Recent Overview of the Use of iPSCs Huntington’s Disease Modeling and Therapy


Abstract
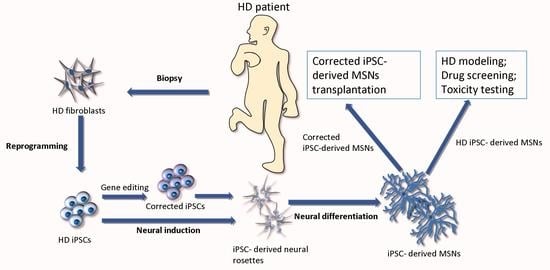
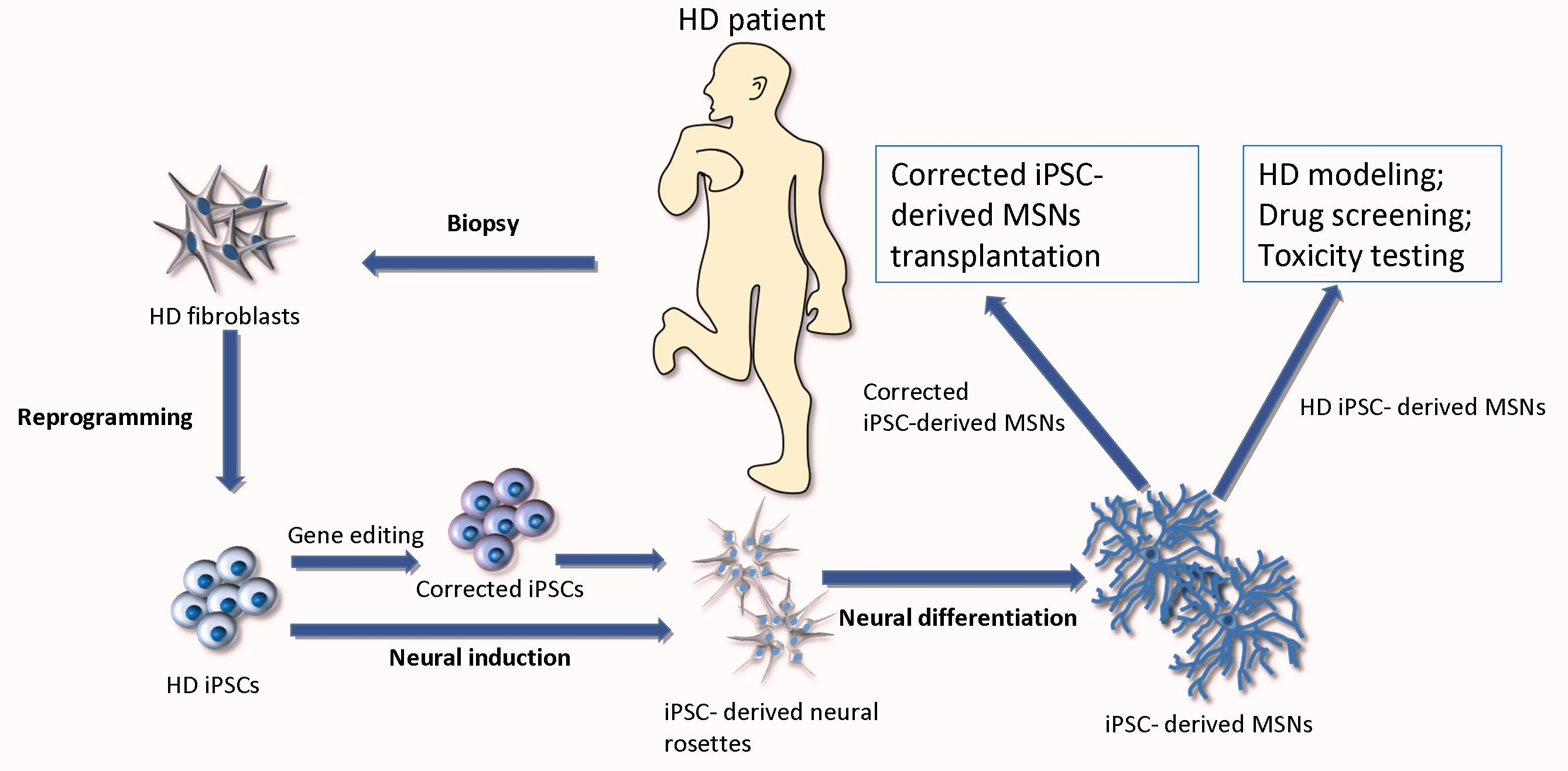
:
1. Introduction
2. Differentiation of iPSCs into MSNs
3. iPSC-Based Modeling of HD
4. iPSC-Derived Brain Organoid Models
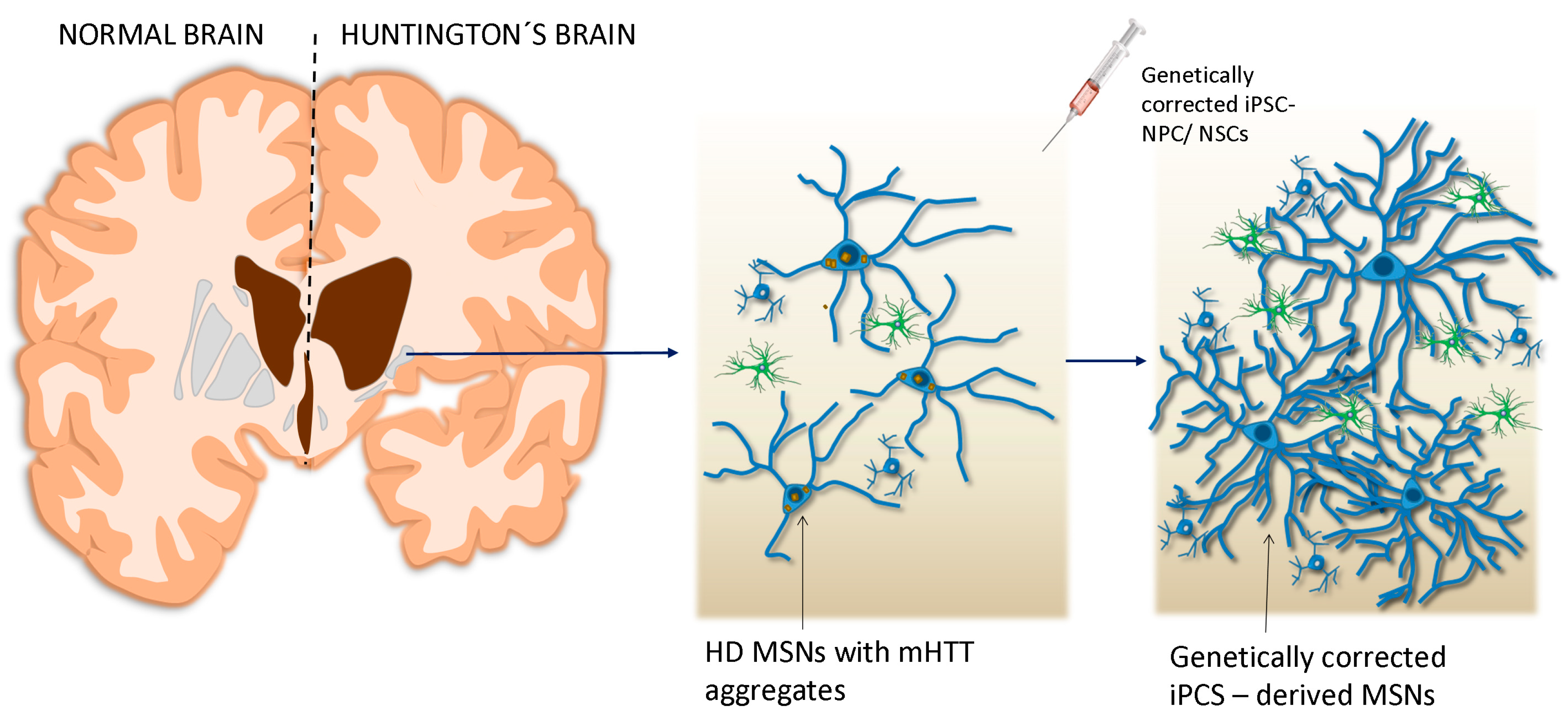
5. Gene Therapy for HD
6. Concluding Remarks
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kaye, J.A.; Finkbeiner, S. Modeling Huntington’s disease with induced pluripotent stem cells. Mol. Cell. Neurosci. 2013, 56, 50–64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Juopperi, T.A.; Kim, W.R.; Chiang, C.H.; Yu, H.; Margolis, R.L.; Ross, C.A.; Ming, G.L.; Song, H. Astrocytes generated from patient induced pluripotent stem cells recapitulate features of Huntington’s disease patient cells. Mol. Brain 2012, 5, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wyant, K.J.; Ridder, A.J.; Dayalu, P. Huntington’s Disease-Update on Treatments. Curr. Neurol. Neurosci. Rep. 2017, 17, 33. [Google Scholar] [CrossRef] [PubMed]
- Bates, G.P.; Dorsey, R.; Gusella, J.F.; Hayden, M.R.; Kay, C.; Leavitt, B.R.; Nance, M.; Ross, C.A.; Scahill, R.I.; Wetzel, R.; et al. Huntington’s disease. Nat. Rev. Dis. Primers 2015, 1, 15005. [Google Scholar] [CrossRef]
- Smith, D.K.; He, M.; Zhang, C.L.; Zheng, J.C. The therapeutic potential of cell identity reprogramming for the treatment of aging-related neurodegenerative disorders. Prog. Neurobiol. 2017, 157, 212–229. [Google Scholar] [CrossRef] [Green Version]
- Bachoud-Lévi, A.C.; Gaura, V.; Brugières, P.; Lefaucheur, J.P.; Boissé, M.F.; Maison, P.; Baudic, S.; Ribeiro, M.J.; Bourdet, C.; Remy, P.; et al. Effect of fetal neural transplants in patients with Huntington’s disease 6 years after surgery: A long-term follow-up study. Lancet Neurol. 2006, 5, 303–309. [Google Scholar] [CrossRef]
- Carter, R.L.; Chan, A.W. Pluripotent stem cells models for Huntington’s disease: Prospects and challenges. J. Genet. Genom. 2012, 39, 253–259. [Google Scholar] [CrossRef] [Green Version]
- Haddad, M.S.; Wenceslau, C.V.; Pompeia, C.; Kerkis, I. Cell-based technologies for Huntington’s disease. Dement. Neuropsychol. 2016, 10, 287–295. [Google Scholar] [CrossRef]
- Chen, Y.; Carter, R.L.; Cho, I.K.; Chan, A.W. Cell-based therapies for Huntington’s disease. Drug Discov. Today 2014, 19, 980–984. [Google Scholar] [CrossRef] [Green Version]
- Bachoud-Lévi, A.C. From open to large-scale randomized cell transplantation trials in Huntington’s disease: Lessons from the multicentric intracerebral grafting in Huntington’s disease trial (MIG-HD) and previous pilot studies. Prog. Brain Res. 2017, 230, 227–261. [Google Scholar]
- Golas, M.M. Human cellular models of medium spiny neuron development and Huntington disease. Life Sci. 2018, 209, 179–196. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.; An, M.C.; Montoro, D.; Ellerby, L.M. Characterization of Human Huntington’s Disease Cell Model from Induced Pluripotent Stem Cells. PLoS Curr. 2010, 2, RRN1193. [Google Scholar] [CrossRef] [PubMed]
- Camnasio, S.; Delli Carri, A.; Lombardo, A.; Grad, I.; Mariotti, C.; Castucci, A.; Rozell, B.; Lo Riso, P.; Castiglioni, V.; Zuccato, C.; et al. The first reported generation of several induced pluripotent stem cell lines from homozygous and heterozygous Huntington’s disease patients demonstrates mutation related enhanced lysosomal activity. Neurobiol. Dis. 2012, 46, 41–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Friedlander, R.M. Using non-coding small RNAs to develop therapies for Huntington’s disease. Gene Ther. 2011, 18, 1139–1149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geater, C.; Hernandez, S.; Thompson, L.; Mattis, V.B. Cellular Models: HD Patient-Derived Pluripotent Stem Cells. Methods Mol. Biol. 2018, 1780, 41–73. [Google Scholar] [PubMed]
- Csobonyeiova, M.; Polak, S.; Zamborsky, R.; Danisovic, L. Recent Progress in the Regeneration of Spinal Cord Injuries by Induced Pluripotent Stem Cells. Int. J. Mol. Sci. 2019, 20, 3838. [Google Scholar] [CrossRef] [Green Version]
- Delli Carri, A.; Onorati, M.; Castiglioni, V.; Faedo, A.; Camnasio, S.; Toselli, M.; Biella., G.; Cattaneo, E. Human pluripotent stem cell differentiation into authentic striatal projection neurons. Stem Cell Rev. Rep. 2013, 9, 461–474. [Google Scholar] [CrossRef]
- Chiu, F.L.; Lin, J.T.; Chuang, C.Y.; Chien, T.; Chen, C.M.; Chen, K.H.; Hsiao, H.Y.; Lin, Y.S.; Chern, Y.; Kuo, H.C. Elucidating the role of the A2A adenosine receptor in neurodegeneration using neurons derived from Huntington’s disease iPSCs. Hum. Mol. Genet. 2015, 24, 6066–6079. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Qiao, F.; Leiferman, P.C.; Ross, A.; Schlenker, E.H.; Wang, H. FOXOs modulate proteasome activity in human-induced pluripotent stem cells of Huntington’s disease and their derived neural cells. Hum. Mol. Genet. 2017, 26, 4416–4428. [Google Scholar] [CrossRef] [Green Version]
- Carter, R.L.; Chen, Y.; Kunkanjanawan, T.; Xu, Y.; Moran, S.P.; Putkhao, K.; Yang, J.; Huang, A.H.; Parnpai, R.; Chan, A.W. Reversal of cellular phenotypes in neural cells derived from Huntington’s disease monkey-induced pluripotent stem cells. Stem Cell Rep. 2014, 3, 585–593. [Google Scholar] [CrossRef] [Green Version]
- Cho, I.K.; Yang, B.; Forest, C.; Qian, L.; Chan, A.W.S. Amelioration of Huntington’s disease phenotype in astrocytes derived from iPSC-derived neural progenitor cells of Huntington’s disease monkeys. PLoS ONE 2019, 14, e0214156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- An, M.C.; Zhang, N.; Scott, G.; Montoro, D.; Wittkop, T.; Mooney, S.; Melov, S.; Ellerby, L.M. Genetic correction of Huntington’s disease phenotypes in induced pluripotent stem cells. Cell Stem Cell 2012, 11, 253–263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nekrasov, E.D.; Vigont, V.A.; Klyushnikov, S.A.; Lebedeva, O.S.; Vassina, E.M.; Bogomazova, A.N.; Chestkov, I.V.; Semashko, T.A.; Kiseleva, E.; Suldina, L.A.; et al. Manifestation of Huntington’s disease pathology in human induced pluripotent stem cell-derived neurons. Mol. Neurodegener. 2016, 11, 27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Le, W. Modeling neurodegenerative diseases in Caenorhabditis elegans. Exp. Neurol. 2013, 250, 94–103. [Google Scholar] [CrossRef] [PubMed]
- Green, E.W.; Giorgini, F. Choosing and using Drosophila models to characterize modifiers of Huntington’s disease. Biochem. Soc. Trans. 2012, 40, 739–745. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.; Wang, C.E.; Zhao, B.; Li, W.; Ouyang, Z.; Liu, Z.; Yang, H.; Fan, P.; O’Neill, A.; Gu, W.; et al. Expression of Huntington’s disease protein results in apoptotic neurons in the brains of cloned transgenic pigs. Hum. Mol. Genet. 2010, 19, 3983–3994. [Google Scholar] [CrossRef]
- Ardan, T.; Baxa, M.; Levinská, B.; Sedláčková, M.; Nguyen, T.D.; Klíma, J.; Juhás, Š.; Juhásová, J.; Šmatlíková, P.; Vochozková, P.; et al. Transgenic minipig model of Huntington’s disease exhibiting gradually progressing neurodegeneration. Dis. Models Mech. 2019, 13, Dmm.041319. [Google Scholar] [CrossRef] [Green Version]
- Figiel, M.; Szlachcic, W.J.; Switonski, P.M.; Gabka, A.; Krzyzosiak, W.J. Mouse models of polyglutamine diseases: Review and data table. Part I. Mol. Neurobiol. 2012, 46, 393–429. [Google Scholar] [CrossRef] [Green Version]
- Switonski, P.M.; Szlachcic, W.J.; Gabka, A.; Krzyzosiak, W.J.; Figiel, M. Mouse models of polyglutamine diseases in therapeutic approaches: Review and data table. Part II. Mol. Neurobiol. 2012, 46, 430–466. [Google Scholar] [CrossRef] [Green Version]
- Yang, S.H.; Cheng, P.H.; Banta, H.; Piotrowska-Nitsche, K.; Yang, J.J.; Cheng, E.C.; Snyder, B.; Larkin, K.; Liu, J.; Orkin, J.; et al. Towards a transgenic model of Huntington’s disease in a non-human primate. Nature 2008, 453, 921–924. [Google Scholar] [CrossRef] [Green Version]
- Chang, R.; Liu, X.; Li, S.; Li, X.J. Transgenic animal models for study of the pathogenesis of Huntington’s disease and therapy. Drug Des. Dev. Ther. 2015, 9, 2179–2188. [Google Scholar]
- Bordoni, M.; Rey, F.; Fantini, V.; Pansarasa, O.; Di Giulio, A.M.; Carelli, S.; Cereda, C. From Neuronal Differentiation of iPSCs to 3D Neuro-Organoids: Modelling and Therapy of Neurodegenerative Diseases. Int. J. Mol. Sci. 2018, 19, 3972. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-Gharaibeh, A.; Culver, R.; Stewart, A.N.; Srinageshwar, B.; Spelde, K.; Frollo, L.; Kolli, N.; Story, D.; Paladugu, L.; Anwar, S.; et al. Induced Pluripotent Stem Cell-Derived Neural Stem Cell Transplantations Reduced Behavioral Deficits and Ameliorated Neuropathological Changes in YAC128 Mouse Model of Huntington’s Disease. Front. Neurosci. 2017, 11, 628. [Google Scholar] [CrossRef]
- Szlachcic, W.J.; Switonski, P.M.; Krzyzosiak, W.J.; Figlerowicz, M.; Figiel, M. Huntington disease iPSCs show early molecular changes in intracellular signaling, the expression of oxidative stress proteins and the p53 pathway. Dis. Models Mech. 2015, 8, 1047–1057. [Google Scholar] [CrossRef] [Green Version]
- Świtońska, K.; Szlachcic, W.J.; Handschuh, L.; Wojciechowski, P.; Marczak, Ł.; Stelmaszczuk, M.; Figlerowicz, M.; Figiel, M. Identification of Altered Developmental Pathways in Human Juvenile HD iPSC With 71Q and 109Q Using Transcriptome Profiling. Front. Cell. Neurosci. 2019, 12, 528. [Google Scholar] [CrossRef]
- Kunkanjanawan, T.; Carter, R.; Ahn, K.S.; Yang, J.; Parnpai, R.; Chan, A.W.S. Induced Pluripotent HD Monkey Stem Cells Derived Neural Cells for Drug Discovery. SLAS Discov. 2017, 22, 696–705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, I.K.; Hunter, C.E.; Ye, S.; Pongos, A.L.; Chan, A.W.S. Combination of stem cell and gene therapy ameliorates symptoms in Huntington’s disease mice. NPJ Regen. Med. 2019, 4, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, X.; Disatnik, M.H.; Monbureau, M.; Shamloo, M.; Mochly-Rosen, D.; Qi, X. Inhibition of mitochondrial fragmentation diminishes Huntington’s disease-associated neurodegeneration. J. Clin. Investig. 2013, 123, 5371–5388. [Google Scholar] [CrossRef] [PubMed]
- Popoli, P.; Blum, D.; Domenici, M.R.; Burnouf, S.; Chern, Y. A critical evaluation of adenosine A2A receptors as potentially “druggable” targets in Huntington’s disease. Curr. Pharm. Des. 2008, 14, 1500–1511. [Google Scholar] [CrossRef]
- Pacitti, D.; Privolizzi, R.; Bax, B.E. Organs to Cells and Cells to Organoids: The Evolution of in vitro Central Nervous System Modelling. Front. Cell. Neurosci. 2019, 13, 129. [Google Scholar] [CrossRef]
- Wu, Y.Y.; Chiu, F.L.; Yeh, C.S.; Kuo, H.C. Opportunities and challenges for the use of induced pluripotent stem cells in modelling neurodegenerative disease. R. Soc. Open Biol. 2019, 9, 180177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Hu, D.; Shang, Y.; Qi, X. Using induced pluripotent stem cell neuronal models to study neurodegenerative diseases. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165431. [Google Scholar] [CrossRef] [PubMed]
- Lancaster, M.A.; Knoblich, J.A. Generation of cerebral organoids from human pluripotent stem cells. Nat. Protoc. 2014, 9, 2329–2340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, D.; Pekkanen-Mattila, M.; Shahsavani, M.; Falk, A.; Teixeira, A.I.; Herland, A. A 3D Alzheimer’s disease culture model and the induction of P21-activated kinase mediated sensing in iPSC derived neurons. Biomaterials 2014, 35, 1420–1428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raja, W.K.; Mungenast, A.E.; Lin, Y.T.; Ko, T.; Abdurrob, F.; Seo, J.; Tsai, L.H. Self-Organizing 3D Human Neural Tissue Derived from Induced Pluripotent Stem Cells Recapitulate Alzheimer’s Disease Phenotypes. PLoS ONE 2016, 11, e0161969. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Son, M.Y.; Sim, H.; Son, Y.S.; Jung, K.B.; Lee, M.O.; Oh, J.H.; Chung, S.K.; Jung, C.R.; Kim, J. Distinctive genomic signature of neural and intestinal organoids from familial Parkinson’s disease patient-derived induced pluripotent stem cells. Neuropathol. Appl. Neurobiol. 2017, 43, 584–603. [Google Scholar] [CrossRef]
- Kim, H.; Park, H.J.; Choi, H.; Chang, Y.; Park, H.; Shin, J.; Kim, J.; Lengner, C.J.; Lee, Y.K.; Kim, J. Modeling G2019S-LRRK2 Sporadic Parkinson’s Disease in 3D Midbrain Organoids. Stem Cell Rep. 2019, 12, 518–531. [Google Scholar] [CrossRef] [Green Version]
- Srikanth, P.; Lagomarsino, V.N.; Muratore, C.R.; Ryu, S.C.; He, A.; Taylor, W.M.; Zhou, C.; Arellano, M.; Young-Pearse, T.L. Shared effects of DISC1 disruption and elevated WNT signaling in human cerebral organoids. Transl. Psychiatry 2018, 8, 77. [Google Scholar] [CrossRef] [Green Version]
- Iefremova, V.; Manikakis, G.; Krefft, O.; Jabali, A.; Weynans, K.; Wilkens, R.; Marsoner, F.; Brändl, B.; Müller, F.J.; Koch, P.; et al. An Organoid-Based Model of Cortical Development Identifies Non-Cell-Autonomous Defects in Wnt Signaling Contributing to Miller-Dieker Syndrome. Cell Rep. 2017, 19, 50–59. [Google Scholar] [CrossRef] [Green Version]
- Lancaster, M.A.; Renner, M.; Martin, C.A.; Wenzel, D.; Bicknell, L.S.; Hurles, M.E.; Homfray, T.; Penninger, J.M.; Jackson, A.P.; Knoblich, J.A. Cerebral organoids model human brain development and microcephaly. Nature 2013, 501, 373–379. [Google Scholar] [CrossRef]
- Conforti, P.; Besusso, D.; Bocchi, V.D.; Faedo, A.; Cesana, E.; Rossetti, G.; Ranzani, V.; Svendsen, C.N.; Thompson, L.M.; Toselli, M.; et al. Faulty neuronal determination and cell polarization are reverted by modulating HD early phenotypes. Proc. Natl. Acad. Sci. USA 2018, 115, E762–E771. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Ooi, J.; Utami, K.H.; Langley, S.R.; Aning, O.A.; Park, D.S.; Renner, M.; Ma, S.; Cheok, C.F.; Knoblich, J.A.; et al. Expanded huntingtin CAG repeats disrupt the balance between neural progenitor expansion and differentiation in human cerebral organoids. bioRxiv 2020, 850586. [Google Scholar] [CrossRef]
- Takebe, T.; Enomura, M.; Yoshizawa, E.; Kimura, M.; Koike, H.; Ueno, Y.; Matsuzaki, T.; Yamazaki, T.; Toyohara, T.; Osafune, K.; et al. Vascularized and Complex Organ Buds from Diverse Tissues via Mesenchymal Cell-Driven Condensation. Cell Stem Cell 2015, 16, 556–565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mansour, A.A.; Gonçalves, J.T.; Bloyd, C.W.; Li, H.; Fernandes, S.; Quang, D.; Johnston, S.; Parylak, S.L.; Jin, X.; Gage, F.H. An in vivo model of functional and vascularized human brain organoids. Nat. Biotechnol. 2018, 36, 432–441. [Google Scholar] [CrossRef] [PubMed]
- Ormel, P.R.; de Sá Vieira, R.; van Bodegraven, E.J.; Karst, H.; Harschnitz, O.; Sneeboer, M.A.M.; Johansen, L.E.; van Dijk, R.E.; Scheefhals, N.; Berdenis van Berlekom, A.; et al. Microglia innately develop within cerebral organoids. Nat. Commun. 2018, 9, 4167. [Google Scholar] [CrossRef] [PubMed]
- Pandya, H.; Shen, M.J.; Ichikawa, D.M.; Sedlock, A.B.; Choi, Y.; Johnson, K.R.; Kim, G.; Brown, M.A.; Elkahloun, A.G.; Maric, D.; et al. Differentiation of human and murine induced pluripotent stem cells to microglia-like cells. Nat. Neurosci. 2017, 20, 753–759. [Google Scholar] [CrossRef]
- Schwartz, M.P.; Hou, Z.; Propson, N.E.; Zhang, J.; Engstrom, C.J.; Santos Costa, V.; Jiang, P.; Nguyen, B.K.; Bolin, J.M.; Daly, W.; et al. Human pluripotent stem cell-derived neural constructs for predicting neural toxicity. Proc. Natl. Acad. Sci. USA 2015, 112, 12516–12521. [Google Scholar] [CrossRef] [Green Version]
- Costamagna, G.; Andreoli, L.; Corti, S.; Faravelli, I. iPSCs-Based Neural 3D Systems: A Multidimensional Approach for Disease Modeling and Drug Discovery. Cells 2019, 8, 1438. [Google Scholar] [CrossRef] [Green Version]
- Tachibana, C.Y. Stem-cell culture moves to the third dimension. Nature 2018, 558, 329–331. [Google Scholar] [CrossRef]
- McBride, J.L.; Boudreau, R.L.; Harper, S.Q.; Staber, P.D.; Monteys, A.M.; Martins, I.; Gilmore, B.L.; Burstein, H.; Peluso, R.W.; Polisky, B.; et al. Artificial miRNAs mitigate shRNA-mediated toxicity in the brain: Implications for the therapeutic development of RNAi. Proc. Natl. Acad. Sci. USA 2008, 105, 5868–5873. [Google Scholar] [CrossRef] [Green Version]
- Drouet, V.; Perrin, V.; Hassig, R.; Dufour, N.; Auregan, G.; Alves, S.; Bonvento, G.; Brouillet, E.; Luthi-Carter, R.; Hantraye, P.; et al. Sustained effects of nonallele-specific Huntingtin silencing. Ann. Neurol. 2009, 65, 276–285. [Google Scholar] [CrossRef] [PubMed]
- Kordasiewicz, H.B.; Stanek, L.M.; Wancewicz, E.V.; Mazur, C.; McAlonis, M.M.; Pytel, K.A.; Artates, J.W.; Weiss, A.; Cheng, S.H.; Shihabuddin, L.S.; et al. Sustained therapeutic reversal of Huntington’s disease by transient repression of huntingtin synthesis. Neuron 2012, 74, 1031–1044. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sah, D.W.; Aronin, N. Oligonucleotide therapeutic approaches for Huntington disease. J. Clin. Investig. 2011, 121, 500–507. [Google Scholar] [CrossRef] [PubMed]
- Carroll, J.B.; Warby, S.C.; Southwell, A.L.; Doty, C.N.; Greenlee, S.; Skotte, N.; Hung, G.; Bennett, C.F.; Freier, S.M.; Hayden, M.R. Potent and selective antisense oligonucleotides targeting single-nucleotide polymorphisms in the Huntington disease gene/allele-specific silencing of mutant huntingtin. Mol. Ther. 2011, 19, 2178–2185. [Google Scholar] [CrossRef] [Green Version]
- Monteys, A.M.; Ebanks, S.A.; Keiser, M.S.; Davidson, B.L. CRISPR/Cas9 Editing of the Mutant Huntingtin Allele In Vitro and In Vivo. Mol. Ther. 2017, 25, 12–23. [Google Scholar] [CrossRef] [Green Version]
- Shin, J.W.; Kim, K.H.; Chao, M.J.; Atwal, R.S.; Gillis, T.; MacDonald, M.E.; Gusella, J.F.; Lee, J.M. Permanent inactivation of Huntington’s disease mutation by personalized allele-specific CRISPR/Cas9. Hum. Mol. Genet. 2016, 25, 4566–4576. [Google Scholar] [CrossRef] [Green Version]
- Yang, S.; Chang, R.; Yang, H.; Zhao, T.; Hong, Y.; Kong, H.E.; Sun, X.; Qin, Z.; Jin, P.; Li, S.; et al. CRISPR/Cas9-mediated gene editing ameliorates neurotoxicity in mouse model of Huntington’s disease. J. Clin. Investig. 2017, 127, 2719–2724. [Google Scholar] [CrossRef] [Green Version]
- Kolli, N.; Lu, M.; Maiti, P.; Rossignol, J.; Dunbar, G.L. CRISPR-Cas9 Mediated Gene-Silencing of the Mutant Huntingtin Gene in an In Vitro Model of Huntington’s Disease. Int. J. Mol. Sci. 2017, 18, 754. [Google Scholar] [CrossRef] [Green Version]
- Tabrizi, S.J.; Ghosh, R.; Leavitt, B.R. Huntingtin Lowering Strategies for Disease Modification in Huntington’s Disease. Neuron 2019, 101, 801–819. [Google Scholar] [CrossRef] [Green Version]
- Kay, C.; Skotte, N.H.; Southwell, A.L.; Hayden, M.R. Personalized gene silencing therapeutics for Huntington disease. Clin. Genet. 2014, 86, 29–36. [Google Scholar] [CrossRef] [Green Version]
- Di Lullo, E.; Kriegstein, A.R. The use of brain organoids to investigate neural development and disease. Nat. Rev. Neurosci. 2017, 18, 573–584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bagley, J.A.; Reumann, D.; Bian, S.; Lévi-Strauss, J.; Knoblich, J.A. Fused cerebral organoids model interactions between brain regions. Nat. Methods 2017, 14, 743–751. [Google Scholar] [CrossRef] [PubMed]
- Gore, A.; Li, Z.; Fung, H.L.; Young, J.E.; Agarwal, S.; Antosiewicz-Bourget, J.; Canto, I.; Giorgetti, A.; Israel, M.A.; Kiskinis, E.; et al. Somatic coding mutations in human induced pluripotent stem cells. Nature 2011, 471, 63–67. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
| Starting Cell Type | Neural Induction | Obtained Cell Type | Final Differentiation | Differentiation Length | Resulting Cell Population | Detected Properties | Reference |
|---|---|---|---|---|---|---|---|
| HD–iPSCs | Induction of EBs (Neural Expansion Medium + N2/B27 + LIF + bFGF) | NSCs | SHH + DKK1 + BDNF + Y27632 + cAMP and valproic acid | 40–42 days | GABA+ MSNs | DARPP32 positivity; increased caspase activity | [12] |
| HD–ES/iPSCs | DMEM/F12 + N2 + SB431542 +Noggin + dorsomorphin | NPCs | SHH + DKK1 + BDNF + N2/B27 + Y27632 | 80 days | MAP2+/GABA+ MSNs | DARPP32 positivity; improved behavioral phenotype in lesioned rats | [17] |
| Human HD–iPSCs | Induction of EBs (DMEM/F1 + N2/B27 + bFGF withdrawal) | NPCs | N2/B27 + NEAA + bFGF | 16 weeks | TUJ1+, MAP2+, and Olig2+ neurons; further cultivation into GABA+ neurons | GABA/GAD65/DARPP-32 positivity; higher rate of DNA damage | [18] |
| Human HD–iPSCs | Induction of EBs | NPCs | SHH/purmorphamin + cAMP + BDNF + GDNF and IGF1 | 60 days | GABA+, TUJ1+, MSNs | DARPP32 positivity; under exposure to menadion–increased cell death; several small aggregate inclusions | [19] |
| HD monkey iPSCs | Induction of neural rosettes (DMEM/F12 + N2/B27 + bFGF+ mLIF) | NPCs | SHH/FGF8 and ascorbic acid | 43 days | GABA+, MAP2+ neurons | elevated expression of HTT; presence of HTT aggregates; higher susceptibility to oxidative stress | [20] |
| HD monkey iPSCs | Neurobasal-A medium + B27+ bFGF + mLIF | NPCs | AZA-C + TSA, BMP2 + B27 | 30 days | astrocytes | presence of nuclear and cytoplasmic HTT aggregates; higher susceptibility to oxidative stress | [21] |
| HD–iPSCs | DMEM/F12 + N2 + LIF + bFGF | NPCs | B27 + SHH, DKK1 + BDNF + Y27632 | 40 days | GABA+ neurons | DARPP32 positivity; mHTT genetic correction of pathogenic HD signalling pathways | [22] |
| HD–iPSCs | DMEM/F12 + N2 + Noggin + Dorsomorphin + bFGF | NPCs | N2/B27 + BDNF + forskolin | 56–57 days | GABA+ MSNs | Increased protein aggregate inclusions | [23] |
| Model Cell Type | Results | Reference |
|---|---|---|
| HD iPSCs–MSNs | - elevated caspase activity upon growth factor deprivation | [12] |
| HD iPSC–MSN | - neuroprotective effect of CGS21680 and APEC  therapeutic potential therapeutic potential | [18] |
| HD iPSC–NPCs | - higher levels of FOXO1 and FOXO4 elevated proteasome activity | [19] |
| iPSC- GABA+ neurons | - under treatment with memantine reversal of HD pathologic events | [20] |
| HD monkey iPSC–astrocytes | - detection of numerous HD related pathologiesm HTT aggregates, inefficient glutamate clearance, suppression of mitochondrial function, abnormal electrophysiology | [21] |
| Corrected HD iPSC–NPCs | - after transplantation into mice model survival and differentiation of cells into the GABAergic neurons | [22] |
| iPSC–NSCs | - after bilateral transplantation into mice striatum improved locomotor function | [33] |
| mice HD iPSCs/human HD iPSCs | - dysregulation of ERK signaling, β-catenin phosphorylation, SOD1 accumulation and p53 expression | [34] |
| Juvenile HD–iPSCs | - high number of significantly dysregulated mRNAs | [35] |
| HD iPSC–MSN | - increased calcium SOC activity; treatment by quinazoline derivative - EVP4593 led to reduced activity of SOC currents and normalization of calcium transport | [23] |
| HD monkey iPSC–NPCs | - under treatment with memantine, Rilizole and Methylene blue the most potent anti-apoptotic drug was Rilizole; the most effective in reduction of mTT aggregates was Methylene blue | [36] |
| Corrected HD monkey iPSC–GABA+ neurons | - after transplantation into mice striatum longer lifespan of HD mice model; improved behavioral and locomotor function | [37] |
| 2D Systems | 3D Organoids | |
|---|---|---|
| Culture method | - cell growth and differentiation on monolayers | - cell differentiation and self-organization within matrigel |
| Cell population | - usually immature cell populations | - improved maturation |
| Duration of differentiation | - fast differentiation process | - slow differentiation process |
| Tissue composition | - lack of tissue microenvironment | - similar cytoarchitecture with in vivo tissue |
| Vascular supply | - no | - limited |
| High-throughput generation | - high | - low |
| Genome editing | - easy | - hard |
| Technical procedure | - mostly easy - less time consuming | - moderate - more time consuming |
| Disease modeling specificity | - moderate | - high |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Csobonyeiova, M.; Polak, S.; Danisovic, L. Recent Overview of the Use of iPSCs Huntington’s Disease Modeling and Therapy. Int. J. Mol. Sci. 2020, 21, 2239. https://doi.org/10.3390/ijms21062239
Csobonyeiova M, Polak S, Danisovic L. Recent Overview of the Use of iPSCs Huntington’s Disease Modeling and Therapy. International Journal of Molecular Sciences. 2020; 21(6):2239. https://doi.org/10.3390/ijms21062239
Chicago/Turabian StyleCsobonyeiova, Maria, Stefan Polak, and Lubos Danisovic. 2020. "Recent Overview of the Use of iPSCs Huntington’s Disease Modeling and Therapy" International Journal of Molecular Sciences 21, no. 6: 2239. https://doi.org/10.3390/ijms21062239
APA StyleCsobonyeiova, M., Polak, S., & Danisovic, L. (2020). Recent Overview of the Use of iPSCs Huntington’s Disease Modeling and Therapy. International Journal of Molecular Sciences, 21(6), 2239. https://doi.org/10.3390/ijms21062239