction of allosteric Abl kinase agonists from antagonists by an NMR-based conformational assay. J. Am. Chem. Soc. 2010, 132, 7043–7048. [Google Scholar] [CrossRef]
- Tong, M.; Pelton, J.G.; Gill, M.L.; Zhang, W.; Picart, F.; Seeliger, M.A. Survey of solution dynamics in Src kinase reveals allosteric cross talk between the ligand binding and regulatory sites. Nat. Commun. 2017, 8, 2160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skora, L.; Mestan, J.; Fabbro, D.; Jahnke, W.; Grzesiek, S. NMR reveals the allosteric opening and closing of Abelson tyrosine kinase by ATP-site and myristoyl pocket inhibitors. Proc. Natl. Acad. Sci. USA 2013, 110, E4437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wylie, A.A.; Schoepfer, J.; Jahnke, W.; Cowan-Jacob, S.W.; Loo, A.; Furet, P.; Marzinzik, A.L.; Pelle, X.; Donovan, J.; Zhu, W.; et al. The allosteric inhibitor ABL001 enables dual targeting of BCR–ABL1. Nature 2017, 543, 733. [Google Scholar] [CrossRef] [PubMed]
- Schnell, J.R.; Dyson, H.J.; Wright, P.E. Structure, Dynamics, and Catalytic Function of Dihydrofolate Reductase. Ann. Rev. Biophys. Biomol. Struct. 2004, 33, 119–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boehr, D.D.; McElheny, D.; Dyson, H.J.; Wright, P.E. Millisecond timescale fluctuations in dihydrofolate reductase are exquisitely sensitive to the bound ligands. Proc. Natl. Acad. Sci. USA 2010, 107, 1373–1378. [Google Scholar] [CrossRef] [Green Version]
- Boehr, D.D.; McElheny, D.; Dyson, H.J.; Wright, P.E. The Dynamic Energy Landscape of Dihydrofolate Reductase Catalysis. Science 2006, 313, 1638. [Google Scholar] [CrossRef]
- Carroll, M.J.; Mauldin, R.V.; Gromova, A.V.; Singleton, S.F.; Collins, E.J.; Lee, A.L. Evidence for dynamics in proteins as a mechanism for ligand dissociation. Nat. Chem. Biol. 2012, 8, 246–252. [Google Scholar] [CrossRef] [Green Version]
- Alderson, T.R.; Roche, J.; Gastall, H.Y.; Dias, D.M.; Pritišanac, I.; Ying, J.; Bax, A.; Benesch, J.L.P.; Baldwin, A.J. Local unfolding of the HSP27 monomer regulates chaperone activity. Nat. Commun. 2019, 10, 1068. [Google Scholar] [CrossRef] [Green Version]
- Freilich, R.; Betegon, M.; Tse, E.; Mok, S.-A.; Julien, O.; Agard, D.A.; Southworth, D.R.; Takeuchi, K.; Gestwicki, J.E. Competing protein-protein interactions regulate binding of Hsp27 to its client protein tau. Nat. Commun. 2018, 9, 4563. [Google Scholar] [CrossRef] [Green Version]
- Libich, D.S.; Tugarinov, V.; Clore, G.M. Intrinsic unfoldase/foldase activity of the chaperonin GroEL directly demonstrated using multinuclear relaxation-based NMR. Proc. Natl. Acad. Sci. USA 2015, 112, 8817–8823. [Google Scholar] [CrossRef] [Green Version]
- Huang, R.; Ripstein, Z.A.; Rubinstein, J.L.; Kay, L.E. Cooperative subunit dynamics modulate p97 function. Proc. Natl. Acad. Sci. USA 2019, 116, 158–167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kitevski-LeBlanc, J.L.; Yuwen, T.; Dyer, P.N.; Rudolph, J.; Luger, K.; Kay, L.E. Investigating the Dynamics of Destabilized Nucleosomes Using Methyl-TROSY NMR. J. Am. Chem. Soc. 2018, 140, 4774–4777. [Google Scholar] [CrossRef] [PubMed]
