Norepinephrine-Induced DNA Damage in Ovarian Cancer Cells

 , , and
, , and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Dose-Dependent Norepinephrine (NE)-Induced DNA Double-Strand Breaks in EOC Cells
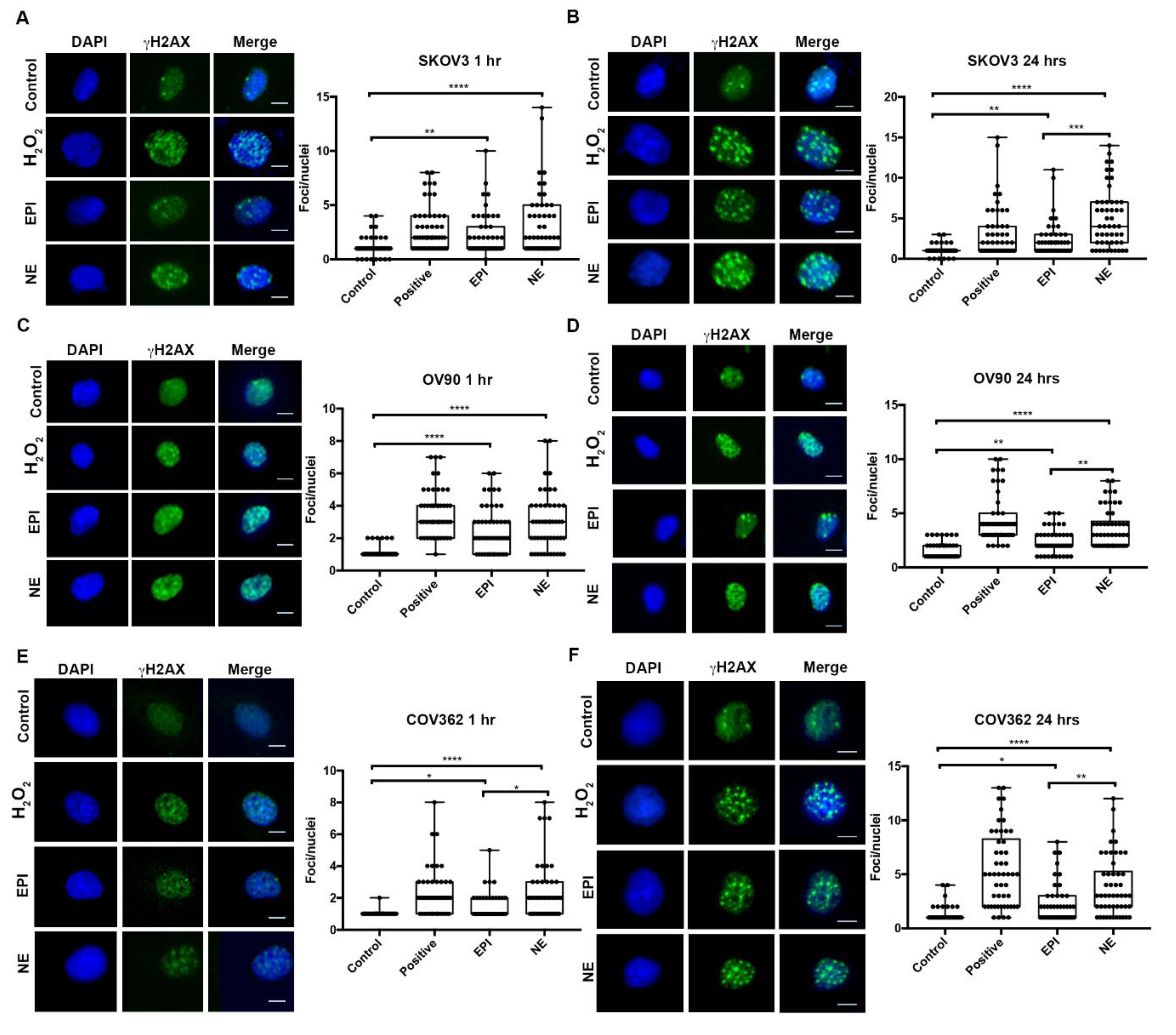
2.2. NE and EPI Exposure Increases γ-H2AX foci Formation in Ovarian Cancer Cells
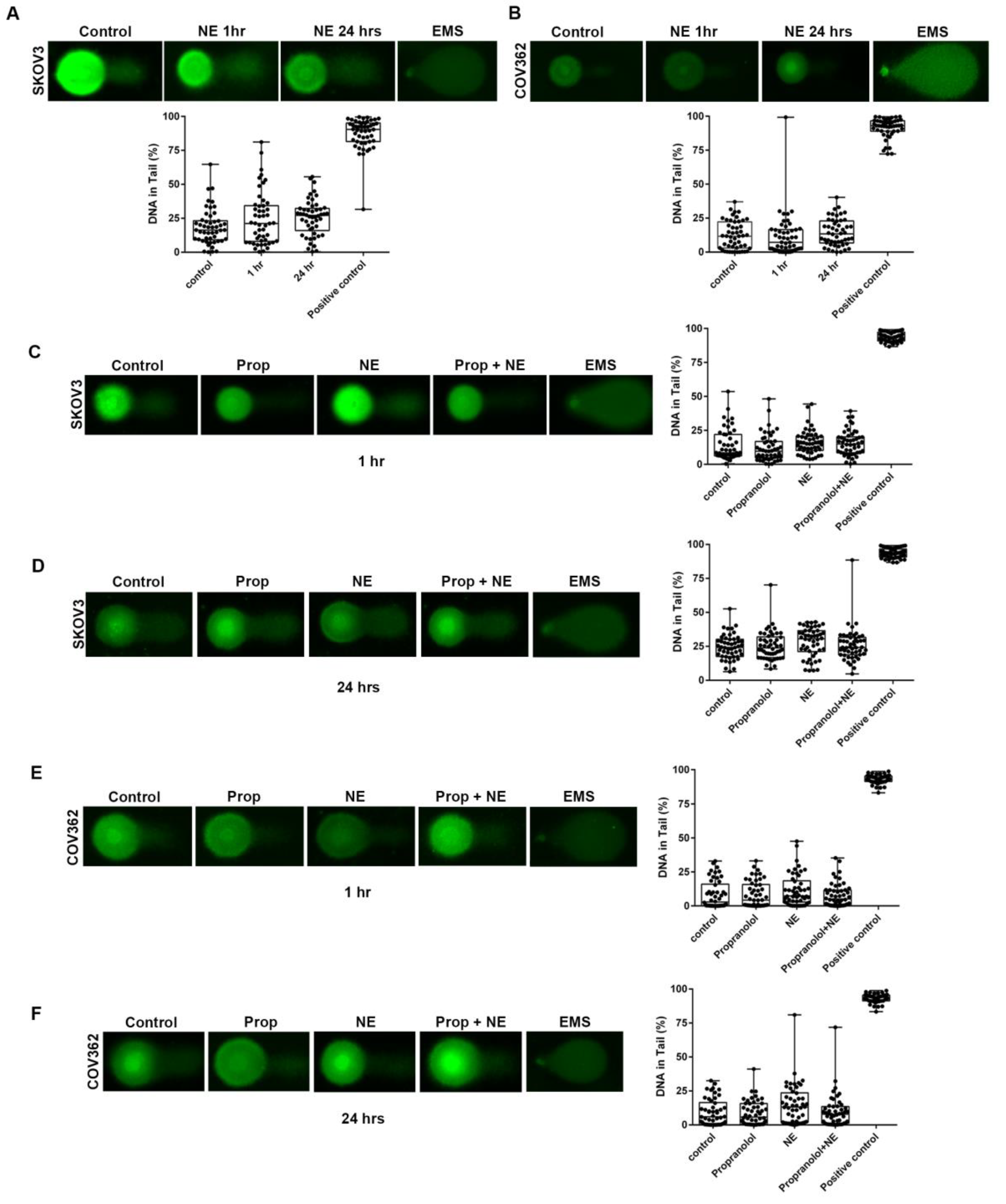
2.3. NE exposure Leads to Double, but not Single, Strand DNA Breaks, in Ovarian Cancer Cells
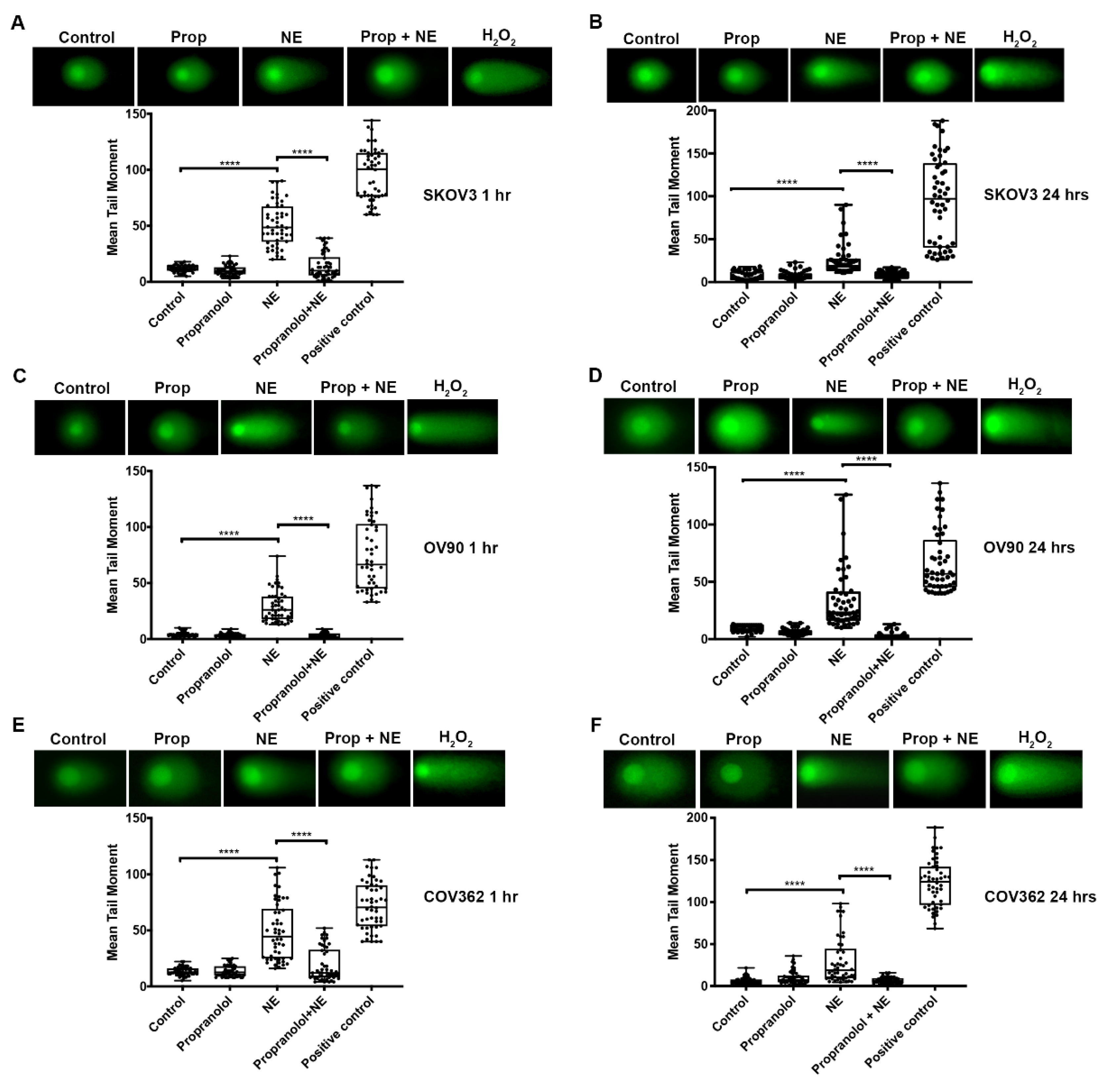
2.4. β-adrenergic Receptor Blockade Abrogates NE-Induced DNA Damage
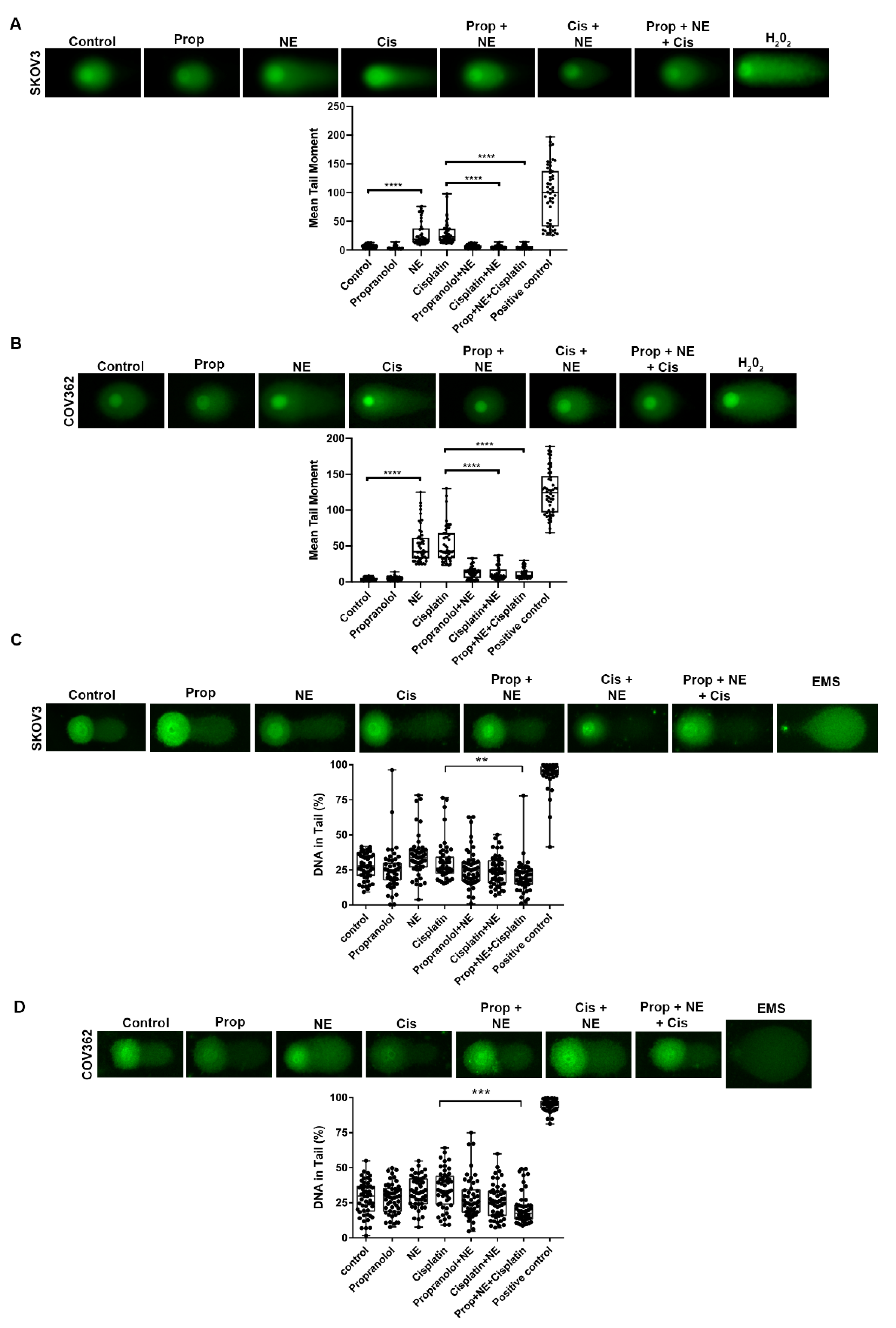
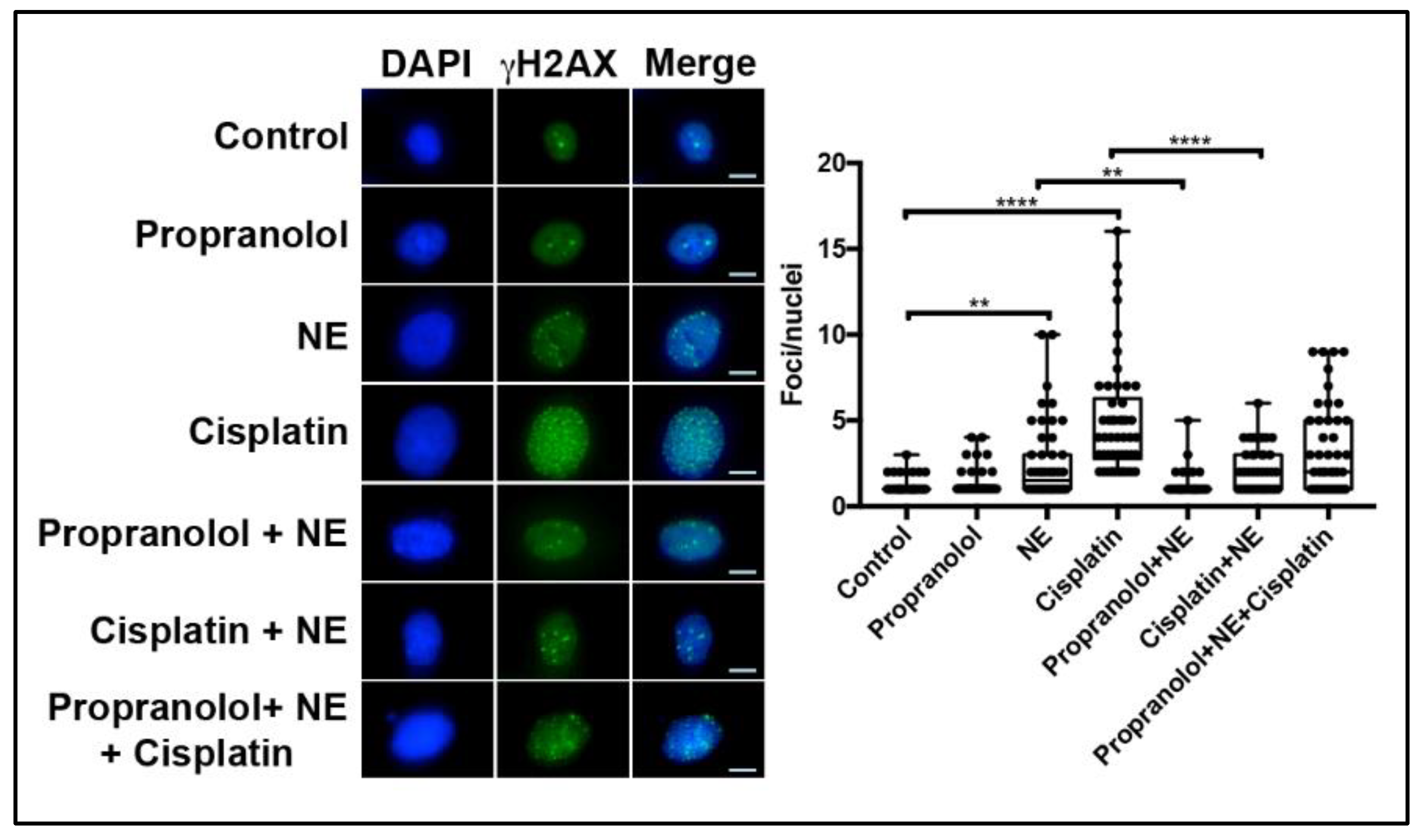
2.5. NE Stimulation Protects Ovarian Cancer Cells from Cisplatin-Induced DNA Damage
3. Discussion
4. Materials and Methods
4.1. Cell lines and Culture Conditions
4.2. Immunofluorescence, Imaging, and Image Analysis
4.3. Neutral Comet Assay
4.4. Alkaline Comet Assay
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| OC | Ovarian cancer |
| NE | Norepinephrine (noradrenaline) |
| EPI | Epinephrine (adrenaline) |
| SSB | Single-strand break |
| DSB | Double-strand break |
| DAPI | 4′,6-diamidino-2-phenylindole |
References
- Agarwal, R.; Kaye, S.B. Ovarian cancer: Strategies for overcoming resistance to chemotherapy. Nat. Rev. Cancer 2003, 3, 502–516. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2017. CA Cancer J. Clin. 2017, 67, 7–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lutgendorf, S.K.; Sood, A.K.; Anderson, B.; McGinn, S.; Maiseri, H.; Dao, M.; Sorosky, J.I.; De Geest, K.; Ritchie, J.; Lubaroff, D.M. Social Support, Psychological Distress, and Natural Killer Cell Activity in Ovarian Cancer. JCO 2005, 23, 7105–7113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lutgendorf, S.K.; De Geest, K.; Bender, D.; Ahmed, A.; Goodheart, M.J.; Dahmoush, L.; Zimmerman, M.B.; Penedo, F.J.; Lucci, J.A., III; Ganjei-Azar, P.; et al. Social Influences on Clinical Outcomes of Patients with Ovarian Cancer. JCO 2012, 30, 2885–2890. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Costanzo, E.S.; Lutgendorf, S.K.; Sood, A.K.; Anderson, B.; Sorosky, J.; Lubaroff, D.M. Psychosocial factors and interleukin-6 among women with advanced ovarian cancer. Cancer 2005, 104, 305–313. [Google Scholar] [CrossRef] [PubMed]
- Diaz, E.S.; Karlan, B.Y.; Li, A.J. Impact of beta blockers on epithelial ovarian cancer survival. Gynecol. Oncol. 2012, 127, 375–378. [Google Scholar] [CrossRef] [PubMed]
- Watkins, J.L.; Thaker, P.H.; Nick, A.M.; Ramondetta, L.M.; Kumar, S.; Urbauer, D.L.; Matsuo, K.; Squires, K.C.; Coleman, R.L.; Lutgendorf, S.K.; et al. Clinical impact of selective and nonselective beta-blockers on survival in patients with ovarian cancer. Cancer 2015, 121, 3444–3451. [Google Scholar] [CrossRef]
- Al-Niaimi, A.; Dickson, E.L.; Albertin, C.; Karnowski, J.; Niemi, C.; Spencer, R.; Shahzad, M.M.K.; Uppal, S.; Saha, S.; Rice, L.; et al. The impact of perioperative β blocker use on patient outcomes after primary cytoreductive surgery in high-grade epithelial ovarian carcinoma. Gynecol. Oncol. 2016, 143, 521–525. [Google Scholar] [CrossRef]
- Ramondetta, L.M.; Hu, W.; Thaker, P.H.; Urbauer, D.L.; Chisholm, G.B.; Westin, S.N.; Sun, Y.; Ramirez, P.T.; Fleming, N.; Sahai, S.K.; et al. Prospective pilot trial with combination of propranolol with chemotherapy in patients with epithelial ovarian cancer and evaluation on circulating immune cell gene expression. Gynecol. Oncol. 2019, 154, 524–530. [Google Scholar] [CrossRef]
- Bodurka-Bevers, D.; Basen-Engquist, K.; Carmack, C.L.; Fitzgerald, M.A.; Wolf, J.K.; de Moor, C.; Gershenson, D.M. Depression, Anxiety, and Quality of Life in Patients with Epithelial Ovarian Cancer. Gynecol. Oncol. 2000, 78, 302–308. [Google Scholar] [CrossRef]
- Lutgendorf, S.K.; DeGeest, K.; Dahmoush, L.; Farley, D.; Penedo, F.; Bender, D.; Goodheart, M.; Buekers, T.E.; Mendez, L.; Krueger, G. Social isolation is associated with elevated tumor norepinephrine in ovarian carcinoma patients. Brain Behav. Immun. 2011, 25, 250–255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mausbach, B.T.; Dimsdale, J.E.; Ziegler, M.G.; Mills, P.J.; Ancoli-Israel, S.; Patterson, T.L.; Grant, I. Depressive Symptoms Predict Norepinephrine Response to a Psychological Stressor Task in Alzheimer’s Caregivers. Psychosom. Med. 2005, 67, 638–642. [Google Scholar] [CrossRef] [PubMed]
- Hughes, J.W.; Watkins, L.; Blumenthal, J.A.; Kuhn, C.; Sherwood, A. Depression and anxiety symptoms are related to increased 24-hour urinary norepinephrine excretion among healthy middle-aged women. J. Psychosom. Res. 2004, 57, 353–358. [Google Scholar] [CrossRef]
- Huang, T.; Tworoger, S.S.; Hecht, J.L.; Rice, M.S.; Sood, A.K.; Kubzansky, L.D.; Poole, E.M. Association of Ovarian Tumor 2-Adrenergic Receptor Status with Ovarian Cancer Risk Factors and Survival. Cancer Epidemiol. Biomark. Prev. 2016, 25, 1587–1594. [Google Scholar] [CrossRef] [Green Version]
- Poole, E.M.; Kubzansky, L.D.; Sood, A.K.; Okereke, O.I.; Tworoger, S.S. A prospective study of phobic anxiety, risk of ovarian cancer, and survival among patients. Cancer Causes Control 2016, 27, 661–668. [Google Scholar] [CrossRef]
- Su, F.; Ouyang, N.; Zhu, P.; Ouyang, N.; Jia, W.; Gong, C.; Ma, X.; Xu, H.; Song, E. Psychological stress induces chemoresistance in breast cancer by upregulating mdr1. Biochem. Biophys. Res. Commun. 2005, 329, 888–897. [Google Scholar] [CrossRef]
- Flint, M.S.; Kim, G.; Hood, B.L.; Bateman, N.W.; Stewart, N.A.; Conrads, T.P. Stress hormones mediate drug resistance to paclitaxel in human breast cancer cells through a CDK-1-dependent pathway. Psychoneuroendocrinology 2009, 34, 1533–1541. [Google Scholar] [CrossRef]
- Kang, Y.; Nagaraja, A.S.; Armaiz-Pena, G.N.; Dorniak, P.L.; Hu, W.; Rupaimoole, R.; Liu, T.; Gharpure, K.M.; Previs, R.A.; Hansen, J.M.; et al. Adrenergic Stimulation of DUSP1 Impairs Chemotherapy Response in Ovarian Cancer. Clin. Cancer Res. 2016, 22, 1713–1724. [Google Scholar] [CrossRef] [Green Version]
- Flint, M.S.; Baum, A.; Chambers, W.H.; Jenkins, F.J. Induction of DNA damage, alteration of DNA repair and transcriptional activation by stress hormones. Psychoneuroendocrinology 2007, 32, 470–479. [Google Scholar] [CrossRef]
- Lutgendorf, S.K.; Cole, S.; Costanzo, E.; Bradley, S.; Coffin, J.; Jabbari, S.; Rainwater, K.; Ritchie, J.M.; Yang, M.; Sood, A.K. Stress-related mediators stimulate vascular endothelial growth factor secretion by two ovarian cancer cell lines. Clin. Cancer Res. 2003, 9, 4514–4521. [Google Scholar]
- Thaker, P.H.; Han, L.Y.; Kamat, A.A.; Arevalo, J.M.; Takahashi, R.; Lu, C.; Jennings, N.B.; Armaiz-Pena, G.; Bankson, J.A.; Ravoori, M.; et al. Chronic stress promotes tumor growth and angiogenesis in a mouse model of ovarian carcinoma. Nat. Med. 2006, 12, 939–944. [Google Scholar] [CrossRef] [PubMed]
- Sood, A.K.; Bhatty, R.; Kamat, A.A.; Landen, C.N.; Han, L.; Thaker, P.H.; Gershenson, D.M.; Lutgendorf, S.; Cole, S.W. Stress Hormone-Mediated Invasion of Ovarian Cancer Cells. Clin. Cancer Res. 2006, 12, 369–375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nilsson, M.B.; Armaiz-Pena, G.; Takahashi, R.; Lin, Y.G.; Trevino, J.; Li, Y.; Jennings, N.; Arevalo, J.; Lutgendorf, S.K.; Gallick, G.E.; et al. Stress Hormones Regulate Interleukin-6 Expression by Human Ovarian Carcinoma Cells through a Src-dependent Mechanism. J. Biol. Chem. 2007, 282, 29919–29926. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Landen, C.N.; Lin, Y.G.; Armaiz-Pena, G.N.; Das, P.D.; Arevalo, J.M.; Kamat, A.A.; Han, L.Y.; Jennings, N.B.; Spannuth, W.A.; Thaker, P.H.; et al. Neuroendocrine Modulation of Signal Transducer and Activator of Transcription-3 in Ovarian Cancer. Cancer Res. 2007, 67, 10389–10396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sood, A.K.; Armaiz-Pena, G.N.; Halder, J.; Nick, A.M.; Stone, R.L.; Hu, W.; Carroll, A.R.; Spannuth, W.A.; Deavers, M.T.; Allen, J.K.; et al. Adrenergic modulation of focal adhesion kinase protects human ovarian cancer cells from anoikis. J. Clin. Investig. 2010, 120, 1515–1523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shahzad, M.M.K.; Arevalo, J.M.; Armaiz-Pena, G.N.; Lu, C.; Stone, R.L.; Moreno-Smith, M.; Nishimura, M.; Lee, J.-W.; Jennings, N.B.; Bottsford-Miller, J.; et al. Stress effects on FosB- and interleukin-8 (IL8)-driven ovarian cancer growth and metastasis. J. Biol. Chem. 2010, 285, 35462–35470. [Google Scholar] [CrossRef] [Green Version]
- Armaiz-Pena, G.N.; Gonzalez-Villasana, V.; Nagaraja, A.S.; Rodriguez-Aguayo, C.; Sadaoui, N.C.; Stone, R.L.; Matsuo, K.; Dalton, H.J.; Previs, R.A.; Jennings, N.B.; et al. Adrenergic regulation of monocyte chemotactic protein 1 leads to enhanced macrophage recruitment and ovarian carcinoma growth. Oncotarget 2015, 6, 4266–4273. [Google Scholar] [CrossRef] [Green Version]
- Reeder, A.; Attar, M.; Nazario, L.; Bathula, C.; Zhang, A.; Hochbaum, D.; Roy, E.; Cooper, K.L.; Oesterreich, S.; Davidson, N.E.; et al. Stress hormones reduce the efficacy of paclitaxel in triple negative breast cancer through induction of DNA damage. Br. J. Cancer 2015, 112, 1461–1470. [Google Scholar] [CrossRef] [Green Version]
- Ames, B.N.; Shigenaga, M.K.; Gold, L.S. DNA lesions, inducible DNA repair, and cell division: Three key factors in mutagenesis and carcinogenesis. Environ. Health Perspect. 1993, 101, 35–44. [Google Scholar]
- Broustas, C.G.; Lieberman, H.B. DNA damage response genes and the development of cancer metastasis. Radiat. Res. 2014, 181, 111–130. [Google Scholar] [CrossRef] [Green Version]
- Tubbs, A.; Nussenzweig, A. Endogenous DNA Damage as a Source of Genomic Instability in Cancer. Cell 2017, 168, 644–656. [Google Scholar] [CrossRef]
- Negrini, S.; Gorgoulis, V.G.; Halazonetis, T.D. Genomic instability—An evolving hallmark of cancer. Nat. Rev. Mol. Cell Biol. 2010, 11, 220–228. [Google Scholar] [CrossRef]
- Tian, K.; Rajendran, R.; Doddananjaiah, M.; Krstic-Demonacos, M.; Schwartz, J.-M. Dynamics of DNA Damage Induced Pathways to Cancer. PLoS ONE 2013, 8, e72303. [Google Scholar] [CrossRef] [Green Version]
- Flint, M.S.; Baum, A.; Episcopo, B.; Knickelbein, K.Z.; Liegey Dougall, A.J.; Chambers, W.H.; Jenkins, F.J. Chronic exposure to stress hormones promotes transformation and tumorigenicity of 3T3 mouse fibroblasts. Stress 2013, 16, 114–121. [Google Scholar] [CrossRef] [Green Version]
- Beaufort, C.M.; Helmijr, J.C.A.; Piskorz, A.M.; Hoogstraat, M.; Ruigrok-Ritstier, K.; Besselink, N.; Murtaza, M.; van IJcken, W.F.J.; Heine, A.A.J.; Smid, M.; et al. Ovarian Cancer Cell Line Panel (OCCP): Clinical Importance of In Vitro Morphological Subtypes. PLoS ONE 2014, 9, e103988. [Google Scholar] [CrossRef]
- Schmidt, C.; Kraft, K. Beta-Endorphin and catecholamine concentrations during chronic and acute stress in intensive care patients. Eur. J. Med. Res. 1996, 1, 528–532. [Google Scholar]
- Lara, H.E.; Dorfman, M.; Venegas, M.; Luza, S.M.; Luna, S.L.; Mayerhofer, A.; Guimaraes, M.A.; Rosa E Silva, A.A.M.; Ramírez, V.D. Changes in sympathetic nerve activity of the mammalian ovary during a normal estrous cycle and in polycystic ovary syndrome: Studies on norepinephrine release. Microsc. Res. Tech. 2002, 59, 495–502. [Google Scholar] [CrossRef]
- Bierhaus, A.; Wolf, J.; Andrassy, M.; Rohleder, N.; Humpert, P.M.; Petrov, D.; Ferstl, R.; von Eynatten, M.; Wendt, T.; Rudofsky, G.; et al. A mechanism converting psychosocial stress into mononuclear cell activation. Proc. Natl. Acad. Sci. USA 2003, 100, 1920–1925. [Google Scholar] [CrossRef] [Green Version]
- Lara, H.E.; Porcile, A.; Espinoza, J.; Romero, C.; Luza, S.M.; Fuhrer, J.; Miranda, C.; Roblero, L. Release of norepinephrine from human ovary: Coupling to steroidogenic response. Endocrine 2001, 15, 187–192. [Google Scholar] [CrossRef]
- Nankova, B.; Kvetnansky, R.; Hiremagalur, B.; Sabban, B.; Rusnak, M.; Sabban, E.L. Immobilization stress elevates gene expression for catecholamine biosynthetic enzymes and some neuropeptides in rat sympathetic ganglia: Effects of adrenocorticotropin and glucocorticoids. Endocrinology 1996, 137, 5597–5604. [Google Scholar] [CrossRef]
- Mullany, L.K.; Wong, K.-K.; Marciano, D.C.; Katsonis, P.; King-Crane, E.R.; Ren, Y.A.; Lichtarge, O.; Richards, J.S. Specific TP53 Mutants Overrepresented in Ovarian Cancer Impact CNV, TP53 Activity, Responses to Nutlin-3a, and Cell Survival. Neoplasia 2015, 17, 789–803. [Google Scholar] [CrossRef] [Green Version]
- Yaginuma, Y.; Westphal, H. Abnormal structure and expression of the p53 gene in human ovarian carcinoma cell lines. Cancer Res. 1992, 52, 4196–4199. [Google Scholar]
- Pulliam, N.; Fang, F.; Ozes, A.R.; Tang, J.; Adewuyi, A.; Keer, H.; Lyons, J.; Baylin, S.B.; Matei, D.; Nakshatri, H.; et al. An Effective Epigenetic-PARP Inhibitor Combination Therapy for Breast and Ovarian Cancers Independent of BRCA Mutations. Clin. Cancer Res. 2018, 24, 3163–3175. [Google Scholar] [CrossRef] [Green Version]
- Lane, D. p53, guardian of the genome. Nature 1992, 358, 15–16. [Google Scholar] [CrossRef]
- Agarwal, M.L.; Taylor, W.R.; Chernov, M.V.; Chernova, O.B.; Stark, G.R. The p53 network. J. Biol. Chem. 1998, 273, 1–4. [Google Scholar] [CrossRef] [Green Version]
- Helleday, T.; Petermann, E.; Lundin, C.; Hodgson, B.; Sharma, R.A. DNA repair pathways as targets for cancer therapy. Nat. Rev. Cancer 2008, 8, 193–204. [Google Scholar] [CrossRef]
- Hakem, R. DNA-damage repair; the good, the bad, and the ugly. EMBO J. 2008, 27, 589–605. [Google Scholar] [CrossRef] [Green Version]
- Rogakou, E.P.; Pilch, D.R.; Orr, A.H.; Ivanova, V.S.; Bonner, W.M. DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J. Biol. Chem. 1998, 273, 5858–5868. [Google Scholar] [CrossRef] [Green Version]
- Jackson, S.P.; Bartek, J. The DNA-damage response in human biology and disease. Nature 2009, 461, 1071–1078. [Google Scholar] [CrossRef] [Green Version]
- Rothkamm, K.; Krüger, I.; Thompson, L.H.; Löbrich, M. Pathways of DNA double-strand break repair during the mammalian cell cycle. Mol. Cell. Biol. 2003, 23, 5706–5715. [Google Scholar] [CrossRef] [Green Version]
- Patel, P.R. Norepinephrine Reduces Reactive Oxygen Species (ROS) and DNA Damage in Ovarian Surface Epithelial Cells. J. Bioanal. Biomed. 2015, 7, 75–80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gülçin, İ. Antioxidant activity of l-adrenaline: A structure–activity insight. Chem. Biol. Interact. 2009, 179, 71–80. [Google Scholar] [CrossRef] [PubMed]
- Kawashima, T.; Ohkubo, K.; Fukuzumi, S. Radical Scavenging Reactivity of Catecholamine Neurotransmitters and the Inhibition Effect for DNA Cleavage. J. Phys. Chem. B 2010, 114, 675–680. [Google Scholar] [CrossRef] [PubMed]
- Djelic, N.; Anderson, D. The effect of the antioxidant catalase on oestrogens, triiodothyronine, and noradrenaline in the Comet assay. Teratog. Carcinog. Mutagen. 2003, 23, 69–81. [Google Scholar] [CrossRef]
- Neri, M.; Cerretani, D.; Fiaschi, A.I.; Laghi, P.F.; Lazzerini, P.E.; Maffione, A.B.; Micheli, L.; Bruni, G.; Nencini, C.; Giorgi, G.; et al. Correlation between cardiac oxidative stress and myocardial pathology due to acute and chronic norepinephrine administration in rats. J. Cell. Mol. Med. 2007, 11, 156–170. [Google Scholar] [CrossRef]
- Spencer, W.A.; Jeyabalan, J.; Kichambre, S.; Gupta, R.C. Oxidatively generated DNA damage after Cu(II) catalysis of dopamine and related catecholamine neurotransmitters and neurotoxins: Role of reactive oxygen species. Free Radic. Biol. Med. 2011, 50, 139–147. [Google Scholar] [CrossRef] [Green Version]
- Saller, S.; Merz-Lange, J.; Raffael, S.; Hecht, S.; Pavlik, R.; Thaler, C.; Berg, D.; Berg, U.; Kunz, L.; Mayerhofer, A. Norepinephrine, Active Norepinephrine Transporter, and Norepinephrine-Metabolism Are Involved in the Generation of Reactive Oxygen Species in Human Ovarian Granulosa Cells. Endocrinology 2012, 153, 1472–1483. [Google Scholar] [CrossRef]
- Helm, C.W.; States, J.C. Enhancing the efficacy of cisplatin in ovarian cancer treatment–could arsenic have a role. J. Ovarian Res. 2009, 2, 2–7. [Google Scholar] [CrossRef] [Green Version]
- Siddik, Z.H. Cisplatin: Mode of cytotoxic action and molecular basis of resistance. Oncogene 2003, 22, 7265–7279. [Google Scholar] [CrossRef] [Green Version]
- Rezaee, M.; Sanche, L.; Hunting, D.J. Cisplatin Enhances the Formation of DNA Single- and Double-Strand Breaks by Hydrated Electrons and Hydroxyl Radicals. Radiat. Res. 2013, 179, 323–331. [Google Scholar] [CrossRef]
- Yu, W.; Chen, Y.; Dubrulle, J.; Stossi, F.; Putluri, V.; Sreekumar, A.; Putluri, N.; Baluya, D.; Lai, S.Y.; Sandulache, V.C. Cisplatin generates oxidative stress which is accompanied by rapid shifts in central carbon metabolism. Sci. Rep. 2018, 8, 4306. [Google Scholar] [CrossRef] [Green Version]
- Duke University Light Microscopy Core Facility. Count Nuclear Foci-ImageJ. Available online: https://microscopy.duke.edu/guides/count-nuclear-foci-ImageJ (accessed on 21 August 2018).
- Gyori, B.M.; Venkatachalam, G.; Thiagarajan, P.S.; Hsu, D.; Clement, M.-V. OpenComet: An automated tool for comet assay image analysis. Redox Biol. 2014, 2, 457–465. [Google Scholar] [CrossRef] [Green Version]





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lamboy-Caraballo, R.; Ortiz-Sanchez, C.; Acevedo-Santiago, A.; Matta, J.; N.A. Monteiro, A.; N. Armaiz-Pena, G. Norepinephrine-Induced DNA Damage in Ovarian Cancer Cells. Int. J. Mol. Sci. 2020, 21, 2250. https://doi.org/10.3390/ijms21062250
Lamboy-Caraballo R, Ortiz-Sanchez C, Acevedo-Santiago A, Matta J, N.A. Monteiro A, N. Armaiz-Pena G. Norepinephrine-Induced DNA Damage in Ovarian Cancer Cells. International Journal of Molecular Sciences. 2020; 21(6):2250. https://doi.org/10.3390/ijms21062250
Chicago/Turabian StyleLamboy-Caraballo, Rocio, Carmen Ortiz-Sanchez, Arelis Acevedo-Santiago, Jaime Matta, Alvaro N.A. Monteiro, and Guillermo N. Armaiz-Pena. 2020. "Norepinephrine-Induced DNA Damage in Ovarian Cancer Cells" International Journal of Molecular Sciences 21, no. 6: 2250. https://doi.org/10.3390/ijms21062250
APA StyleLamboy-Caraballo, R., Ortiz-Sanchez, C., Acevedo-Santiago, A., Matta, J., N.A. Monteiro, A., & N. Armaiz-Pena, G. (2020). Norepinephrine-Induced DNA Damage in Ovarian Cancer Cells. International Journal of Molecular Sciences, 21(6), 2250. https://doi.org/10.3390/ijms21062250