Recent Advances in Practical Methods for Liver Cell Biology: A Short Overview

 ,
, 

 and
and
Abstract
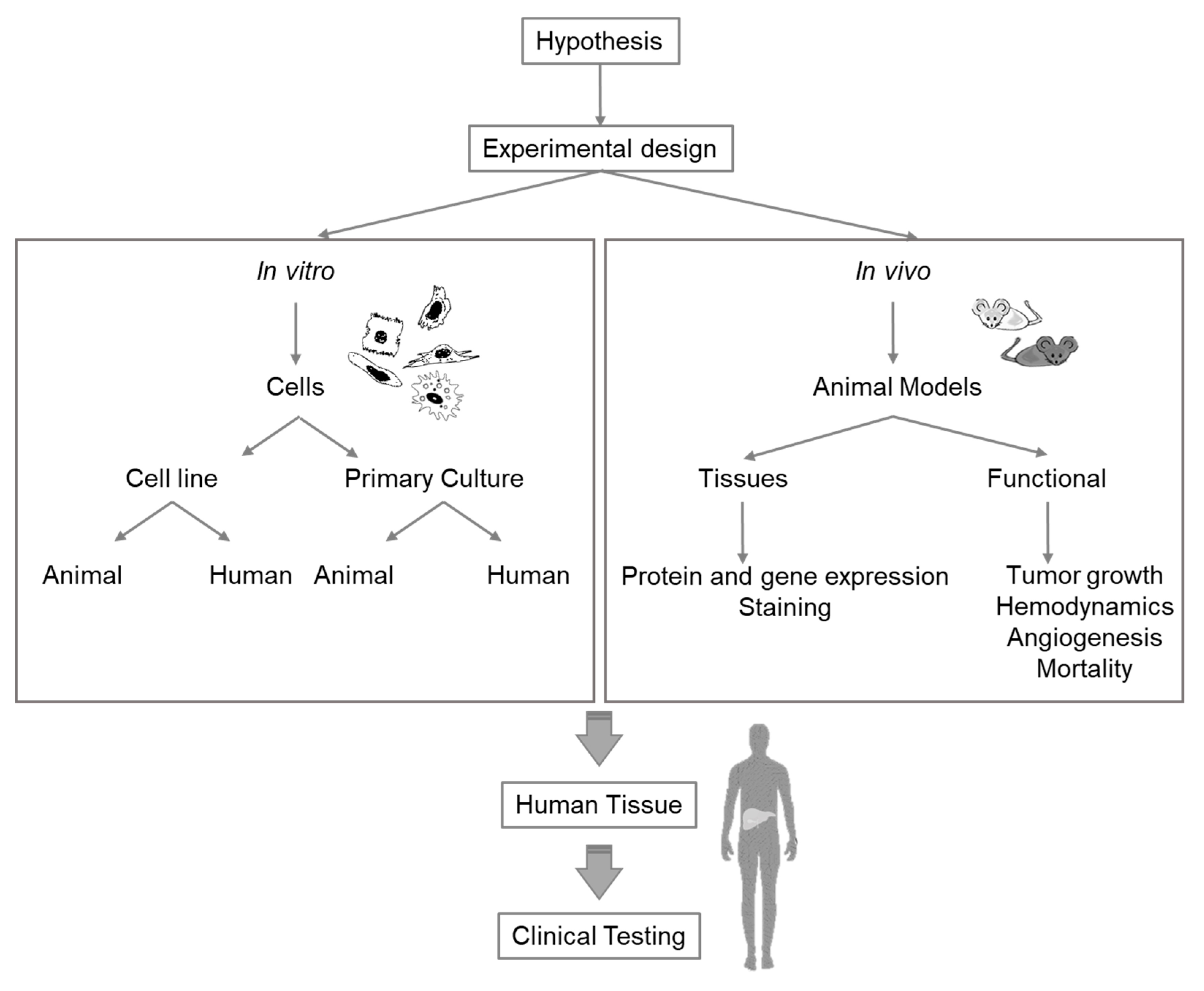
:1. Introduction
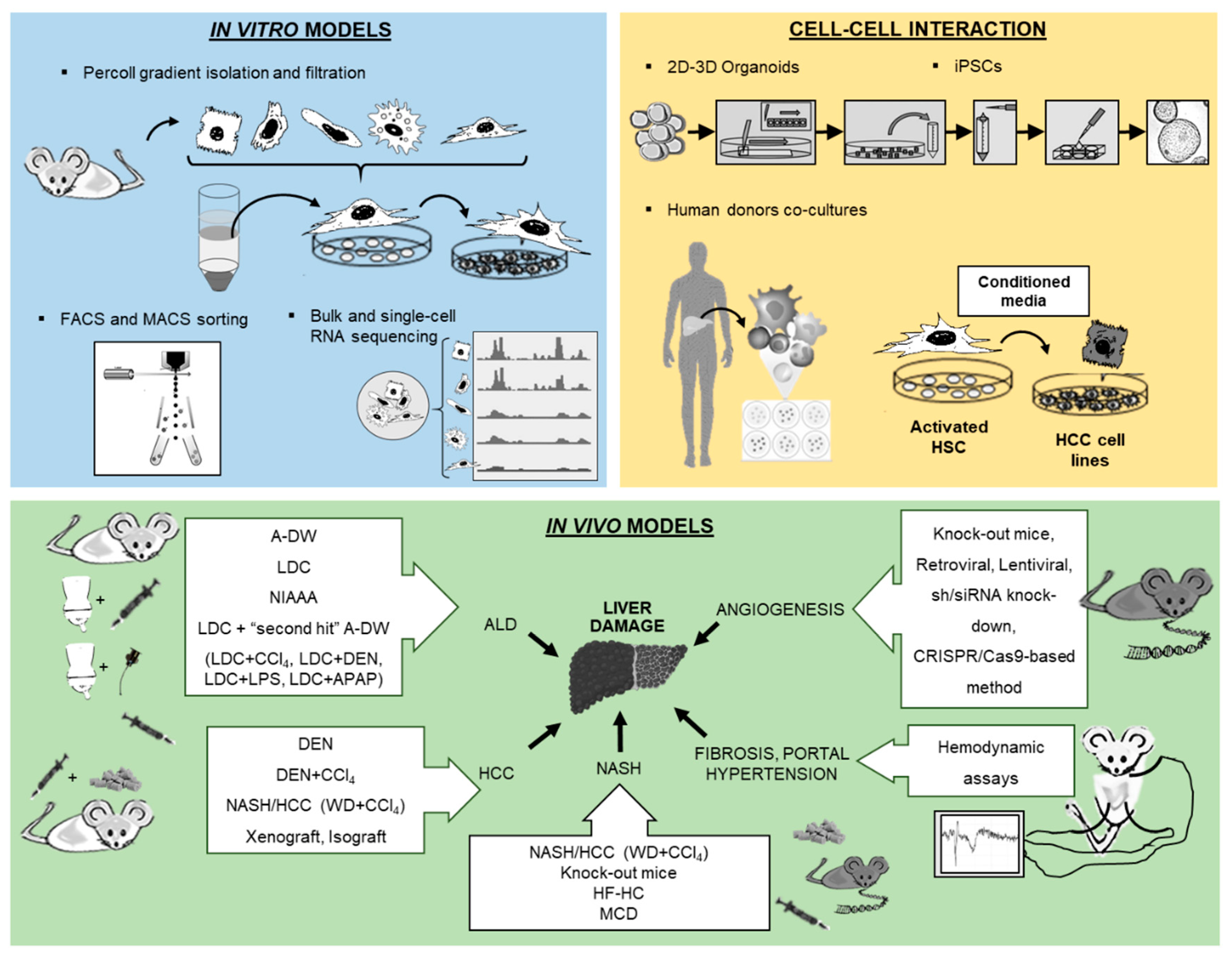
2. In Vitro Models
2.1. Isolation and Culture of Hepatocytes
2.2. HSCs Isolation and Immortalized HSCs Lines
2.3. Analysis of Liver Cells from Different Human Donors and Co-Culture Models
2.4. Isolation and Characterization of Liver Macrophages
2.5. Cellular Models in Biliary Research
3. In Vivo Models
3.1. Angiogenesis and Gene Expression Inhibition
3.2. The Leading Models of Experimental ALD
3.3. Mouse Models for Non-Alcoholic Fatty Liver Disease
3.4. Mouse Models for Hepatocellular Carcinoma
3.5. In Vivo Setup of Liver Disease Models for Surgery and Hemodynamic Assessments
4. Conclusion
Supplementary Materials
Funding
Conflicts of Interest
Abbreviations
| A-DW | Alcohol in drinking water |
| ALD | Alcoholic liver disease |
| APAP | Acetaminophen |
| BAC | Blood alcohol concentration |
| BEC | Biliary epithelial cells |
| CCL4 | Carbon tetrachloride |
| CDAA | Choline-deficient l-amino acid-defined |
| CFTR | Cystic fibrosis transmembrane conductance regulator |
| DEN | N-nitrosodiethylamine |
| FACS | Fluorescence-activated cell sorting |
| FELASA | Federation for Laboratory Animal Science Associations |
| GGT | Γ-Glutamyl-transpeptidase |
| HCC | Hepatocellular carcinoma |
| HFD | High-fat diet |
| HR | Heart rate |
| HSCs | Hepatic stellate cells |
| HVPG | Hepatic venous pressure gradient |
| I.m. | Intramuscularly |
| I.p. | Intraperitoneally |
| IBDU | Intrahepatic bile duct units |
| IEI | Intragastric ethanol infusion |
| iPSCs | Induced pluripotent stem cells |
| KCs | Kupffer cells |
| LDC | Lieber–DeCarli |
| LPS | Lipopolysaccharide |
| MACS | Magnetic activated cell sorting |
| MAP | Mean arterial pressure |
| NAFLD | Non-alcoholic fatty liver disease |
| NASH | Non-alcoholic steatohepatitis |
| NEMO | Nuclear factor (NF)-κB essential modulator |
| NIAAA | Chronic-plus-binge alcohol feeding model |
| PAPPA | Pregnancy-associated plasma protein A |
| PP | Portal pressure |
| PVBF | Portal vein blood flow |
| SMABF | Superior mesenteric artery blood flow |
| WD | Western diet |
References
- Endo-Umeda, K.; Makishima, M. Liver X Receptors Regulate Cholesterol Metabolism and Immunity in Hepatic Nonparenchymal Cells. Int. J. Mol. Sci. 2019, 20, 5045. [Google Scholar] [CrossRef] [Green Version]
- Rauterberg, J.; Voss, B.; Pott, G.; Gerlach, U. Connective tissue components of the normal and fibrotic liver. I. Structure, local distribution and metabolism of connective tissue components in the normal liver and changes in chronic liver diseases. Klin. Wochenschr. 1981, 59, 767–779. [Google Scholar] [CrossRef]
- Yoo, K.-S.; Lim, W.T.; Choi, H.S. Biology of Cholangiocytes: From Bench to Bedside. Gut Liver 2016, 10, 687–698. [Google Scholar] [CrossRef]
- Krenkel, O.; Hundertmark, J.; Abdallah, A.T.; Kohlhepp, M.; Puengel, T.; Roth, T.; Branco, D.P.P.; Mossanen, J.C.; Luedde, T.; Trautwein, C.; et al. Myeloid cells in liver and bone marrow acquire a functionally distinct inflammatory phenotype during obesity-related steatohepatitis. Gut 2019, 69, 551–563. [Google Scholar] [CrossRef]
- Sampaziotis, F.; Justin, A.W.; Tysoe, O.C.; Sawiak, S.; Godfrey, E.M.; Upponi, S.S.; Gieseck, R.L.; de Brito, M.C.; Berntsen, N.L.; Gómez-Vázquez, M.J.; et al. Reconstruction of the mouse extrahepatic biliary tree using primary human extrahepatic cholangiocyte organoids. Nat. Med. 2017, 23, 954–963. [Google Scholar] [CrossRef] [PubMed]
- Tsukamoto, H.; Reidelberger, R.D.; French, S.W.; Largman, C. Long-term cannulation model for blood sampling and intragastric infusion in the rat. Am. J. Physiol. - Regul. Integr. Comp. Physiol. 1984, 247, 595–599. [Google Scholar] [CrossRef] [PubMed]
- Brol, M.J.; Rösch, F.; Schierwagen, R.; Magdaleno, F.; Uschner, F.E.; Manekeller, S.; Queck, A.; Schwarzkopf, K.; Odenthal, M.; Drebber, U.; et al. Combination of CCl4 with alcoholic and metabolic injuries mimics human liver fibrosis. Am. J. Physiol. Gastrointest. Liver Physiol. 2019, 317, G182–G194. [Google Scholar] [CrossRef] [PubMed]
- Bartneck, M.; Schrammen, P.L.; Möckel, D.; Govaere, O.; Liepelt, A.; Krenkel, O.; Ergen, C.; McCain, M.V.; Eulberg, D.; Luedde, T.; et al. The CCR2+ Macrophage Subset Promotes Pathogenic Angiogenesis for Tumor Vascularization in Fibrotic Livers. Cell. Mol. Gastroenterol. Hepatol. 2019, 7, 371–390. [Google Scholar] [CrossRef] [Green Version]