- Choy, M.S.; Li, Y.; Machado, L.E.S.F.; Kunze, M.B.A.; Connors, C.R.; Wei, X.; Lindorff-Larsen, K.; Page, R.; Peti, W. Conformational Rigidity and Protein Dynamics at Distinct Timescales Regulate PTP1B Activity and Allostery. Mol. Cell 2017, 65, 644–658.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whittier, S.K.; Hengge, A.C.; Loria, J.P. Conformational motions regulate phosphoryl transfer in related protein tyrosine phosphatases. Science 2013, 341, 899–903. [Google Scholar] [CrossRef] [Green Version]
- Lisi, G.P.; East, K.W.; Batista, V.S.; Loria, J.P. Altering the allosteric pathway in IGPS suppresses millisecond motions and catalytic activity. Proc. Natl. Acad. Sci. USA 2017, 114, E3414–E3423. [Google Scholar] [CrossRef] [Green Version]
- Singh, H.; Vasa, S.K.; Jangra, H.; Rovó, P.; Päslack, C.; Das, C.K.; Zipse, H.; Schäfer, L.V.; Linser, R. Fast Microsecond Dynamics of the Protein–Water Network in the Active Site of Human Carbonic Anhydrase II Studied by Solid-State NMR Spectroscopy. J. Am. Chem. Soc. 2019, 141, 19276–19288. [Google Scholar] [CrossRef] [Green Version]
- East, K.W.; Newton, J.C.; Morzan, U.N.; Narkhede, Y.B.; Acharya, A.; Skeens, E.; Jogl, G.; Batista, V.S.; Palermo, G.; Lisi, G.P. Allosteric Motions of the CRISPR–Cas9 HNH Nuclease Probed by NMR and Molecular Dynamics. J. Am. Chem. Soc. 2020, 142, 1348–1358. [Google Scholar] [CrossRef]
- Kim, T.H.; Mehrabi, P.; Ren, Z.; Sljoka, A.; Ing, C.; Bezginov, A.; Ye, L.; Pomès, R.; Prosser, R.S.; Pai, E.F. The role of dimer asymmetry and protomer dynamics in enzyme catalysis. Science 2017, 355, eaag2355. [Google Scholar] [CrossRef]
- Takeuchi, K.; Imai, M.; Shimada, I. Conformational equilibrium defines the variable induction of the multidrug-binding transcriptional repressor QacR. Proc. Natl. Acad. Sci. USA 2019, 116, 19963–19972. [Google Scholar] [CrossRef] [Green Version]
- Bah, A.; Vernon, R.M.; Siddiqui, Z.; Krzeminski, M.; Muhandiram, R.; Zhao, C.; Sonenberg, N.; Kay, L.E.; Forman-Kay, J.D. Folding of an intrinsically disordered protein by phosphorylation as a regulatory switch. Nature 2015, 519, 106–109. [Google Scholar] [CrossRef]
- Delaforge, E.; Kragelj, J.; Tengo, L.; Palencia, A.; Milles, S.; Bouvignies, G.; Salvi, N.; Blackledge, M.; Jensen, M.R. Deciphering the Dynamic Interaction Profile of an Intrinsically Disordered Protein by NMR Exchange Spectroscopy. J. Am. Chem. Soc. 2018, 140, 1148–1158. [Google Scholar] [CrossRef] [PubMed]
- Borgia, A.; Borgia, M.B.; Bugge, K.; Kissling, V.M.; Heidarsson, P.O.; Fernandes, C.B.; Sottini, A.; Soranno, A.; Buholzer, K.J.; Nettels, D.; et al. Extreme disorder in an ultrahigh-affinity protein complex. Nature 2018, 555, 61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berlow, R.B.; Dyson, H.J.; Wright, P.E. Hypersensitive termination of the hypoxic response by a disordered protein switch. Nature 2017, 543, 447. [Google Scholar] [CrossRef] [PubMed]
- Mizukoshi, Y.; Takeuchi, K.; Arutaki, M.; Tokunaga, Y.; Takizawa, T.; Hanzawa, H.; Shimada, I. Improvement of Ligand Affinity and Thermodynamic Properties by NMR-Based Evaluation of Local Dynamics and Surface Complementarity in the Receptor-Bound State. Angew. Chem. Int. Ed. 2016, 55, 14606–14609. [Google Scholar] [CrossRef] [PubMed]
