Various Stages of Immune Synapse Formation Are Differently Dependent on the Strength of the TCR Stimulus

 , , ,
, , ,  and
and 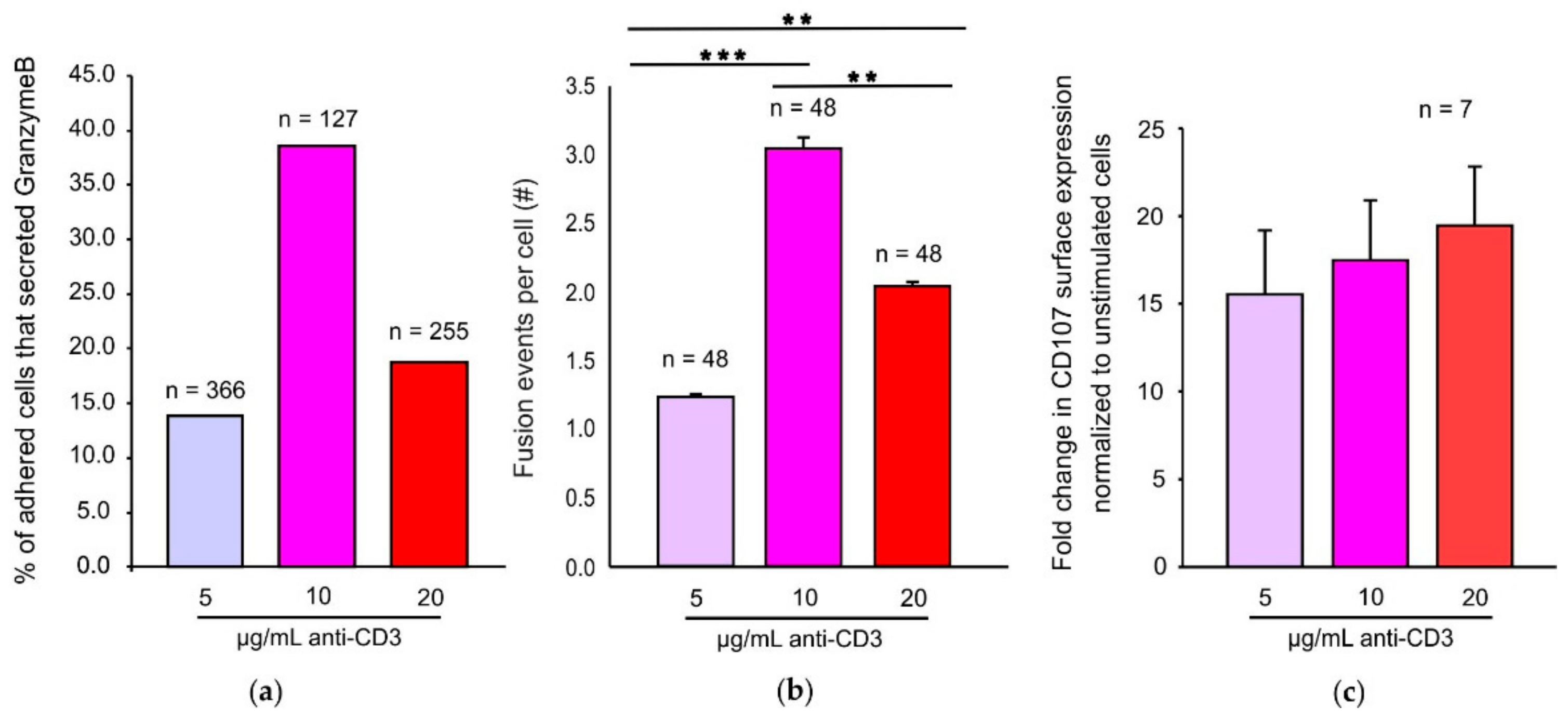{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. An Optimum TCR Trigger is Required for Maximal CG Fusion Efficiency
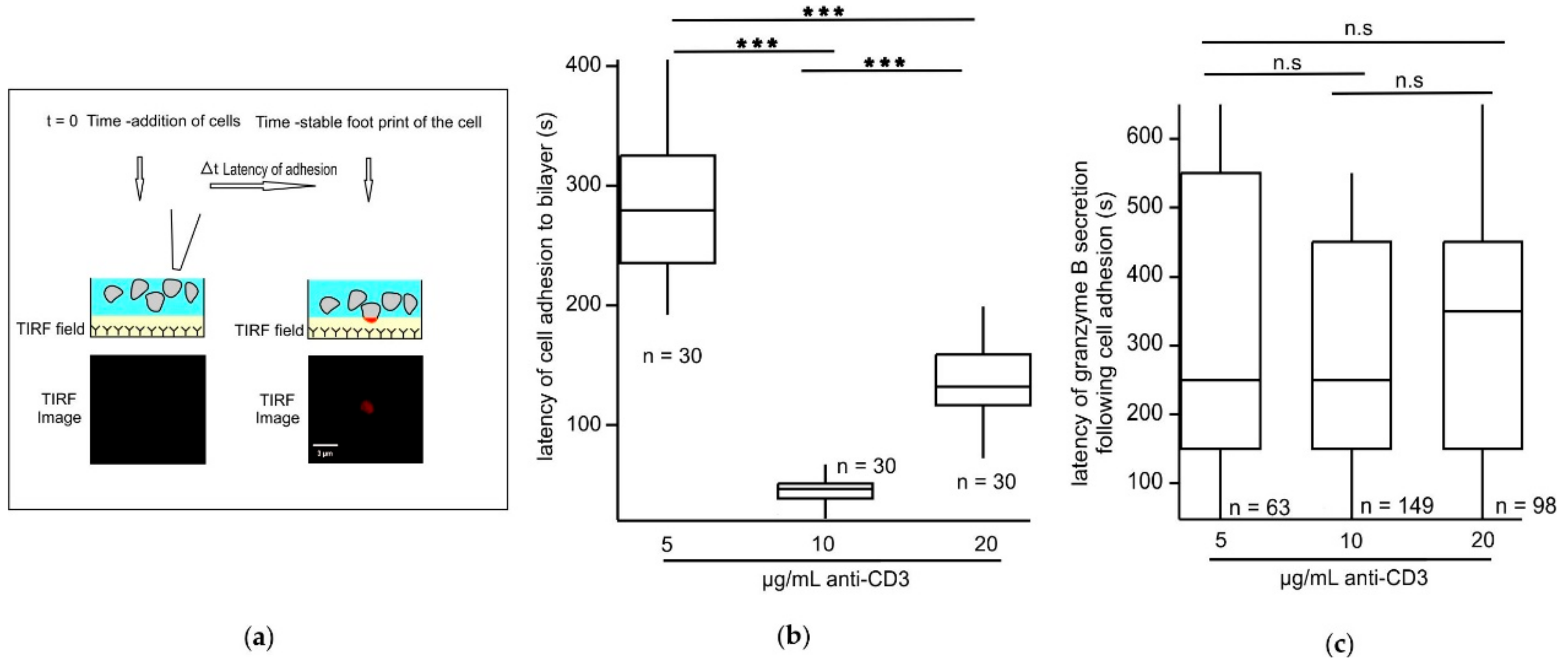
2.2. An Optimum TCR Trigger Strength Enables CTLs to Adhere Significantly Faster to Stimulatory Bilayers in Comparison to Lower or Higher Trigger Strengths
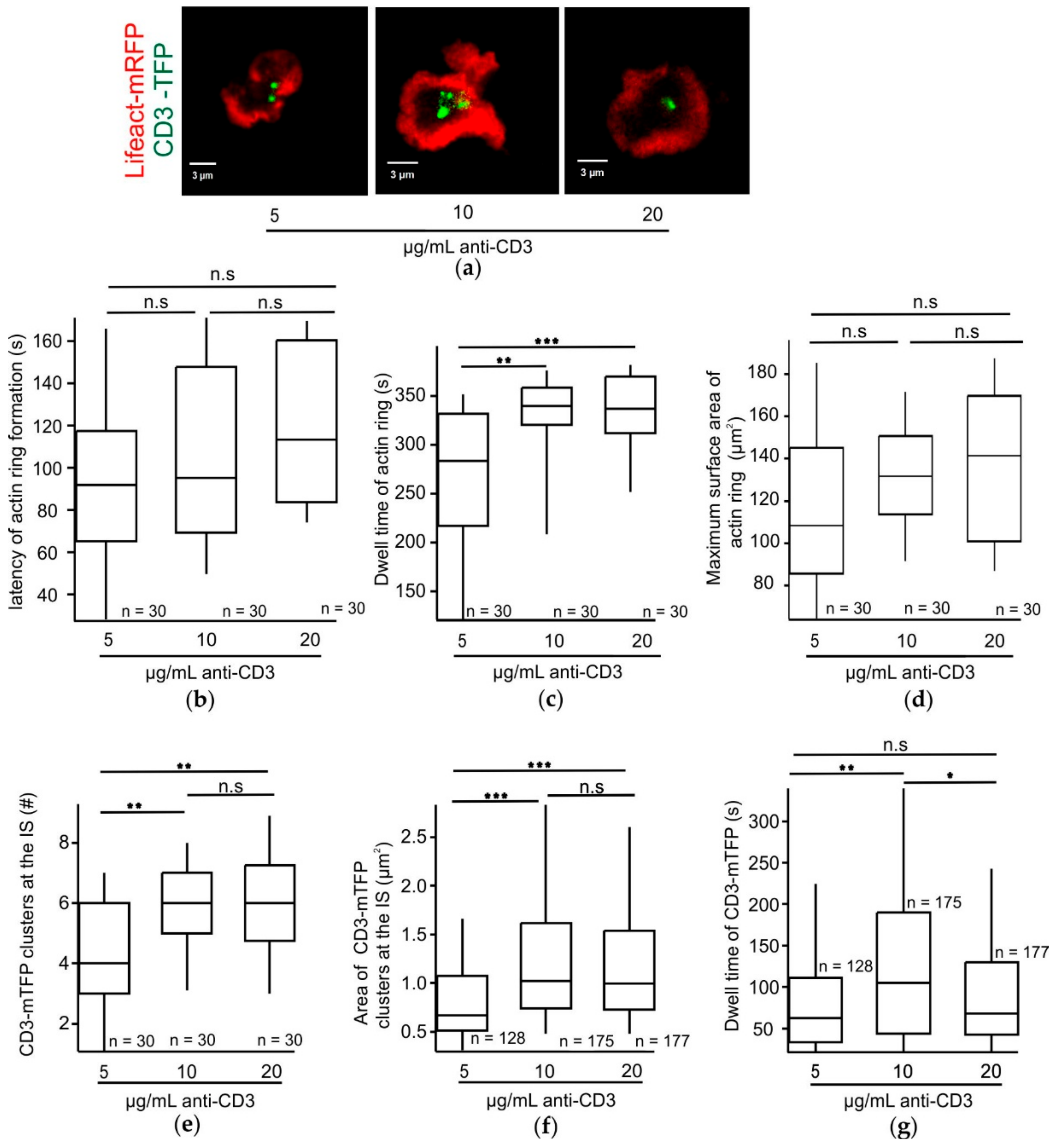
2.3. While the Dwell Time of Actin Rings Proportionally Increases with the Strength of the TCR Stimulus, the Dwell Time of CD3-mTFP Clusters at the IS Requires an Optimum TCR Stimulus in an Effector CTL Synapse
2.4. Modes of Exocytosis of Cytotoxic Granules at Immune Synapses Are Independent of the Strength of the TCR Stimulus
3. Discussion
4. Materials and Methods
4.1. Mice
4.2. Cells
4.3. Electroporation of CTLs
4.4. Plasmids
4.5. Lipid Bilayer Preparation
4.6. Total Internal Reflection Fluorescence Microscopy
4.7. Imaging Analysis
4.8. Degranulation Assay
4.9. Statistics
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| SMAC | Supramolecular activation center |
| CG | Cytotoxic granule |
| GzmB | Granzyme B |
| CTL | Cytotoxic T lymphocytes |
| TIRFM | Total internal reflection fluorescence microscopy |
References
- De La Roche, M.; Asano, Y.; Griffiths, G.M. Origins of the cytolytic synapse. Nat. Rev. Immunol. 2016, 16, 421–432. [Google Scholar] [CrossRef] [PubMed]
- Huse, M. The T-cell-receptor signaling network. J. Cell Sci. 2009, 122, 1269–1273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwarz, E.C.; Qu, B.; Hoth, M. Calcium, cancer and killing: The role of calcium in killing cancer cells by cytotoxic T lymphocytes and natural killer cells. Biochim. et Biophys. Acta (BBA) Bioenerg. 2013, 1833, 1603–1611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kabanova, A.; Zurli, V.; Baldari, C.T. Signals Controlling Lytic Granule Polarization at the Cytotoxic Immune Synapse. Front. Immunol. 2018, 9, 307. [Google Scholar] [CrossRef]
- Hashimoto-Tane, A.; Sakuma, M.; Ike, H.; Yokosuka, T.; Kimura, Y.; Ohara, O.; Saito, T. Micro–adhesion rings surrounding TCR microclusters are essential for T cell activation. J. Cell Boil. 2016, 214, 2141OIA136. [Google Scholar] [CrossRef]
- Jankowska, K.I.; Williamson, E.K.; Roy, N.H.; Blumenthal, D.; Chandra, V.; Baumgart, T.; Burkhardt, J.K. Integrins Modulate T Cell Receptor Signaling by Constraining Actin Flow at the Immunological Synapse. Front. Immunol. 2018, 9, 25. [Google Scholar] [CrossRef] [Green Version]
- Ritter, A.T.; Asano, Y.; Stinchcombe, J.C.; Dieckmann, N.; Chen, B.-C.; Gawden-Bone, C.; Van Engelenburg, S.; Legant, W.; Gao, L.; Davidson, M.W.; et al. Actin depletion initiates events leading to granule secretion at the immunological synapse. Immun. 2015, 42, 864–876. [Google Scholar] [CrossRef] [Green Version]
- Le Floc’h, A.; Tanaka, Y.; Bantilan, N.S.; Voisinne, G.; Altan-Bonnet, G.; Fukui, Y.; Huse, M. Annular PIP3 accumulation controls actin architecture and modulates cytotoxicity at the immunological synapse. J. Exp. Med. 2013, 210(12), 2721–2737. [Google Scholar]
- Ritter, A.T.; Kapnick, S.M.; Murugesan, S.; Schwartzberg, P.L.; Griffiths, G.M.; Lippincott-Schwartz, J. Cortical actin recovery at the immunological synapse leads to termination of lytic granule secretion in cytotoxic T lymphocytes. Proc. Natl. Acad. Sci. USA 2017, 114, E6585–E6594. [Google Scholar] [CrossRef] [Green Version]
- Das, V.; Nal, B.; Dujeancourt, A.; Thoulouze, M.-I.; Galli, T.; Roux, P.; Dautry-Varsat, A.; Alcover, A. Activation-induced polarized recycling targets T cell antigen receptors to the immunological synapse; involvement of SNARE complexes. Immunity 2004, 20, 577–588. [Google Scholar] [CrossRef] [Green Version]
- Friedl, P.; Boer, A.T.D.; Gunzer, M. Tuning immune responses: Diversity and adaptation of the immunological synapse. Nat. Rev. Immunol. 2005, 5, 532–545. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.-F.; Bzeih, H.; Chitirala, P.; Ravichandran, K.; Sleiman, M.; Krause, E.; Hahn, U.; Pattu, V.; Rettig, J. Preparing the lethal hit: Interplay between exo- and endocytic pathways in cytotoxic T lymphocytes. Cell. Mol. Life Sci. 2016, 74, 399–408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, H.-F.; Bzeih, H.; Schirra, C.; Chitirala, P.; Halimani, M.; Cordat, E.; Krause, E.; Rettig, J.; Pattu, V. Endocytosis of Cytotoxic Granules Is Essential for Multiple Killing of Target Cells by T Lymphocytes. J. Immunol. 2016, 197, 2473–2484. [Google Scholar] [CrossRef] [PubMed]
- Wiedemann, A.; Depoil, D.; Faroudi, M.; Valitutti, S. Cytotoxic T lymphocytes kill multiple targets simultaneously via spatiotemporal uncoupling of lytic and stimulatory synapses. Proc. Natl. Acad. Sci. USA 2006, 103, 10985–10990. [Google Scholar] [CrossRef] [Green Version]
- Dustin, M.L. Insights into Function of the Immunological Synapse from Studies with Supported Planar Bilayers. Hantaviruses 2009, 340, 1–24. [Google Scholar]
- Dustin, M.L.; Starr, T.; Varma, R.; Thomas, V.K. Supported Planar Bilayers for Study of the Immunological Synapse. Curr. Protoc. Immunol. 2007, 76. [Google Scholar] [CrossRef] [PubMed]
- Torres, A.J.; Contento, R.L.; Gordo, S.; Wucherpfennig, K.W.; Love, J.C. Functional single-cell analysis of T-cell activation by supported lipid bilayer-tethered ligands on arrays of nanowells. Lab Chip 2012, 13, 90–99. [Google Scholar] [CrossRef] [Green Version]
- Jenkins, M.R.; Tsun, A.; Stinchcombe, J.C.; Griffiths, G.M. The Strength of T Cell Receptor Signal Controls the Polarization of Cytotoxic Machinery to the Immunological Synapse. Immunity 2009, 31, 621–631. [Google Scholar] [CrossRef] [Green Version]
- Kaizuka, Y.; Douglass, A.D.; Varma, R.; Dustin, M.L.; Vale, R.D. Mechanisms for segregating T cell receptor and adhesion molecules during immunological synapse formation in Jurkat T cells. Proc. Natl. Acad. Sci. USA 2007, 104, 20296–20301. [Google Scholar] [CrossRef] [Green Version]
- Riedl, J.; Crevenna, A.; Kessenbrock, K.; Yu, J.H.; Neukirchen, R.; Bista, M.; Bradke, F.; Jenne, D.; A Holak, T.; Werb, Z.; et al. Lifeact: A versatile marker to visualize F-actin. Nat. Methods 2008, 5, 605–607. [Google Scholar] [CrossRef]
- Marshall, M.; Pattu, V.; Halimani, M.; Maier-Peuschel, M.; Müller, M.-L.; Becherer, U.; Hong, W.; Hoth, M.; Tschernig, T.; Bryceson, Y.T.; et al. VAMP8-dependent fusion of recycling endosomes with the plasma membrane facilitates T lymphocyte cytotoxicity. J. Cell Boil. 2015, 210, 135–151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, S.; Renden, R.; Von Gersdorff, H. Synaptic vesicle endocytosis: Fast and slow modes of membrane retrieval. Trends Neurosci. 2008, 31, 559–568. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matti, U.; Pattu, V.; Halimani, M.; Schirra, C.; Krause, E.; Liu, Y.; Weins, L.; Chang, H.F.; Guzman, R.; Olausson, J.; et al. Synaptobrevin2 is the v-SNARE required for cytotoxic T-lymphocyte lytic granule fusion. Nat. Commun. 2013, 4, 1439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, Y.; Rosendale, M.; Campbell, R.E.; Perrais, D. pHuji, a pH-sensitive red fluorescent protein for imaging of exo- and endocytosis. J. Cell Biol. 2014, 207, 419–432. [Google Scholar] [CrossRef] [Green Version]
- Schutz, G.J.; Huppa, J.B. How drag sharpens a T cell’s view on antigen. Proc. Natl. Acad. Sci. USA 2019, 116, 16669–16671. [Google Scholar] [CrossRef] [Green Version]
- Basu, R.; Whitlock, B.M.; Husson, J.; Le Floc’H, A.; Jin, W.; Oyler-Yaniv, A.; Dotiwala, F.; Giannone, G.; Hivroz, C.; Biais, N.; et al. Cytotoxic T cells use mechanical force to potentiate target cell killing. Cell 2016, 165, 100–110. [Google Scholar] [CrossRef] [Green Version]
- Saitakis, M.; Dogniaux, S.; Goudot, C.; Bufi, N.; Asnacios, S.; Maurin, M.; Randriamampita, C.; Asnacios, A.; Hivroz, C. Different TCR-induced T lymphocyte responses are potentiated by stiffness with variable sensitivity. eLife 2017, 6, 6. [Google Scholar] [CrossRef]
- Bryceson, Y.T.; Fauriat, C.; Nunes, J.M.D.E.; Wood, S.M.; Björkström, N.K.; Long, E.O.; Ljunggren, H.-G. Functional analysis of human NK cells by flow cytometry. Breast Cancer 2010, 612, 335–352. [Google Scholar]
- Zhou, X.; Friedmann, K.S.; Lyrmann, H.; Zhou, Y.; Schoppmeyer, R.; Knörck, A.; Mang, S.; