Molecular T-Cell Repertoire Analysis as Source of Prognostic and Predictive Biomarkers for Checkpoint Blockade Immunotherapy





Abstract
:1. Introduction
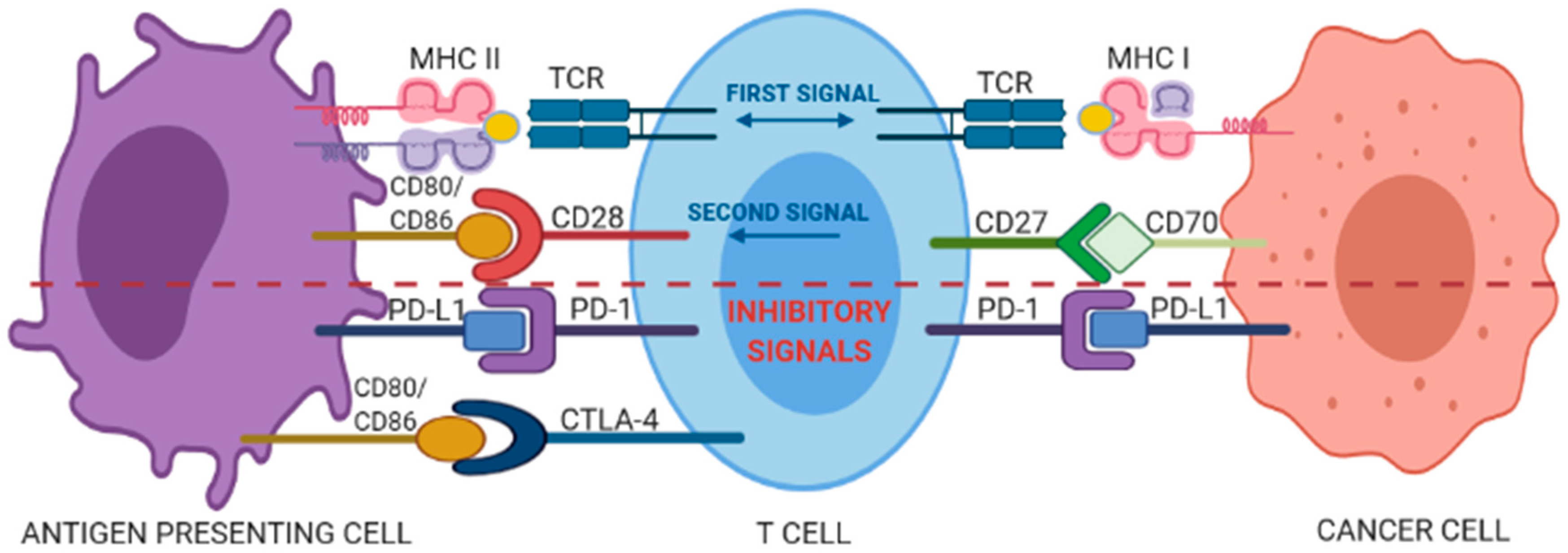
2. Checkpoint Blockade Immunotherapy
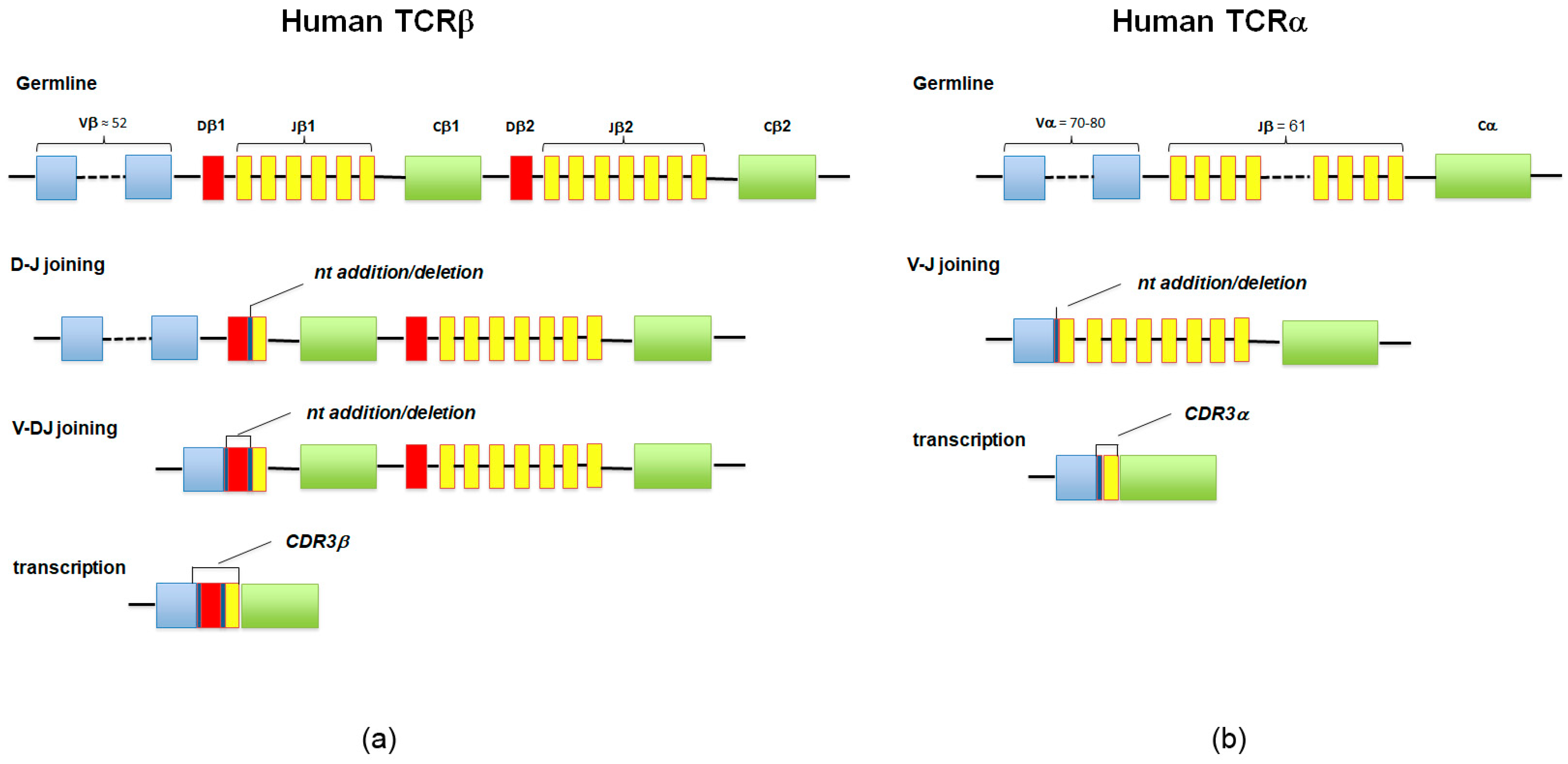
3. The Immune Repertoire
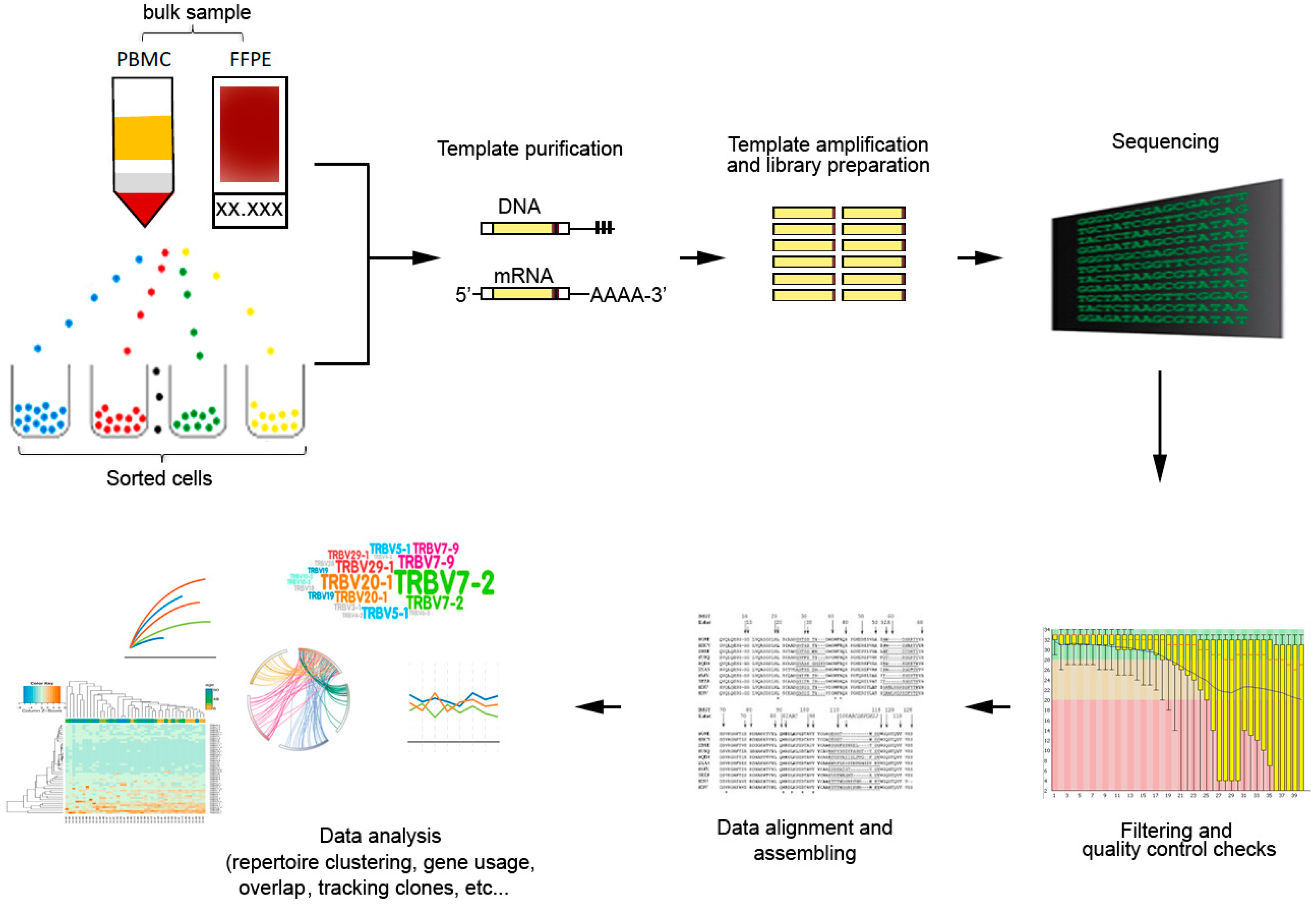
4. Methodologies for TCR Repertoire Analysis
5. Analysis of Immune Repertoire
6. The Impact of CBI on TCR Repertoire
6.1. Metastatic Melanoma
6.2. Lung Cancer
6.3. Squamous Cell Carcinoma
6.4. Prostate and Urothelial Cancers
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| APC | Antigen presenting cell |
| CBI | checkpoints blockade immunotherapy |
| CDR3 | complementarity-determining region 3 |
| HTS | high-throughput sequencing |
| MHC | major histocompatibility complex |
| NSCLC | non–small-cell lung cancer |
| PCR | polymerase chain reaction |
| RACE | rapid amplification of cDNA ends |
| SSC | squamous cell carcinoma |
| TCR | T cell receptor |
| UMIs | unique molecular identifiers |
| VDJ | variable (V), diversity (D), and joining (J) gene segments |
References
- Topalian, S.L.; Hodi, F.S.; Brahmer, J.R.; Gettinger, S.N.; Smith, D.C.; McDermott, D.F.; Powderly, J.D.; Sosman, J.A.; Atkins, M.B.; Leming, P.D.; et al. Five-year survival and correlates among patients with advanced melanoma, renal cell carcinoma, or non-small cell lung cancer treated with nivolumab. Jama Oncol. 2019. Available online: https://jamanetwork.com/journals/jamaoncology/fullarticle/2738775 (accessed on 25 July 2019).
- Ribas, A.; Wolchok, J.D. Cancer immunotherapy using checkpoint blockade. Science 2018, 359, 1350–1355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Topalian, S.L.; Taube, J.M.; Anders, R.A.; Pardoll, D.M. Mechanism-driven biomarkers to guide immune checkpoint blockade in cancer therapy. Nat. Rev. Cancer 2016, 16, 275–287. [Google Scholar] [CrossRef] [PubMed]
- Ribas, A.; Tumeh, P.C. The future of cancer therapy: Selecting patients likely to respond to pd1/l1 blockade. Clin. Cancer Res.: Off. J. Am. Assoc. Cancer Res. 2014, 20, 4982–4984. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masucci, G.V.; Cesano, A.; Hawtin, R.; Janetzki, S.; Zhang, J.; Kirsch, I.; Dobbin, K.K.; Alvarez, J.; Robbins, P.B.; Selvan, S.R.; et al. Validation of biomarkers to predict response to immunotherapy in cancer: Volume i-pre-analytical and analytical validation. J. Immunother. Cancer 2016, 4, 76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sewell, A.K. Why must t cells be cross-reactive? Nat. Rev. Immunol. 2012, 12, 669–677. [Google Scholar] [CrossRef] [PubMed]
- Drake, C.G.; Lipson, E.J.; Brahmer, J.R. Breathing new life into immunotherapy: Review of melanoma, lung and kidney cancer. Nat. Rev. Clin. Oncol. 2014, 11, 24–37. [Google Scholar] [CrossRef]
- Woodsworth, D.J.; Castellarin, M.; Holt, R.A. Sequence analysis of t-cell repertoires in health and disease. Genome Med. 2013, 5, 98. [Google Scholar] [CrossRef] [Green Version]
- Vesely, M.D.; Kershaw, M.H.; Schreiber, R.D.; Smyth, M.J. Natural innate and adaptive immunity to cancer. Annu. Rev. Immunol. 2011, 29, 235–271. [Google Scholar] [CrossRef] [Green Version]
- Zitvogel, L.; Tesniere, A.; Kroemer, G. Cancer despite immunosurveillance: Immunoselection and immunosubversion. Nat. Rev. Immunol. 2006, 6, 715–727. [Google Scholar] [CrossRef]
- Pardoll, D.M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 2012, 12, 252–264. [Google Scholar] [CrossRef] [Green Version]
- Oosterwegel, M.A.; Greenwald, R.J.; Mandelbrot, D.A.; Lorsbach, R.B.; Sharpe, A.H. Ctla-4 and t cell activation. Curr. Opin. Immunol. 1999, 11, 294–300. [Google Scholar] [CrossRef]
- Qureshi, O.S.; Zheng, Y.; Nakamura, K.; Attridge, K.; Manzotti, C.; Schmidt, E.M.; Baker, J.; Jeffery, L.E.; Kaur, S.; Briggs, Z.; et al. Trans-endocytosis of cd80 and cd86: A molecular basis for the cell-extrinsic function of ctla-4. Science 2011, 332, 600–603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Freeman, G.J.; Long, A.J.; Iwai, Y.; Bourque, K.; Chernova, T.; Nishimura, H.; Fitz, L.J.; Malenkovich, N.; Okazaki, T.; Byrne, M.C.; et al. Engagement of the pd-1 immunoinhibitory receptor by a novel b7 family member leads to negative regulation of lymphocyte activation. J. Exp. Med. 2000, 192, 1027–1034. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wherry, E.J. T cell exhaustion. Nat. Immunol. 2011, 12, 492–499. [Google Scholar] [CrossRef]
- Mazzarella, L.; Duso, B.A.; Trapani, D.; Belli, C.; D’Amico, P.; Ferraro, E.; Viale, G.; Curigliano, G. The evolving landscape of ’next-generation’ immune checkpoint inhibitors: A review. Eur. J. Cancer 2019, 117, 14–31. [Google Scholar] [CrossRef]
- Leach, D.R.; Krummel, M.F.; Allison, J.P. Enhancement of antitumor immunity by ctla-4 blockade. Science 1996, 271, 1734–1736. [Google Scholar] [CrossRef] [Green Version]
- Egen, J.G.; Ouyang, W.; Wu, L.C. Human anti-tumor immunity: Insights from immunotherapy clinical trials. Immunity 2020, 52, 36–54. [Google Scholar] [CrossRef]
- Singh, S.; Hassan, D.; Aldawsari, H.M.; Molugulu, N.; Shukla, R.; Kesharwani, P. Immune checkpoint inhibitors: A promising anticancer therapy. Drug Discov. Today 2020, 25, 223–229. [Google Scholar] [CrossRef]
- Chen, L.; Han, X. Anti-pd-1/pd-l1 therapy of human cancer: Past, present, and future. J. Clin. Investig. 2015, 125, 3384–3391. [Google Scholar] [CrossRef] [Green Version]
- Gunturi, A.; McDermott, D.F. Nivolumab for the treatment of cancer. Expert Opin. Investig. Drugs 2015, 24, 253–260. [Google Scholar] [CrossRef]
- Hodi, F.S.; O’Day, S.J.; McDermott, D.F.; Weber, R.W.; Sosman, J.A.; Haanen, J.B.; Gonzalez, R.; Robert, C.; Schadendorf, D.; Hassel, J.C.; et al. Improved survival with ipilimumab in patients with metastatic melanoma. N. Engl. J. Med. 2010, 363, 711–723. [Google Scholar] [CrossRef] [PubMed]
- Powles, T.; Eder, J.P.; Fine, G.D.; Braiteh, F.S.; Loriot, Y.; Cruz, C.; Bellmunt, J.; Burris, H.A.; Petrylak, D.P.; Teng, S.L.; et al. Mpdl3280a (anti-pd-l1) treatment leads to clinical activity in metastatic bladder cancer. Nature 2014, 515, 558–562. [Google Scholar] [CrossRef] [PubMed]
- Weber, J.S.; D’Angelo, S.P.; Minor, D.; Hodi, F.S.; Gutzmer, R.; Neyns, B.; Hoeller, C.; Khushalani, N.I.; Miller, W.H., Jr.; Lao, C.D.; et al. Nivolumab versus chemotherapy in patients with advanced melanoma who progressed after anti-ctla-4 treatment (checkmate 037): A randomised, controlled, open-label, phase 3 trial. Lancet Oncol. 2015, 16, 375–384. [Google Scholar] [CrossRef]
- Puri, S.; Shafique, M. Combination checkpoint inhibitors for treatment of non-small-cell lung cancer: An update on dual anti-ctla-4 and anti-pd-1/pd-l1 therapies. Drugs Context 2020, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ju, X.; Zhang, H.; Zhou, Z.; Wang, Q. Regulation of pd-l1 expression in cancer and clinical implications in immunotherapy. Am. J. Cancer Res. 2020, 10, 1–11. [Google Scholar]
- Schummer, P.; Schilling, B.; Gesierich, A. Long-term outcomes in braf-mutated melanoma treated with combined targeted therapy or immune checkpoint blockade: Are we approaching a true cure? Am. J. Clin. Dermatol. 2020. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.; Chen, W.; Xu, Z.P.; Gu, W. Pd-l1 distribution and perspective for cancer immunotherapy-blockade, knockdown, or inhibition. Front. Immunol. 2019, 10, 2022. [Google Scholar] [CrossRef] [Green Version]
- Kalbasi, A.; Ribas, A. Tumour-intrinsic resistance to immune checkpoint blockade. Nat. Rev. Immunol. 2020, 20, 25–39. [Google Scholar] [CrossRef]
- Bai, J.; Gao, Z.; Li, X.; Dong, L.; Han, W.; Nie, J. Regulation of pd-1/pd-l1 pathway and resistance to pd-1/pd-l1 blockade. Oncotarget 2017, 8, 110693–110707. [Google Scholar] [CrossRef] [Green Version]
- Barrueto, L.; Caminero, F.; Cash, L.; Makris, C.; Lamichhane, P.; Deshmukh, R.R. Resistance to checkpoint inhibition in cancer immunotherapy. Transl. Oncol. 2020, 13, 100738. [Google Scholar] [CrossRef]
- Xu, J.L.; Davis, M.M. Diversity in the cdr3 region of v(h) is sufficient for most antibody specificities. Immunity 2000, 13, 37–45. [Google Scholar] [CrossRef] [Green Version]
- Turner, S.J.; Doherty, P.C.; McCluskey, J.; Rossjohn, J. Structural determinants of t-cell receptor bias in immunity. Nat. Rev. Immunol. 2006, 6, 883–894. [Google Scholar] [CrossRef] [PubMed]
- Kirsch, I.; Vignali, M.; Robins, H. T-cell receptor profiling in cancer. Mol. Oncol. 2015, 9, 2063–2070. [Google Scholar] [CrossRef] [PubMed]
- Bradley, P.; Thomas, P.G. Using t cell receptor repertoires to understand the principles of adaptive immune recognition. Annu. Rev. Immunol. 2019, 37, 547–570. [Google Scholar] [CrossRef]
- Jenkins, M.K.; Chu, H.H.; McLachlan, J.B.; Moon, J.J. On the composition of the preimmune repertoire of t cells specific for peptide-major histocompatibility complex ligands. Annu. Rev. Immunol. 2010, 28, 275–294. [Google Scholar] [CrossRef]
- Lythe, G.; Callard, R.E.; Hoare, R.L.; Molina-Paris, C. How many tcr clonotypes does a body maintain? J. Theor. Biol. 2016, 389, 214–224. [Google Scholar] [CrossRef] [Green Version]
- Greiff, V.; Miho, E.; Menzel, U.; Reddy, S.T. Bioinformatic and statistical analysis of adaptive immune repertoires. Trends Immunol. 2015, 36, 738–749. [Google Scholar] [CrossRef]
- Rosati, E.; Dowds, C.M.; Liaskou, E.; Henriksen, E.K.K.; Karlsen, T.H.; Franke, A. Overview of methodologies for t-cell receptor repertoire analysis. BMC Biotechnol. 2017, 17, 61. [Google Scholar] [CrossRef]
- Mimmi, S.; Vecchio, E.; Iaccino, E.; Rossi, M.; Lupia, A.; Albano, F.; Chiurazzi, F.; Fiume, G.; Pisano, A.; Ceglia, S.; et al. Evidence of shared epitopic reactivity among independent b-cell clones in chronic lymphocytic leukemia patients. Leukemia 2016, 30, 2419–2422. [Google Scholar] [CrossRef] [Green Version]
- Warren, R.L.; Freeman, J.D.; Zeng, T.; Choe, G.; Munro, S.; Moore, R.; Webb, J.R.; Holt, R.A. Exhaustive t-cell repertoire sequencing of human peripheral blood samples reveals signatures of antigen selection and a directly measured repertoire size of at least 1 million clonotypes. Genome Res. 2011, 21, 790–797. [Google Scholar] [CrossRef] [Green Version]
- Greiff, V.; Menzel, U.; Haessler, U.; Cook, S.C.; Friedensohn, S.; Khan, T.A.; Pogson, M.; Hellmann, I.; Reddy, S.T. Quantitative assessment of the robustness of next-generation sequencing of antibody variable gene repertoires from immunized mice. bmc Immunol. 2014, 15, 40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lynch, J.N.; Donermeyer, D.L.; Weber, K.S.; Kranz, D.M.; Allen, P.M. Subtle changes in tcralpha cdr1 profoundly increase the sensitivity of cd4 t cells. Mol. Immunol. 2013, 53, 283–294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Birnbaum, M.E.; Mendoza, J.L.; Sethi, D.K.; Dong, S.; Glanville, J.; Dobbins, J.; Ozkan, E.; Davis, M.M.; Wucherpfennig, K.W.; Garcia, K.C. Deconstructing the peptide-mhc specificity of t cell recognition. Cell 2014, 157, 1073–1087. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okino, S.T.; Kong, M.; Sarras, H.; Wang, Y. Evaluation of bias associated with high-multiplex, target-specific pre-amplification. Biomol. Detect. Quantif. 2016, 6, 13–21. [Google Scholar] [CrossRef] [Green Version]
- Boyd, S.D.; Marshall, E.L.; Merker, J.D.; Maniar, J.M.; Zhang, L.N.; Sahaf, B.; Jones, C.D.; Simen, B.B.; Hanczaruk, B.; Nguyen, K.D.; et al. Measurement and clinical monitoring of human lymphocyte clonality by massively parallel vdj pyrosequencing. Sci. Transl. Med. 2009, 1, 12ra23. [Google Scholar] [CrossRef] [Green Version]
- Freeman, J.D.; Warren, R.L.; Webb, J.R.; Nelson, B.H.; Holt, R.A. Profiling the t-cell receptor beta-chain repertoire by massively parallel sequencing. Genome Res. 2009, 19, 1817–1824. [Google Scholar] [CrossRef] [Green Version]
- Kivioja, T.; Vaharautio, A.; Karlsson, K.; Bonke, M.; Enge, M.; Linnarsson, S.; Taipale, J. Counting absolute numbers of molecules using unique molecular identifiers. Nat. Methods 2011, 9, 72–74. [Google Scholar] [CrossRef]
- Ma, K.Y.; He, C.; Wendel, B.S.; Williams, C.M.; Xiao, J.; Yang, H.; Jiang, N. Immune repertoire sequencing using molecular identifiers enables accurate clonality discovery and clone size quantification. Front. Immunol. 2018, 9, 33. [Google Scholar] [CrossRef]
- Oakes, T.; Heather, J.M.; Best, K.; Byng-Maddick, R.; Husovsky, C.; Ismail, M.; Joshi, K.; Maxwell, G.; Noursadeghi, M.; Riddell, N.; et al. Quantitative characterization of the t cell receptor repertoire of naive and memory subsets using an integrated experimental and computational pipeline which is robust, economical, and versatile. Front. Immunol. 2017, 8, 1267. [Google Scholar] [CrossRef] [Green Version]
- Uddin, I.; Woolston, A.; Peacock, T.; Joshi, K.; Ismail, M.; Ronel, T.; Husovsky, C.; Chain, B. Quantitative analysis of the t cell receptor repertoire. Methods Enzymol. 2019, 629, 465–492. [Google Scholar]
- Bolotin, D.A.; Mamedov, I.Z.; Britanova, O.V.; Zvyagin, I.V.; Shagin, D.; Ustyugova, S.V.; Turchaninova, M.A.; Lukyanov, S.; Lebedev, Y.B.; Chudakov, D.M. Next generation sequencing for tcr repertoire profiling: Platform-specific features and correction algorithms. Eur. J. Immunol. 2012, 42, 3073–3083. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giudicelli, V.; Duroux, P.; Ginestoux, C.; Folch, G.; Jabado-Michaloud, J.; Chaume, D.; Lefranc, M.P. Imgt/ligm-db, the imgt comprehensive database of immunoglobulin and t cell receptor nucleotide sequences. Nucleic Acids Res. 2006, 34, D781–D784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Yang, X.; Zhang, Y.; Zhang, Y.; Wang, M.; Ou, J.X.; Zhu, Y.; Zeng, H.; Wu, J.; Lan, C.; et al. Tools for fundamental analysis functions of tcr repertoires: A systematic comparison. Brief. Bioinform. 2019. Available online: https://academic.oup.com/bib/advance-article/doi/10.1093/bib/bbz092/5586920. (accessed on 18 October 2019).
- Alamyar, E.; Duroux, P.; Lefranc, M.P.; Giudicelli, V. Imgt((r)) tools for the nucleotide analysis of immunoglobulin (ig) and t cell receptor (tr) v-(d)-j repertoires, polymorphisms, and ig mutations: Imgt/v-quest and imgt/highv-quest for ngs. Methods Mol. Biol. 2012, 882, 569–604. [Google Scholar]
- Bolotin, D.A.; Poslavsky, S.; Mitrophanov, I.; Shugay, M.; Mamedov, I.Z.; Putintseva, E.V.; Chudakov, D.M. Mixcr: Software for comprehensive adaptive immunity profiling. Nat. Methods 2015, 12, 380–381. [Google Scholar] [CrossRef] [PubMed]
- Bolotin, D.A.; Shugay, M.; Mamedov, I.Z.; Putintseva, E.V.; Turchaninova, M.A.; Zvyagin, I.V.; Britanova, O.V.; Chudakov, D.M. Mitcr: Software for t-cell receptor sequencing data analysis. Nat. Methods 2013, 10, 813–814. [Google Scholar] [CrossRef]
- Giraud, M.; Salson, M.; Duez, M.; Villenet, C.; Quief, S.; Caillault, A.; Grardel, N.; Roumier, C.; Preudhomme, C.; Figeac, M. Fast multiclonal clusterization of v(d)j recombinations from high-throughput sequencing. BMC Genom. 2014, 15, 409. [Google Scholar] [CrossRef] [Green Version]
- Kuchenbecker, L.; Nienen, M.; Hecht, J.; Neumann, A.U.; Babel, N.; Reinert, K.; Robinson, P.N. Imseq—A fast and error aware approach to immunogenetic sequence analysis. Bioinformatics 2015, 31, 2963–2971. [Google Scholar] [CrossRef] [Green Version]
- Gerritsen, B.; Pandit, A.; Andeweg, A.C.; de Boer, R.J. Rtcr: A pipeline for complete and accurate recovery of t cell repertoires from high throughput sequencing data. Bioinformatics 2016, 32, 3098–3106. [Google Scholar] [CrossRef] [Green Version]
- Hung, S.J.; Chen, Y.L.; Chu, C.H.; Lee, C.C.; Chen, W.L.; Lin, Y.L.; Lin, M.C.; Ho, C.L.; Liu, T. Trig: A robust alignment pipeline for non-regular t-cell receptor and immunoglobulin sequences. BMC Bioinform. 2016, 17, 433. [Google Scholar] [CrossRef] [Green Version]
- immunarch, R package version 0.5.5.; An R Package for Painless Bioinformatics Analysis of T-cell and B-cell Immune Repertoire Data. Available online: https://zenodo.org/record/3383240#.XoBv3i2B3jE (accessed on 30 March 2020).
- Sidhom, J.W.; Bessell, C.A.; Havel, J.J.; Kosmides, A.; Chan, T.A.; Schneck, J.P. Immunomap: A bioinformatics tool for t-cell repertoire analysis. Cancer Immunol. Res. 2018, 6, 151–162. [Google Scholar] [CrossRef] [Green Version]
- Shugay, M.; Bagaev, D.V.; Turchaninova, M.A.; Bolotin, D.A.; Britanova, O.V.; Putintseva, E.V.; Pogorelyy, M.V.; Nazarov, V.I.; Zvyagin, I.V.; Kirgizova, V.I.; et al. Vdjtools: Unifying post-analysis of t cell receptor repertoires. Plos Comput. Biol. 2015, 11, e1004503. [Google Scholar] [CrossRef] [PubMed]
- LymphoSeq, R package version 1.14.0.; Analyze high-throughput sequencing of t and b cell receptors. Available online: https://bioconductor.org/packages/devel/bioc/vignettes/LymphoSeq/inst/doc/LymphoSeq.html (accessed on 30 March 2020).
- Laydon, D.J.; Melamed, A.; Sim, A.; Gillet, N.A.; Sim, K.; Darko, S.; Kroll, J.S.; Douek, D.C.; Price, D.A.; Bangham, C.R.; et al. Quantification of htlv-1 clonality and tcr diversity. PLoS Comput. Biol. 2014, 10, e1003646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mora, T.W.; Walczak, A. Quantifying lymphocyte receptor diversity. arXiv 2016, arXiv:604.00487v00481. Available online: https://arxiv.org/pdf/1604.00487.pdf (accessed on 2 April 2016).
- Robins, H.S.; Campregher, P.V.; Srivastava, S.K.; Wacher, A.; Turtle, C.J.; Kahsai, O.; Riddell, S.R.; Warren, E.H.; Carlson, C.S. Comprehensive assessment of t-cell receptor beta-chain diversity in alphabeta t cells. Blood 2009, 114, 4099–4107. [Google Scholar] [CrossRef] [PubMed]
- Carlson, C.S.; Emerson, R.O.; Sherwood, A.M.; Desmarais, C.; Chung, M.W.; Parsons, J.M.; Steen, M.S.; LaMadrid-Herrmannsfeldt, M.A.; Williamson, D.W.; Livingston, R.J.; et al. Using synthetic templates to design an unbiased multiplex pcr assay. Nat. Commun. 2013, 4, 2680. [Google Scholar] [CrossRef] [Green Version]
- Thomas, P.G.; Handel, A.; Doherty, P.C.; La Gruta, N.L. Ecological analysis of antigen-specific ctl repertoires defines the relationship between naive and immune t-cell populations. Proc. Natl. Acad. Sci. USA 2013, 110, 1839–1844. [Google Scholar] [CrossRef] [Green Version]
- Robert, L.; Tsoi, J.; Wang, X.; Emerson, R.; Homet, B.; Chodon, T.; Mok, S.; Huang, R.R.; Cochran, A.J.; Comin-Anduix, B.; et al. Ctla4 blockade broadens the peripheral t-cell receptor repertoire. Clin. Cancer Res.: Off. J. Am. Assoc. Cancer Res. 2014, 20, 2424–2432. [Google Scholar] [CrossRef] [Green Version]
- Cha, E.; Klinger, M.; Hou, Y.; Cummings, C.; Ribas, A.; Faham, M.; Fong, L. Improved survival with t cell clonotype stability after anti-ctla-4 treatment in cancer patients. Sci. Transl. Med. 2014, 6, 238ra270. [Google Scholar] [CrossRef] [Green Version]
- Tumeh, P.C.; Harview, C.L.; Yearley, J.H.; Shintaku, I.P.; Taylor, E.J.; Robert, L.; Chmielowski, B.; Spasic, M.; Henry, G.; Ciobanu, V.; et al. Pd-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 2014, 515, 568–571. [Google Scholar] [CrossRef]
- Snyder, A.; Nathanson, T.; Funt, S.A.; Ahuja, A.; Buros Novik, J.; Hellmann, M.D.; Chang, E.; Aksoy, B.A.; Al-Ahmadie, H.; Yusko, E.; et al. Contribution of systemic and somatic factors to clinical response and resistance to pd-l1 blockade in urothelial cancer: An exploratory multi-omic analysis. PLoS Med. 2017, 14, e1002309. [Google Scholar] [CrossRef]
- Forde, P.M.; Chaft, J.E.; Smith, K.N.; Anagnostou, V.; Cottrell, T.R.; Hellmann, M.D.; Zahurak, M.; Yang, S.C.; Jones, D.R.; Broderick, S.; et al. Neoadjuvant pd-1 blockade in resectable lung cancer. N. Engl. J. Med. 2018, 378, 1976–1986. [Google Scholar] [CrossRef]
- Yusko, E.; Vignali, M.; Wilson, R.K.; Mardis, E.R.; Hodi, F.S.; Horak, C.; Chang, H.; Woods, D.M.; Robins, H.; Weber, J. Association of tumor microenvironment t-cell repertoire and mutational load with clinical outcome after sequential checkpoint blockade in melanoma. Cancer Immunol. Res. 2019, 7, 458–465. [Google Scholar] [CrossRef] [PubMed]
- Postow, M.A.; Manuel, M.; Wong, P.; Yuan, J.; Dong, Z.; Liu, C.; Perez, S.; Tanneau, I.; Noel, M.; Courtier, A.; et al. Peripheral t cell receptor diversity is associated with clinical outcomes following ipilimumab treatment in metastatic melanoma. J. Immunother. Cancer 2015, 3, 23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hogan, S.A.; Courtier, A.; Cheng, P.F.; Jaberg-Bentele, N.F.; Goldinger, S.M.; Manuel, M.; Perez, S.; Plantier, N.; Mouret, J.F.; Nguyen-Kim, T.D.L.; et al. Peripheral blood tcr repertoire profiling may facilitate patient stratification for immunotherapy against melanoma. Cancer Immunol. Res. 2019, 7, 77–85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hopkins, A.C.; Yarchoan, M.; Durham, J.N.; Yusko, E.C.; Rytlewski, J.A.; Robins, H.S.; Laheru, D.A.; Le, D.T.; Lutz, E.R.; Jaffee, E.M. T cell receptor repertoire features associated with survival in immunotherapy-treated pancreatic ductal adenocarcinoma. JCI Insight 2018, 3, e122092. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roh, W.; Chen, P.L.; Reuben, A.; Spencer, C.N.; Prieto, P.A.; Miller, J.P.; Gopalakrishnan, V.; Wang, F.; Cooper, Z.A.; Reddy, S.M.; et al. Integrated molecular analysis of tumor biopsies on sequential ctla-4 and pd-1 blockade reveals markers of response and resistance. Sci. Transl. Med. 2017, 9, eaah3560. [Google Scholar] [CrossRef] [Green Version]
- Subudhi, S.K.; Aparicio, A.; Gao, J.; Zurita, A.J.; Araujo, J.C.; Logothetis, C.J.; Tahir, S.A.; Korivi, B.R.; Slack, R.S.; Vence, L.; et al. Clonal expansion of cd8 t cells in the systemic circulation precedes development of ipilimumab-induced toxicities. Proc. Natl. Acad. Sci. USA 2016, 113, 11919–11924. [Google Scholar] [CrossRef] [Green Version]
- Han, J.; Duan, J.; Bai, H.; Wang, Y.; Wan, R.; Wang, X.; Chen, S.; Tian, Y.; Wang, D.; Fei, K.; et al. Tcr repertoire diversity of peripheral pd-1(+)cd8(+) t cells predicts clinical outcomes after immunotherapy in patients with non-small cell lung cancer. Cancer Immunol. Res. 2020, 8, 146–154. [Google Scholar] [CrossRef]
- Khunger, A.; Rytlewski, J.A.; Fields, P.; Yusko, E.C.; Tarhini, A.A. The impact of ctla-4 blockade and interferon-alpha on clonality of t-cell repertoire in the tumor microenvironment and peripheral blood of metastatic melanoma patients. Oncoimmunology 2019, 8, e1652538. [Google Scholar] [CrossRef] [Green Version]
- Looney, T.J.; Topacio-Hall, D.; Lowman, G.; Conroy, J.; Morrison, C.; Oh, D.; Fong, L.; Zhang, L. Tcr convergence in individuals treated with immune checkpoint inhibition for cancer. Front. Immunol. 2019, 10, 2985. [Google Scholar] [CrossRef] [Green Version]
- Neller, M.A.; Burrows, J.M.; Rist, M.J.; Miles, J.J.; Burrows, S.R. High frequency of herpesvirus-specific clonotypes in the human t cell repertoire can remain stable over decades with minimal turnover. J. Virol. 2013, 87, 697–700. [Google Scholar] [CrossRef] [Green Version]
- Anagnostou, V.; Forde, P.M.; White, J.R.; Niknafs, N.; Hruban, C.; Naidoo, J.; Marrone, K.; Sivakumar, I.K.A.; Bruhm, D.C.; Rosner, S.; et al. Dynamics of tumor and immune responses during immune checkpoint blockade in non-small cell lung cancer. Cancer Res. 2019, 79, 1214–1225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dash, P.; Fiore-Gartland, A.J.; Hertz, T.; Wang, G.C.; Sharma, S.; Souquette, A.; Crawford, J.C.; Clemens, E.B.; Nguyen, T.H.O.; Kedzierska, K.; et al. Quantifiable predictive features define epitope-specific t cell receptor repertoires. Nature 2017, 547, 89–93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glanville, J.; Huang, H.; Nau, A.; Hatton, O.; Wagar, L.E.; Rubelt, F.; Ji, X.; Han, A.; Krams, S.M.; Pettus, C.; et al. Identifying specificity groups in the t cell receptor repertoire. Nature 2017, 547, 94–98. [Google Scholar] [CrossRef] [PubMed]
- Danilova, L.; Anagnostou, V.; Caushi, J.X.; Sidhom, J.W.; Guo, H.; Chan, H.Y.; Suri, P.; Tam, A.; Zhang, J.; Asmar, M.E.; et al. The mutation-associated neoantigen functional expansion of specific t cells (manafest) assay: A sensitive platform for monitoring antitumor immunity. Cancer Immunol. Res. 2018, 6, 888–899. [Google Scholar] [CrossRef] [Green Version]
- Sims, J.S.; Grinshpun, B.; Feng, Y.; Ung, T.H.; Neira, J.A.; Samanamud, J.L.; Canoll, P.; Shen, Y.; Sims, P.A.; Bruce, J.N. Diversity and divergence of the glioma-infiltrating t-cell receptor repertoire. Proc. Natl. Acad. Sci. USA 2016, 113, E3529–E3537. [Google Scholar] [CrossRef] [Green Version]
- Huang, A.C.; Postow, M.A.; Orlowski, R.J.; Mick, R.; Bengsch, B.; Manne, S.; Xu, W.; Harmon, S.; Giles, J.R.; Wenz, B.; et al. T-cell invigoration to tumour burden ratio associated with anti-pd-1 response. Nature 2017, 545, 60–65. [Google Scholar] [CrossRef] [Green Version]
- Schumacher, T.N.; Schreiber, R.D. Neoantigens in cancer immunotherapy. Science 2015, 348, 69–74. [Google Scholar] [CrossRef] [Green Version]
- McGranahan, N.; Furness, A.J.; Rosenthal, R.; Ramskov, S.; Lyngaa, R.; Saini, S.K.; Jamal-Hanjani, M.; Wilson, G.A.; Birkbak, N.J.; Hiley, C.T.; et al. Clonal neoantigens elicit t cell immunoreactivity and sensitivity to immune checkpoint blockade. Science 2016, 351, 1463–1469. [Google Scholar] [CrossRef] [Green Version]
- Riaz, N.; Morris, L.; Havel, J.J.; Makarov, V.; Desrichard, A.; Chan, T.A. The role of neoantigens in response to immune checkpoint blockade. Int. Immunol. 2016, 28, 411–419. [Google Scholar] [CrossRef] [Green Version]
- Gros, A.; Robbins, P.F.; Yao, X.; Li, Y.F.; Turcotte, S.; Tran, E.; Wunderlich, J.R.; Mixon, A.; Farid, S.; Dudley, M.E.; et al. Pd-1 identifies the patient-specific cd8(+) tumor-reactive repertoire infiltrating human tumors. J. Clin. Investig. 2014, 124, 2246–2259. [Google Scholar] [CrossRef]
- Zhu, W.; Germain, C.; Liu, Z.; Sebastian, Y.; Devi, P.; Knockaert, S.; Brohawn, P.; Lehmann, K.; Damotte, D.; Validire, P.; et al. A high density of tertiary lymphoid structure b cells in lung tumors is associated with increased cd4(+) t cell receptor repertoire clonality. Oncoimmunology 2015, 4, e1051922. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Zhang, B.; Yang, Y.; Zhu, J.; Cheng, S.; Mao, Y.; Feng, L.; Xiao, T. Characterization of distinct t cell receptor repertoires in tumor and distant non-tumor tissues from lung cancer patients. Genom. Proteom. Bioinform. 2019, 17, 287–296. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Ji, Z.; Caushi, J.X.; El Asmar, M.; Anagnostou, V.; Cottrell, T.R.; Chan, H.Y.; Suri, P.; Guo, H.; Merghoub, T.; et al. Compartmental analysis of t-cell clonal dynamics as a function of pathologic response to neoadjuvant pd-1 blockade in resectable non-small cell lung cancer. Clin. Cancer Res. 2020, 26, 1327–1337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yost, K.E.; Satpathy, A.T.; Wells, D.K.; Qi, Y.; Wang, C.; Kageyama, R.; McNamara, K.L.; Granja, J.M.; Sarin, K.Y.; Brown, R.A.; et al. Clonal replacement of tumor-specific t cells following pd-1 blockade. Nat. Med. 2019, 25, 1251–1259. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Company | Kit/Service | Starting Material | Library Preparation | Chains | Sequencing Platform |
|---|---|---|---|---|---|
| ThermoFisher Sci. | Oncomine TCR Beta | DNA/RNA | Multiplex PCR primers FR1-C | β | Iontorrent |
| Takara | SMARTer Human TCRα/β Profiling Kit | RNA | 5′ RACE | α/β | ILLUMINA |
| Adaptive Biotechnologies | ImmunoSEQ | DNA | Multiplex-PCR primers V-J | α/β/δ/γ | ILLUMINA |
| BGI (Copenhagen N, Denmark) | IR-SEQ | RNA | Multiplex PCR or 5′ RACE | α/β | ILLUMINA |
| CD Genomics (New York, USA) | Immune Repertoire Sequencing | DNARNA | Multiplex PCR or 5′ RACE | α/β | ILLUMINA |
| iRepertoire, Inc. (Huntsville, USA) | DNARNA | Multiplex PCR primers V-J or V-C | α/β/δ/γ | ILLUMINA |
| Tools | Data Format | PCR/Sequencing Error Correction | Accessibility 1 | Reference |
|---|---|---|---|---|
| IMGT/HighV-Quest | FASTA | NO | Web | [55] |
| MiXCR | FASTA/FASTQ | YES | SA | [56] |
| MiTCR | FASTQ | YES | SA | [57] |
| Vidjil | FASTA/FASTQ | YES | Web/SA | [58] |
| IMSEQ | FASTA/FASTQ | YES | SA | [59] |
| RTCR | FASTQ | YES | SA | [60] |
| TRIg | FASTA | NO | SA | [61] |
| Reference | Disease | CBI | TCR Repertoire Metrics |
|---|---|---|---|
| Robert, L. et al. [71] | melanoma | CTLA4 (tremelimumab) | richness, Shannon diversity index, Pielou’s evenness index |
| Cha, et al. [72] | melanoma, prostate | CTLA4 (ipilimumab) | top 25th percentile clonotypes, Morisita’s distance |
| Tumeh, P.C. et al. [73] | melanoma | PD-1 (pembrolizumab) | Shannon entropy, 1-normalized entropy |
| Snyder, A. et al. [74] | urothelial | PD-L1 (atezolizumab) | Shannon entropy, 1-normalized entropy |
| Forde, P.M. et al. [75] | NSCLC1 | PD-1 (nivolumab) | 1-normalized entropy |
| Yusko, E. et al. [76] | melanoma | PD-1/CTLA4 (nivo/ipilimumab) | 1-normalized entropy |
| Postow, M.A. et al. [77] | melanoma | CTLA4 (ipilimumab) | richness, evenness index |
| Hogan, S.A. et al. [78] | melanoma | PD-1/CTLA4 | diversity evenness score (DE50) |
| Hopkins, A. et al. [79] | pancreatic ductal adenocarcinoma | CTLA4 (ipilimumab) | Morisita’s distance, (1-normalized entropy) |
| Roh, W. et al. [80] | melanoma | PD-1/CTLA4 (nivo/ipilimumab) | Shannon entropy, TCR clonality |
| Subudhi, S.H et al. [81] | prostate | CTLA4 (ipilimumab) | Shannon entropy, 1-normalized entropy |
| Han, J. et al. [82] | NSCLS | PD-1/PD-L1 | Shannon entropy, 1-normalized entropy |
| Khunger, A. et al. [83] | melanoma | CTLA (tremelimumab) | 1- Pielou’s Evenness, Morisita’s distance |
| Looney, T.J. et al. [84] | Clear cells, melanoma, prostate | CTLA | Shannon entropy, TCR Convergence |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aversa, I.; Malanga, D.; Fiume, G.; Palmieri, C. Molecular T-Cell Repertoire Analysis as Source of Prognostic and Predictive Biomarkers for Checkpoint Blockade Immunotherapy. Int. J. Mol. Sci. 2020, 21, 2378. https://doi.org/10.3390/ijms21072378
Aversa I, Malanga D, Fiume G, Palmieri C. Molecular T-Cell Repertoire Analysis as Source of Prognostic and Predictive Biomarkers for Checkpoint Blockade Immunotherapy. International Journal of Molecular Sciences. 2020; 21(7):2378. https://doi.org/10.3390/ijms21072378
Chicago/Turabian StyleAversa, Ilenia, Donatella Malanga, Giuseppe Fiume, and Camillo Palmieri. 2020. "Molecular T-Cell Repertoire Analysis as Source of Prognostic and Predictive Biomarkers for Checkpoint Blockade Immunotherapy" International Journal of Molecular Sciences 21, no. 7: 2378. https://doi.org/10.3390/ijms21072378
APA StyleAversa, I., Malanga, D., Fiume, G., & Palmieri, C. (2020). Molecular T-Cell Repertoire Analysis as Source of Prognostic and Predictive Biomarkers for Checkpoint Blockade Immunotherapy. International Journal of Molecular Sciences, 21(7), 2378. https://doi.org/10.3390/ijms21072378