- Brady, J.P.; Farber, P.J.; Sekhar, A.; Lin, Y.-H.; Huang, R.; Bah, A.; Nott, T.J.; Chan, H.S.; Baldwin, A.J.; Forman-Kay, J.D.; et al. Structural and hydrodynamic properties of an intrinsically disordered region of a germ cell-specific protein on phase separation. Proc. Natl. Acad. Sci. USA 2017, 114, E8194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akke, M.; Brueschweiler, R.; Palmer, A.G. NMR order parameters and free energy: An analytical approach and its application to cooperative calcium(2+) binding by calbindin D9k. J. Am. Chem. Soc. 1993, 115, 9832–9833. [Google Scholar] [CrossRef]
- Akke, M. Conformational dynamics and thermodynamics of protein-ligand binding studied by NMR relaxation. Biochem. Soc. Trans. 2012, 40, 419–423. [Google Scholar] [CrossRef]
- Wand, A.J. The dark energy of proteins comes to light: Conformational entropy and its role in protein function revealed by NMR relaxation. Curr. Opin. Struct. Biol. 2012, 23, 75–81. [Google Scholar] [CrossRef] [Green Version]
- Igumenova, T.I.; Frederick, K.K.; Wand, A.J. Characterization of the Fast Dynamics of Protein Amino Acid Side Chains Using NMR Relaxation in Solution. Chem. Rev. 2006, 106, 1672–1699. [Google Scholar] [CrossRef]
- Wand, A.J. Dynamic activation of protein function: A view emerging from NMR spectroscopy. Nat. Struct. Biol. 2001, 8, 926–931. [Google Scholar] [CrossRef]
- Wand, A.J.; Sharp, K.A. Measuring Entropy in Molecular Recognition by Proteins. Annu. Rev. Biophys. 2018, 47, 41–61. [Google Scholar] [CrossRef] [PubMed]
- Frederick, K.K.; Marlow, M.S.; Valentine, K.G.; Wand, A.J. Conformational entropy in molecular recognition by proteins. Nature 2007, 448, 325–329. [Google Scholar] [CrossRef] [PubMed]
- Marlow, M.S.; Dogan, J.; Frederick, K.K.; Valentine, K.G.; Wand, A.J. The role of conformational entropy in molecular recognition by calmodulin. Nat. Chem. Biol. 2010, 6, 352–358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caro, J.A.; Harpole, K.W.; Kasinath, V.; Lim, J.; Granja, J.; Valentine, K.G.; Sharp, K.A.; Wand, A.J. Entropy in molecular recognition by proteins. Proc. Natl. Acad. Sci. USA 2017, 114, 6563–6568. [Google Scholar] [CrossRef] [Green Version]
- Kasinath, V.; Sharp, K.A.; Wand, A.J. Microscopic insights into the NMR relaxation-based protein conformational entropy meter. J. Am. Chem. Soc. 2013, 135, 15092–15100. [Google Scholar] [CrossRef] [Green Version]
- Tzeng, S.R.; Kalodimos, C.G. Dynamic activation of an allosteric regulatory protein. Nature 2009, 462, 368–372. [Google Scholar] [CrossRef]
- Diehl, C.; Genheden, S.; Modig, K.; Ryde, U.; Akke, M. Conformational entropy changes upon lactose binding to the carbohydrate recognition domain of galectin-3. J. Biomol. NMR 2009, 45, 157–169. [Google Scholar] [CrossRef]
- Takeuchi, K.; Tokunaga, Y.; Imai, M.; Takahashi, H.; Shimada, I. Dynamic multidrug recognition by multidrug transcriptional repressor LmrR. Sci. Rep. 2014, 4, 6922. [Google Scholar] [CrossRef]