- Tsuchida, T.; Lee, Y.A.; Fujiwara, N.; Ybanez, M.; Allen, B.; Martins, S.; Fiel, M.I.; Goossens, N.; Chou, H.I.; Hoshida, Y.; et al. A simple diet- and chemical-induced murine NASH model with rapid progression of steatohepatitis, fibrosis and liver cancer. J. Hepatol. 2018, 69, 385–395. [Google Scholar] [CrossRef]
- Geerts, A.; Niki, T.; Hellemans, K.; De Craemer, D.; Van Den Berg, K.; Lazou, J.-M.; Stange, G.; Van De Winkel, M.; De Bleser, P. Purification of rat hepatic stellate cells by side scatter-activated cell sorting. Hepatology 1998, 27, 590–598. [Google Scholar] [CrossRef]
- Weiskirchen, S.; Tag, C.G.; Sauer-Lehnen, S.; Tacke, F.; Weiskirchen, R. Isolation and culture of primary murine hepatic stellate cells. Methods Mol. Biol. 2017, 1627, 165–191. [Google Scholar] [PubMed]
- Tacke, F.; Weiskirchen, R. Update on hepatic stellate cells: Pathogenic role in liver fibrosis and novel isolation techniques. Expert Rev. Gastroenterol. Hepatol. 2012, 6, 67–80. [Google Scholar] [CrossRef] [PubMed]
- Mossanen, J.C.; Krenkel, O.; Ergen, C.; Govaere, O.; Liepelt, A.; Puengel, T.; Heymann, F.; Kalthoff, S.; Lefebvre, E.; Eulberg, D.; et al. Chemokine (C-C motif) receptor 2-positive monocytes aggravate the early phase of acetaminophen-induced acute liver injury. Hepatology 2016, 64, 1667–1682. [Google Scholar] [CrossRef] [PubMed]
- Mennone, A.; Alvaro, D.; Cho, W.; Boyer, J.L. Isolation of small polarized bile duct units. Proc. Natl. Acad. Sci. USA 1995, 92, 6527–6531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, L.; Faris, R.A.; Hixson, D.C. Characterization of a mature bile duct antigen expressed on a subpopulation of biliary ductular cells but absent from oval cells. Hepatology 1993, 18, 357–366. [Google Scholar] [CrossRef] [PubMed]
- Mühlbauer, M.; Weiss, T.S.; Thasler, W.E.; Gelbmann, C.M.; Schnabl, B.; Schölmerich, J.; Hellerbrand, C. LPS-mediated NFkappaB activation varies between activated human hepatic stellate cells from different donors. Biochem. Biophys. Res. Commun. 2004, 325, 191–197. [Google Scholar] [CrossRef]
- Hellerbrand, C. Hepatic stellate cells--the pericytes in the liver. Pflugers Arch. 2013, 465, 775–778. [Google Scholar] [CrossRef]
- Amann, T.; Bataille, F.; Spruss, T.; Mühlbauer, M.; Gäbele, E.; Schölmerich, J.; Kiefer, P.; Bosserhoff, A.K.; Hellerbrand, C. Activated hepatic stellate cells promote tumorigenicity of hepatocellular carcinoma. Cancer Sci. 2009, 100, 646–653. [Google Scholar] [CrossRef]
- Meyer, T.; Koch, A.; Ebert, E.-V.; Czech, B.; Mueller, M.; Bosserhoff, A.; Lang, S.A.; Hellerbrand, C. Effect of melanoma cells on proliferation and migration of activated hepatic stellate cells in vitro. Pathol. Res. Pract. 2017, 213, 400–404. [Google Scholar] [CrossRef]
- Wobser, H.; Dorn, C.; Weiss, T.S.; Amann, T.; Bollheimer, C.; Büttner, R.; Schölmerich, J.; Hellerbrand, C. Lipid accumulation in hepatocytes induces fibrogenic activation of hepatic stellate cells. Cell Res. 2009, 19, 996–1005. [Google Scholar] [CrossRef] [Green Version]
- Krenkel, O.; Puengel, T.; Govaere, O.; Abdallah, A.T.; Mossanen, J.C.; Kohlhepp, M.; Liepelt, A.; Lefebvre, E.; Luedde, T.; Hellerbrand, C.; et al. Therapeutic inhibition of inflammatory monocyte recruitment reduces steatohepatitis and liver fibrosis. Hepatology 2018, 67, 1270–1283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scott, C.L.; T’Jonck, W.; Martens, L.; Todorov, H.; Sichien, D.; Soen, B.; Bonnardel, J.; De Prijck, S.; Vandamme, N.; Cannoodt, R.; et al. The Transcription Factor ZEB2 Is Required to Maintain the Tissue-Specific Identities of Macrophages. Immunity 2018, 49, 312–325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- MacParland, S.A.; Liu, J.C.; Ma, X.-Z.; Innes, B.T.; Bartczak, A.M.; Gage, B.K.; Manuel, J.; Khuu, N.; Echeverri, J.; Linares, I.; et al. Single cell RNA sequencing of human liver reveals distinct intrahepatic macrophage populations. Nat. Commun. 2018, 9, 4383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lavon, N.; Yanuka, O.; Benvenisty, N. Differentiation and isolation of hepatic-like cells from human embryonic stem cells. Differentiation. 2004, 72, 230–238. [Google Scholar] [CrossRef]
- Schwartz, R.E.; Linehan, J.L.; Painschab, M.S.; Hu, W.-S.; Verfaillie, C.M.; Kaufman, D.S. Defined conditions for development of functional hepatic cells from human embryonic stem cells. Stem Cells Dev. 2005, 14, 643–655. [Google Scholar] [CrossRef] [Green Version]