- Lee, C.-J.; Liang, X.; Wu, Q.; Najeeb, J.; Zhao, J.; Gopalaswamy, R.; Titecat, M.; Sebbane, F.; Lemaitre, N.; Toone, E.J.; et al. Drug design from the cryptic inhibitor envelope. Nat. Commun. 2016, 7, 10638. [Google Scholar] [CrossRef] [Green Version]
- Namanja, A.T.; Wang, X.J.; Xu, B.; Mercedes-Camacho, A.Y.; Wilson, B.D.; Wilson, K.A.; Etzkorn, F.A.; Peng, J.W. Toward flexibility-activity relationships by NMR spectroscopy: Dynamics of Pin1 ligands. J. Am. Chem. Soc. 2010, 132, 5607–5609. [Google Scholar] [CrossRef] [Green Version]
- Zintsmaster, J.S.; Wilson, B.D.; Peng, J.W. Dynamics of ligand binding from 13C NMR relaxation dispersion at natural abundance. J. Am. Chem. Soc. 2008, 130, 14060–14061. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tokunaga, Y.; Takeuchi, K.; Shimada, I. Forbidden Coherence Transfer of 19F Nuclei to Quantitatively Measure the Dynamics of a CF3-Containing Ligand in Receptor-Bound States. Molecules 2017, 22, 1492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ikeya, T.; Hanashima, T.; Hosoya, S.; Shimazaki, M.; Ikeda, S.; Mishima, M.; Guntert, P.; Ito, Y. Improved in-cell structure determination of proteins at near-physiological concentration. Sci. Rep. 2016, 6, 38312. [Google Scholar] [CrossRef] [PubMed]
- Danielsson, J.; Mu, X.; Lang, L.; Wang, H.; Binolfi, A.; Theillet, F.X.; Bekei, B.; Logan, D.T.; Selenko, P.; Wennerstrom, H.; et al. Thermodynamics of protein destabilization in live cells. Proc. Natl. Acad. Sci. USA 2015, 112, 12402–12407. [Google Scholar] [CrossRef] [Green Version]
- Hansel, R.; Luh, L.M.; Corbeski, I.; Trantirek, L.; Dotsch, V. In-cell NMR and EPR spectroscopy of biomacromolecules. Angew. Chem. Int. Ed. Engl. 2014, 53, 10300–10314. [Google Scholar] [CrossRef]
- Felli, I.C.; Gonnelli, L.; Pierattelli, R. In-cell(3)C NMR spectroscopy for the study of intrinsically disordered proteins. Nat. Protoc. 2014, 9, 2005–2016. [Google Scholar] [CrossRef]
- Tanaka, T.; Ikeya, T.; Kamoshida, H.; Suemoto, Y.; Mishima, M.; Shirakawa, M.; Guntert, P.; Ito, Y. High-Resolution Protein 3D Structure Determination in Living Eukaryotic Cells. Angew. Chem. Int. Ed. Engl. 2019, 58, 7284–7288. [Google Scholar] [CrossRef]
- Inomata, K.; Ohno, A.; Tochio, H.; Isogai, S.; Tenno, T.; Nakase, I.; Takeuchi, T.; Futaki, S.; Ito, Y.; Hiroaki, H.; et al. High-resolution multi-dimensional NMR spectroscopy of proteins in human cells. Nature 2009, 458, 106–109. [Google Scholar] [CrossRef]
- Mochizuki, A.; Saso, A.; Zhao, Q.; Kubo, S.; Nishida, N.; Shimada, I. Balanced Regulation of Redox Status of Intracellular Thioredoxin Revealed by in-Cell NMR. J. Am. Chem. Soc. 2018, 140, 3784–3790. [Google Scholar] [CrossRef]
- Takeuchi, K.; Arthanari, H.; Imai, M.; Wagner, G.; Shimada, I. Nitrogen-detected TROSY yields comparable sensitivity to proton-detected TROSY for non-deuterated, large proteins under physiological salt conditions. J. Biomol. NMR 2016, 64, 143–151. [Google Scholar] [CrossRef] [Green Version]
- Takeuchi, K.; Arthanari, H.; Wagner, G. Perspective: Revisiting the field dependence of TROSY sensitivity. J.Biomol. NMR 2016, 66, 221–225. [Google Scholar] [CrossRef] [Green Version]
- Takeuchi, K.; Arthanari, H.; Shimada, I.; Wagner, G. Nitrogen detected TROSY at high field yields high resolution and sensitivity for protein NMR. J. Biomol. NMR 2015, 63, 323–331. [Google Scholar] [CrossRef] [Green Version]
- Boeszoermenyi, A.; Chhabra, S.; Dubey, A.; Radeva, D.L.; Burdzhiev, N.T.; Chanev, C.D.; Petrov, O.I.; Gelev, V.M.; Zhang, M.; Anklin, C.; et al. Aromatic 19F-13C TROSY: A background-free approach to probe biomolecular structure, function, and dynamics. Nat. Methods 2019, 16, 333–340. [Google Scholar] [CrossRef]
- Bernado, P.; Shimizu, N.; Zaccai, G.; Kamikubo, H.; Sugiyama, M. Solution scattering approaches to dynamical ordering in biomolecular systems. Biochim. Biophys. Acta Gen. Subj. 2018, 1862, 253–274. [Google Scholar] [CrossRef] [PubMed]
- Rout, M.P.; Sali, A. Principles for Integrative Structural Biology Studies. Cell 2019, 177, 1384–1403. [Google Scholar] [CrossRef] [PubMed]
- van den Bedem, H.; Fraser, J.S. Integrative, dynamic structural biology at atomic resolution—It’s about time. Nat. Methods 2015, 12, 307–318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, Y. Membrane protein structural biology in the era of single particle cryo-EM. Curr. Opin. Struct. Biol. 2018, 52, 58–63. [Google Scholar] [CrossRef]
- Cerofolini, L.; Fragai, M.; Ravera, E.; Diebolder, C.A.; Renault, L.; Calderone, V. Integrative Approaches in Structural Biology: A More Complete Picture from the Combination of Individual Techniques. Biomolecules 2019, 9, 370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lengyel, J.; Hnath, E.; Storms, M.; Wohlfarth, T. Towards an integrative structural biology approach: Combining Cryo-TEM, X-ray crystallography, and NMR. J. Struct. Funct. Genom. 2014, 15, 117–124. [Google Scholar] [CrossRef] [Green Version]
- Purdy, M.D.; Bennett, B.C.; McIntire, W.E.; Khan, A.K.; Kasson, P.M.; Yeager, M. Function and dynamics of macromolecular complexes explored by integrative structural and computational biology. Curr. Opin. Struct. Biol. 2014, 27, 138–148. [Google Scholar] [CrossRef]



© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tokunaga, Y.; Viennet, T.; Arthanari, H.; Takeuchi, K. Spotlight on the Ballet of Proteins: The Structural Dynamic Properties of Proteins Illuminated by Solution NMR. Int. J. Mol. Sci. 2020, 21, 1829. https://doi.org/10.3390/ijms21051829
Tokunaga Y, Viennet T, Arthanari H, Takeuchi K. Spotlight on the Ballet of Proteins: The Structural Dynamic Properties of Proteins Illuminated by Solution NMR. International Journal of Molecular Sciences. 2020; 21(5):1829. https://doi.org/10.3390/ijms21051829
Chicago/Turabian StyleTokunaga, Yuji, Thibault Viennet, Haribabu Arthanari, and Koh Takeuchi. 2020. "Spotlight on the Ballet of Proteins: The Structural Dynamic Properties of Proteins Illuminated by Solution NMR" International Journal of Molecular Sciences 21, no. 5: 1829. https://doi.org/10.3390/ijms21051829
APA StyleTokunaga, Y., Viennet, T., Arthanari, H., & Takeuchi, K. (2020). Spotlight on the Ballet of Proteins: The Structural Dynamic Properties of Proteins Illuminated by Solution NMR. International Journal of Molecular Sciences, 21(5), 1829. https://doi.org/10.3390/ijms21051829