- Ghodsizadeh, A.; Taei, A.; Totonchi, M.; Seifinejad, A.; Gourabi, H.; Pournasr, B.; Aghdami, N.; Malekzadeh, R.; Almadani, N.; Salekdeh, G.H.; et al. Generation of liver disease-specific induced pluripotent stem cells along with efficient differentiation to functional hepatocyte-like cells. Stem cell Rev. reports 2010, 6, 622–632. [Google Scholar] [CrossRef]
- Fiorotto, R.; Amenduni, M.; Mariotti, V.; Fabris, L.; Spirli, C.; Strazzabosco, M. Liver diseases in the dish: iPSC and organoids as a new approach to modeling liver diseases. Biochim. Biophys. acta. Mol. basis Dis. 2019, 1865, 920–928. [Google Scholar] [CrossRef]
- Godoy, P.; Hewitt, N.J.; Albrecht, U.; Andersen, M.E.; Ansari, N.; Bhattacharya, S.; Bode, J.G.; Bolleyn, J.; Borner, C.; Böttger, J.; et al. Recent advances in 2D and 3D in vitro systems using primary hepatocytes, alternative hepatocyte sources and non-parenchymal liver cells and their use in investigating mechanisms of hepatotoxicity, cell signaling and ADME. Arch. Toxicol. 2013, 87, 1315–1530. [Google Scholar] [CrossRef] [Green Version]
- Fernández, M.; Semela, D.; Bruix, J.; Colle, I.; Pinzani, M.; Bosch, J. Angiogenesis in liver disease. J. Hepatol. 2009, 50, 604–620. [Google Scholar] [CrossRef]
- Garcia-Pras, E.; Gallego, J.; Coch, L.; Mejias, M.; Fernandez-Miranda, G.; Pardal, R.; Bosch, J.; Mendez, R.; Fernandez, M. Role and therapeutic potential of vascular stem/progenitor cells in pathological neovascularisation during chronic portal hypertension. Gut 2017, 66, 1306–1320. [Google Scholar] [CrossRef]
- Guo, F.; Zheng, K.; Benedé-Ubieto, R.; Cubero, F.J.; Nevzorova, Y.A. The Lieber-DeCarli Diet-A Flagship Model for Experimental Alcoholic Liver Disease. Alcohol. Clin. Exp. Res. 2018, 42, 1828–1840. [Google Scholar] [CrossRef] [PubMed]
- Liquid Diet Technique of ethanol administration: 1989 update. Alcohol Alcohol. 1989, 24, 197–211.
- Bertola, A.; Mathews, S.; Ki, S.H.; Wang, H.; Gao, B. Mouse model of chronic and binge ethanol feeding (the NIAAA model). Nat. Protoc. 2013, 8, 627–637. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roychowdhury, S.; Chiang, D.J.; McMullen, M.R.; Nagy, L.E. Moderate, chronic ethanol feeding exacerbates carbon-tetrachloride-induced hepatic fibrosis via hepatocyte-specific hypoxia inducible factor 1α. Pharmacol. Res. Perspect. 2014, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schierwagen, R.; Maybüchen, L.; Zimmer, S.; Hittatiya, K.; Bäck, C.; Klein, S.; Uschner, F.E.; Reul, W.; Boor, P.; Nickenig, G.; et al. Seven weeks of Western diet in apolipoprotein-E-deficient mice induce metabolic syndrome and non-alcoholic steatohepatitis with liver fibrosis. Sci. Rep. 2015, 5. [Google Scholar] [CrossRef]
- Handa, P.; Morgan-Stevenson, V.; Maliken, B.D.; Nelson, J.E.; Washington, S.; Westerman, M.; Yeh, M.M.; Kowdley, K.V. Iron overload results in hepatic oxidative stress, immune cell activation, and hepatocellular ballooning injury, leading to nonalcoholic steatohepatitis in genetically obese mice. Am. J. Physiol. Gastrointest. Liver Physiol. 2016, 310, G117–G127. [Google Scholar] [CrossRef]
- Marí, M.; Caballero, F.; Colell, A.; Morales, A.; Caballeria, J.; Fernandez, A.; Enrich, C.; Fernandez-Checa, J.C.; García-Ruiz, C. Mitochondrial free cholesterol loading sensitizes to TNF- and Fas-mediated steatohepatitis. Cell Metab. 2006, 4, 185–198. [Google Scholar] [CrossRef]
- Tacke, F. Targeting hepatic macrophages to treat liver diseases. J. Hepatol. 2017, 66, 1300–1312. [Google Scholar] [CrossRef]
- Königshofer, P.; Brusilovskaya, K.; Schwabl, P.; Reiberger, T. Animal models of portal hypertension. Biochim. Biophys. Acta Mol. Basis Dis. 2019, 1865, 1019–1030. [Google Scholar] [CrossRef]
- Baeck, C.; Wehr, A.; Karlmark, K.R.; Heymann, F.; Vucur, M.; Gassler, N.; Huss, S.; Klussmann, S.; Eulberg, D.; Luedde, T.; et al. Pharmacological inhibition of the chemokine CCL2 (MCP-1) diminishes liver macrophage infiltration and steatohepatitis in chronic hepatic injury. Gut 2012, 61, 416–426. [Google Scholar] [CrossRef]
- Von Montfort, C.; Matias, N.; Fernandez, A.; Fucho, R.; Conde de la Rosa, L.; Martinez-Chantar, M.L.; Mato, J.M.; Machida, K.; Tsukamoto, H.; Murphy, M.P.; et al. Mitochondrial GSH determines the toxic or therapeutic potential of superoxide scavenging in steatohepatitis. J. Hepatol. 2012, 57, 852–859. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Herck, M.A.; Vonghia, L.; Francque, S.M. Animal models of nonalcoholic fatty liver disease—a starter’s guide. Nutrients 2017, 9, 1072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Longato, L. Non-alcoholic fatty liver disease (NAFLD): A tale of fat and sugar? Fibrogenes. Tissue Repair 2013, 6, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lindström, P. The physiology of obese-hyperglycemic mice [ob/ob mice]. ScientificWorldJournal. 2007, 7, 666–685. [Google Scholar] [CrossRef] [Green Version]
- Liedtke, C.; Luedde, T.; Sauerbruch, T.; Scholten, D.; Streetz, K.; Tacke, F.; Tolba, R.; Trautwein, C.; Trebicka, J.; Weiskirchen, R. Experimental liver fibrosis research: Update on animal models, legal issues and translational aspects. Fibrogenes. Tissue Repair. 2013, 6, 19. [Google Scholar] [CrossRef] [Green Version]
- Luedde, T.; Beraza, N.; Kotsikoris, V.; van Loo, G.; Nenci, A.; De Vos, R.; Roskams, T.; Trautwein, C.; Pasparakis, M. Deletion of NEMO/IKKgamma in liver parenchymal cells causes steatohepatitis and hepatocellular carcinoma. Cancer Cell 2007, 11, 119–132. [Google Scholar] [CrossRef]
- Smit, J.J.M.; Schinkel, A.H.; Elferink, R.P.J.O.; Groen, A.K.; Wagenaar, E.; van Deemter, L.; Mol, C.A.A.M.; Ottenhoff, R.; van der Lugt, N.M.T.; van Roon, M.A.; et al. Homozygous disruption of the murine MDR2 P-glycoprotein gene leads to a complete absence of phospholipid from bile and to liver disease. Cell 1993, 75, 451–462. [Google Scholar] [CrossRef]
- Tolba, R.; Kraus, T.; Liedtke, C.; Schwarz, M.; Weiskirchen, R. Diethylnitrosamine (DEN)-induced carcinogenic liver injury in mice. Lab. Anim. 2015, 49, 59–69. [Google Scholar] [CrossRef] [Green Version]
- Uehara, T.; Pogribny, I.P.; Rusyn, I. The DEN and CCl4 -Induced Mouse Model of Fibrosis and Inflammation-Associated Hepatocellular Carcinoma. Curr. Protoc. Pharmacol. 2014, 66, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Klein, S.; Hinüber, C.; Hittatiya, K.; Schierwagen, R.; Uschner, F.E.; Strassburg, C.P.; Fischer, H.-P.; Spengler, U.; Trebicka, J. Novel Rat Model of Repetitive Portal Venous Embolization Mimicking Human Non-Cirrhotic Idiopathic Portal Hypertension. PLoS ONE 2016, 11. [Google Scholar] [CrossRef] [Green Version]
- Königshofer, P.; Brusilovskaya, K.; Schwabl, P.; Podesser, B.K.; Trauner, M.; Reiberger, T. Invasive Hemodynamic Characterization of the Portal-hypertensive Syndrome in Cirrhotic Rats. J. Vis. Exp. 2018. [Google Scholar] [CrossRef] [PubMed]
- Klein, S.; Rick, J.; Lehmann, J.; Schierwagen, R.; Schierwagen, I.G.; Verbeke, L.; Hittatiya, K.; Uschner, F.E.; Manekeller, S.; Strassburg, C.P.; et al. Janus-kinase-2 relates directly to portal hypertension and to complications in rodent and human cirrhosis. Gut 2017, 66, 145–155. [Google Scholar] [CrossRef] [PubMed]
- Mejias, M.; Garcia-Pras, E.; Tiani, C.; Miquel, R.; Bosch, J.; Fernandez, M. Beneficial effects of sorafenib on splanchnic, intrahepatic, and portocollateral circulations in portal hypertensive and cirrhotic rats. Hepatology 2009, 49, 1245–1256. [Google Scholar] [CrossRef] [PubMed]
- Calderone, V.; Gallego, J.; Fernandez-Miranda, G.; Garcia-Pras, E.; Maillo, C.; Berzigotti, A.; Mejias, M.; Bava, F.-A.; Angulo-Urarte, A.; Graupera, M.; et al. Sequential Functions of CPEB1 and CPEB4 Regulate Pathologic Expression of Vascular Endothelial Growth Factor and Angiogenesis in Chronic Liver Disease. Gastroenterology 2016, 150, 982–997. [Google Scholar] [CrossRef] [Green Version]
- Maillo, C.; Martín, J.; Sebastián, D.; Hernández-Alvarez, M.; García-Rocha, M.; Reina, O.; Zorzano, A.; Fernandez, M.; Méndez, R. Circadian- and UPR-dependent control of CPEB4 mediates a translational response to counteract hepatic steatosis under ER stress. Nat. Cell Biol. 2017, 19, 94–105. [Google Scholar] [CrossRef]
- Coch, L.; Mejias, M.; Berzigotti, A.; Garcia-Pras, E.; Gallego, J.; Bosch, J.; Mendez, R.; Fernandez, M. Disruption of negative feedback loop between vasohibin-1 and vascular endothelial growth factor decreases portal pressure, angiogenesis, and fibrosis in cirrhotic rats. Hepatology 2014, 60, 633–647. [Google Scholar] [CrossRef]
- Blouin, A.; Bolender, R.P.; Weibel, E.R. Distribution of organelles and membranes between hepatocytes and nonhepatocytes in the rat liver parenchyma. A stereological study. J. Cell Biol. 1977, 72, 441–455. [Google Scholar] [CrossRef]
- Schulze, R.J.; Schott, M.B.; Casey, C.A.; Tuma, P.L.; McNiven, M.A. The cell biology of the hepatocyte: A membrane trafficking machine. J. Cell Biol. 2019, 218, 2096–2112. [Google Scholar] [CrossRef]
- Seglen, P.O. Preparation of Isolated Rat Liver Cells. Methods Cell Biol. 1976, 13, 29–83. [Google Scholar]
- Kegel, V.; Deharde, D.; Pfeiffer, E.; Zeilinger, K.; Seehofer, D.; Damm, G. Protocol for isolation of primary human hepatocytes and corresponding major populations of non-parenchymal liver cells. J. Vis. Exp. 2016, 2016. [Google Scholar] [CrossRef] [Green Version]
- Donato, M.T.; Bolonio, M.; Cabezas, E.; Pelechá, M.; Pareja, E.; Domènech, A.; Castell, J.V.; Gómez-Lechón, M.J.; Tolosa, L. Improved in vivo efficacy of clinical-grade cryopreserved human hepatocytes in mice with acute liver failure. Cytotherapy 2020, 22, 114–121. [Google Scholar] [CrossRef] [PubMed]
- Pournasr, B.; Duncan, S.A. Modeling inborn errors of hepatic metabolism using induced pluripotent stem cells. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 1994–1999. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corbett, J.L.; Duncan, S.A. iPSC-Derived Hepatocytes as a Platform for Disease Modeling and Drug Discovery. Front. Med. 2019, 6, 265. [Google Scholar] [CrossRef] [PubMed]
- Ferreri, A.J.M.; Illerhaus, G.; Zucca, E.; Cavalli, F. Flows and flaws in primary central nervous system lymphoma. Nat. Rev. Clin. Oncol. 2010, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schon, H.T.; Bartneck, M.; Borkham-Kamphorst, E.; Nattermann, J.; Lammers, T.; Tacke, F.; Weiskirchen, R. Pharmacological intervention in hepatic stellate cell activation and hepatic fibrosis. Front. Pharmacol. 2016, 7, 33. [Google Scholar] [CrossRef] [Green Version]
- Knook, D.L.; Seffelaar, A.M.; de Leeuw, A.M. Fat-storing cells of the rat liver. Their isolation and purification. Exp. Cell Res. 1982, 139, 468–471. [Google Scholar] [CrossRef]
- De Minicis, S.; Seki, E.; Uchinami, H.; Kluwe, J.; Zhang, Y.; Brenner, D.A.; Schwabe, R.F. Gene expression profiles during hepatic stellate cell activation in culture and in vivo. Gastroenterology 2007, 132, 1937–1946. [Google Scholar] [CrossRef]
- Feaver, R.E.; Cole, B.K.; Lawson, M.J.; Hoang, S.A.; Marukian, S.; Blackman, B.R.; Figler, R.A.; Sanyal, A.J.; Wamhoff, B.R.; Dash, A. Development of an in vitro human liver system for interrogating nonalcoholic steatohepatitis. JCI insight 2016, 1. [Google Scholar] [CrossRef]
- Mühlbauer, M.; Bosserhoff, A.K.; Hartmann, A.; Thasler, W.E.; Weiss, T.S.; Herfarth, H.; Lock, G.; Schölmerich, J.; Hellerbrand, C. A novel MCP-1 gene polymorphism is associated with hepatic MCP-1 expression and severity of HCV-related liver disease. Gastroenterology 2003, 125, 1085–1093. [Google Scholar] [CrossRef]
- Engelmann, J.C.; Amann, T.; Ott-Rötzer, B.; Nützel, M.; Reinders, Y.; Reinders, J.; Thasler, W.E.; Kristl, T.; Teufel, A.; Huber, C.G.; et al. Causal Modeling of Cancer-Stromal Communication Identifies PAPPA as a Novel Stroma-Secreted Factor Activating NFκB Signaling in Hepatocellular Carcinoma. PLoS Comput. Biol. 2015, 11. [Google Scholar] [CrossRef]
- Krenkel, O.; Tacke, F. Liver macrophages in tissue homeostasis and disease. Nat. Rev. Immunol. 2017, 17, 306–321. [Google Scholar] [CrossRef] [PubMed]
- Ramachandran, P.; Dobie, R.; Wilson-Kanamori, J.R.; Dora, E.F.; Henderson, B.E.P.; Luu, N.T.; Portman, J.R.; Matchett, K.P.; Brice, M.; Marwick, J.A.; et al. Resolving the fibrotic niche of human liver cirrhosis at single-cell level. Nature 2019, 575, 512–518. [Google Scholar] [CrossRef] [PubMed]
- Bartneck, M.; Warzecha, K.T.; Tag, C.G.; Sauer-Lehnen, S.; Heymann, F.; Trautwein, C.; Weiskirchen, R.; Tacke, F. Isolation and time lapse microscopy of highly pure hepatic stellate cells. Anal. Cell. Pathol. (Amst) 2015, 2015, 417023. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonnardel, J.; T’Jonck, W.; Gaublomme, D.; Browaeys, R.; Scott, C.L.; Martens, L.; Vanneste, B.; De Prijck, S.; Nedospasov, S.A.; Kremer, A.; et al. Stellate Cells, Hepatocytes, and Endothelial Cells Imprint the Kupffer Cell Identity on Monocytes Colonizing the Liver Macrophage Niche. Immunity 2019, 51, 638–654.e9. [Google Scholar] [CrossRef] [Green Version]
- Taylor, M.E.; Snelling, T.; Smith, D.F.; Drickamer, K. Absence of a human ortholog of rodent Kupffer cell galactose-binding receptor encoded by the CLEC4f gene. Glycobiology 2019, 29, 332–345. [Google Scholar] [CrossRef]
- Strazzabosco, M.; Fabris, L. Functional Anatomy of Normal Bile Ducts. Anat. Rec. Adv. Integr. Anat. Evol. Biol. 2008, 291, 653–660. [Google Scholar] [CrossRef] [Green Version]
- Fabris, L.; Spirli, C.; Cadamuro, M.; Fiorotto, R.; Strazzabosco, M. Emerging concepts in biliary repair and fibrosis. Am. J. Physiol. Gastrointest. Liver Physiol. 2017, 313, G102–G116. [Google Scholar] [CrossRef] [Green Version]
- Spirlì, C.; Fabris, L.; Duner, E.; Fiorotto, R.; Ballardini, G.; Roskams, T.; Larusso, N.F.; Sonzogni, A.; Okolicsanyi, L.; Strazzabosco, M. Cytokine-stimulated nitric oxide production inhibits adenylyl cyclase and cAMP-dependent secretion in cholangiocytes. Gastroenterology 2003, 124, 737–753. [Google Scholar] [CrossRef] [Green Version]
- Spirlì, C.; Fiorotto, R.; Song, L.; Santos-Sacchi, J.; Okolicsanyi, L.; Masier, S.; Rocchi, L.; Vairetti, M.P.; De Bernard, M.; Melero, S.; et al. Glibenclamide stimulates fluid secretion in rodent cholangiocytes through a cystic fibrosis transmembrane conductance regulator-independent mechanism. Gastroenterology 2005, 129, 220–233. [Google Scholar] [CrossRef]
- Spirlì, C.; Nathanson, M.H.; Fiorotto, R.; Duner, E.; Denson, L.A.; Sanz, J.M.; Di Virgilio, F.; Okolicsanyi, L.; Casagrande, F.; Strazzabosco, M. Proinflammatory cytokines inhibit secretion in rat bile duct epithelium. Gastroenterology 2001, 121, 156–169. [Google Scholar] [CrossRef]
- Fabris, L.; Cadamuro, M.; Fiorotto, R.; Roskams, T.; Spirlì, C.; Melero, S.; Sonzogni, A.; Joplin, R.E.; Okolicsanyi, L.; Strazzabosco, M. Effects of angiogenic factor overexpression by human and rodent cholangiocytes in polycystic liver diseases. Hepatology 2006, 43, 1001–1012. [Google Scholar] [CrossRef] [PubMed]
- Fiorotto, R.; Villani, A.; Kourtidis, A.; Scirpo, R.; Amenduni, M.; Geibel, P.J.; Cadamuro, M.; Spirli, C.; Anastasiadis, P.Z.; Strazzabosco, M. The cystic fibrosis transmembrane conductance regulator controls biliary epithelial inflammation and permeability by regulating Src tyrosine kinase activity. Hepatology 2016, 64, 2118–2134. [Google Scholar] [CrossRef] [PubMed]
- Joplin, R.; Strain, A.J.; Neuberger, J.M. Biliary epithelial cells from the liver of patients with primary biliary cirrhosis: Isolation, characterization, and short-term culture. J. Pathol. 1990, 162, 255–260. [Google Scholar] [CrossRef] [PubMed]
- Strain, A.J.; Wallace, L.; Joplin, R.; Daikuhara, Y.; Ishii, T.; Kelly, D.A.; Neuberger, J.M. Characterization of biliary epithelial cells isolated from needle biopsies of human liver in the presence of hepatocyte growth factor. Am. J. Pathol. 1995, 146, 537–545. [Google Scholar] [PubMed]
- Fiorotto, R.; Amenduni, M.; Mariotti, V.; Fabris, L.; Spirli, C.; Strazzabosco, M. Src kinase inhibition reduces inflammatory and cytoskeletal changes in ΔF508 human cholangiocytes and improves cystic fibrosis transmembrane conductance regulator correctors efficacy. Hepatology 2018, 67, 972–988. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernandez, M. Molecular pathophysiology of portal hypertension. Hepatology 2015, 61, 1406–1415. [Google Scholar] [CrossRef]
- Nowak-Sliwinska, P.; Alitalo, K.; Allen, E.; Anisimov, A.; Aplin, A.C.; Auerbach, R.; Augustin, H.G.; Bates, D.O.; van Beijnum, J.R.; Bender, R.H.F.; et al. Consensus guidelines for the use and interpretation of angiogenesis assays. Angiogenesis 2018, 21, 425–532. [Google Scholar] [CrossRef] [Green Version]
- Reiberger, T.; Angermayr, B.; Schwabl, P.; Rohr-Udilova, N.; Mitterhauser, M.; Gangl, A.; Peck-Radosavljevic, M. Sorafenib attenuates the portal hypertensive syndrome in partial portal vein ligated rats. J. Hepatol. 2009, 51, 865–873. [Google Scholar] [CrossRef]
- Gallego, J.; Garcia-Pras, E.; Mejias, M.; Pell, N.; Schaeper, U.; Fernandez, M. Therapeutic siRNA targeting endothelial KDR decreases portosystemic collateralization in portal hypertension. Sci. Rep. 2017, 7, 14791. [Google Scholar] [CrossRef] [Green Version]
- Mejias, M.; Coch, L.; Berzigotti, A.; Garcia-Pras, E.; Gallego, J.; Bosch, J.; Fernandez, M. Antiangiogenic and antifibrogenic activity of pigment epithelium-derived factor (PEDF) in bile duct-ligated portal hypertensive rats. Gut 2015, 64, 657–666. [Google Scholar] [CrossRef]
- Asrani, S.K.; Devarbhavi, H.; Eaton, J.; Kamath, P.S. Burden of liver diseases in the world. J. Hepatol. 2019, 70, 151–171. [Google Scholar] [CrossRef] [PubMed]
- D’Souza El-Guindy, N.B.; Kovacs, E.J.; De Witte, P.; Spies, C.; Littleton, J.M.; de Villiers, W.J.S.; Lott, A.J.; Plackett, T.P.; Lanzke, N.; Meadows, G.G. Laboratory models available to study alcohol-induced organ damage and immune variations: Choosing the appropriate model. Alcohol. Clin. Exp. Res. 2010, 34, 1489–1511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brandon-Warner, E.; Schrum, L.W.; Schmidt, C.M.; McKillop, I.H. Rodent models of alcoholic liver disease: Of mice and men. Alcohol 2012, 46, 715–725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delire, B.; Stärkel, P.; Leclercq, I. Animal Models for Fibrotic Liver Diseases: What We Have, What We Need, and What Is under Development. J. Clin. Transl. Hepatol. 2015, 3, 53–66. [Google Scholar] [PubMed]
- Itagaki, H.; Shimizu, K.; Morikawa, S.; Ogawa, K.; Ezaki, T. Morphological and functional characterization of non-alcoholic fatty liver disease induced by a methionine-choline-deficient diet in C57BL/6 mice. Int. J. Clin. Exp. Pathol. 2013, 6, 2683–2696. [Google Scholar] [PubMed]
- Tølbøl, K.S.; Stierstorfer, B.; Rippmann, J.F.; Veidal, S.S.; Rigbolt, K.T.G.; Schönberger, T.; Gillum, M.P.; Hansen, H.H.; Vrang, N.; Jelsing, J.; et al. Disease Progression and Pharmacological Intervention in a Nutrient-Deficient Rat Model of Nonalcoholic Steatohepatitis. Dig. Dis. Sci. 2019, 64, 1238–1256. [Google Scholar] [CrossRef] [Green Version]
- Kashireddy, P.V.; Rao, M.S. Lack of peroxisome proliferator-activated receptor α in mice enhances methionine and choline deficient diet-induced steatohepatitis. Hepatol. Res. 2004, 30, 104–110. [Google Scholar] [CrossRef]
- De Minicis, S.; Agostinelli, L.; Rychlicki, C.; Sorice, G.P.; Saccomanno, S.; Candelaresi, C.; Giaccari, A.; Trozzi, L.; Pierantonelli, I.; Mingarelli, E.; et al. HCC development is associated to peripheral insulin resistance in a mouse model of NASH. PLoS ONE 2014, 9. [Google Scholar] [CrossRef]
- Yin, H.-Q.; Kim, M.; Kim, J.-H.; Kong, G.; Lee, M.-O.; Kang, K.-S.; Yoon, B.-I.; Kim, H.-L.; Lee, B.-H. Hepatic gene expression profiling and lipid homeostasis in mice exposed to steatogenic drug, tetracycline. Toxicol. Sci. 2006, 94, 206–216. [Google Scholar] [CrossRef] [Green Version]
- Nath, S.; Ghosh, S.K.; Choudhury, Y. A murine model of type 2 diabetes mellitus developed using a combination of high fat diet and multiple low doses of streptozotocin treatment mimics the metabolic characteristics of type 2 diabetes mellitus in humans. J. Pharmacol. Toxicol. Methods 2017, 84, 20–30. [Google Scholar] [CrossRef]
- Caviglia, J.M.; Schwabe, R.F. Mouse models of liver cancer. Methods Mol. Biol. 2015, 1267, 165–183. [Google Scholar] [PubMed]
- Dapito, D.H.; Mencin, A.; Gwak, G.Y.; Pradere, J.P.; Jang, M.K.; Mederacke, I.; Caviglia, J.M.; Khiabanian, H.; Adeyemi, A.; Bataller, R.; et al. Promotion of Hepatocellular Carcinoma by the Intestinal Microbiota and TLR4. Cancer Cell 2012, 21, 504–516. [Google Scholar] [CrossRef] [Green Version]
- Dow, M.; Pyke, R.M.; Tsui, B.Y.; Alexandrov, L.B.; Nakagawa, H.; Taniguchi, K.; Seki, E.; Harismendy, O.; Shalapour, S.; Karin, M.; et al. Integrative genomic analysis of mouse and human hepatocellular carcinoma. Proc. Natl. Acad. Sci. U. S. A. 2018, 115, E9879–E9888. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.-S.; Chu, I.-S.; Mikaelyan, A.; Calvisi, D.F.; Heo, J.; Reddy, J.K.; Thorgeirsson, S.S. Application of comparative functional genomics to identify best-fit mouse models to study human cancer. Nat. Genet. 2004, 36, 1306–1311. [Google Scholar] [CrossRef] [PubMed]
- Pimpin, L.; Cortez-Pinto, H.; Negro, F.; Corbould, E.; Lazarus, J.V.; Webber, L.; Sheron, N. EASL HEPAHEALTH Steering Committee Burden of liver disease in Europe: Epidemiology and analysis of risk factors to identify prevention policies. J. Hepatol. 2018, 69, 718–735. [Google Scholar] [CrossRef]
- Reiberger, T.; Mandorfer, M. Beta adrenergic blockade and decompensated cirrhosis. J. Hepatol. 2017, 66, 849–859. [Google Scholar] [CrossRef] [Green Version]
- Directive 2010/63/EU of the European Parliament and of the Council on the protection of animals used for scientific purposes. Official Journal of the European Union. 2010. L276/33–L276/79 (22 September 2010). Available online: http://eur-lex.europa.eu/LexUriServ (accessed on 5 November 2019).
- EARA Statement supporting European Directive 2010/63/EU on the protection of animals used for scientific purposes n.d. Available online: http://www.ehnheart.org/projects/1037:eara-statement-supporting-european-directive-201063eu-on-the-protection-of-animals-used-for-scient (accessed on 5 November 2019).
- Mähler, M.; Berar, M.; Feinstein, R.; Gallagher, A.; Illgen-Wilcke, B.; Pritchett-Corning, K.; Raspa, M. FELASA recommendations for the health monitoring of mouse, rat, hamster, guinea pig and rabbit colonies in breeding and experimental units. Lab. Anim. 2014, 48, 178–192. [Google Scholar]
- Russell, W.M.S.; Burch, R. The Principles of Humane Experimental Technique; Methuen: London, UK, 1959. [Google Scholar]
- Gargiulo, S.; Greco, A.; Gramanzini, M.; Esposito, S.; Affuso, A.; Brunetti, A.; Vesce, G. Mice Anesthesia, Analgesia, and Care, Part I: Anesthetic Considerations in Preclinical Research. ILAR J. 2012, 53, E55–E69. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
| Model | Method | Application | References |
|---|---|---|---|
| In vitro | FACS and MACS sorting | HSC | [10,11,12] |
| Macrophages (KCs and monocytes) | [13] | ||
| Percoll gradient isolation and filtration | Cholangiocytes | [14,15] | |
| Human donor co-cultures | HSC | [16,17,18,19,20] | |
| Hepatocytes | [20] | ||
| Bulk sequencing and single-cell RNA | Macrophages (KCs and monocytes) | [13,21,22,23] | |
| iPSCs | Hepatocytes | [24,25,26] | |
| Cholangiocytes | [27] | ||
| Organoids | 3D liver organoids | Hepatocytes | [28] |
| Cholangiocytes | [27] | ||
| 2D and 3D spheroids for angiogenesis | Endothelial cells Angiogenesis | [29,30] | |
| In vivo | A-DW, LDC, NIAAA LDC + “second hit” (LDC + CCl4, LDC + DEN, LDC + LPS, LDC + APAP) | Alcoholic liver disease | [6,7,31,32,33,34] |
| NASH/HCC (WD + CCl4) Knock-out mice HFD HF-HC MCD | Non-alcoholic liver disease | [1,7,9,35,36,37,38,39,40,41,42,43,44] | |
| Xenograft Isograft DEN DEN + CCL4 NASH/HCC (WD + CCl4) | Hepatocarcinoma | [7,9,45,46,47,48,49] | |
| Hemodynamic assays Monitoring protocols HVPG CT- or MRI-based cross imaging | Portal hypertension | [39,50,51,52] | |
| Knock-out mice, retroviral, lentiviral, sh/siRNA knock-down, CRISPR/Cas9-based method | Angiogenesis | [53,54,55,56] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Torres, S.; Abdullah, Z.; Brol, M.J.; Hellerbrand, C.; Fernandez, M.; Fiorotto, R.; Klein, S.; Königshofer, P.; Liedtke, C.; Lotersztajn, S.; et al. Recent Advances in Practical Methods for Liver Cell Biology: A Short Overview. Int. J. Mol. Sci. 2020, 21, 2027. https://doi.org/10.3390/ijms21062027
Torres S, Abdullah Z, Brol MJ, Hellerbrand C, Fernandez M, Fiorotto R, Klein S, Königshofer P, Liedtke C, Lotersztajn S, et al. Recent Advances in Practical Methods for Liver Cell Biology: A Short Overview. International Journal of Molecular Sciences. 2020; 21(6):2027. https://doi.org/10.3390/ijms21062027
Chicago/Turabian StyleTorres, Sandra, Zeinab Abdullah, Maximilian J Brol, Claus Hellerbrand, Mercedes Fernandez, Romina Fiorotto, Sabine Klein, Philipp Königshofer, Christian Liedtke, Sophie Lotersztajn, and et al. 2020. "Recent Advances in Practical Methods for Liver Cell Biology: A Short Overview" International Journal of Molecular Sciences 21, no. 6: 2027. https://doi.org/10.3390/ijms21062027
APA StyleTorres, S., Abdullah, Z., Brol, M. J., Hellerbrand, C., Fernandez, M., Fiorotto, R., Klein, S., Königshofer, P., Liedtke, C., Lotersztajn, S., Nevzorova, Y. A., Schierwagen, R., Reiberger, T., Uschner, F. E., Tacke, F., Weiskirchen, R., & Trebicka, J. (2020). Recent Advances in Practical Methods for Liver Cell Biology: A Short Overview. International Journal of Molecular Sciences, 21(6), 2027. https://doi.org/10.3390/ijms21062027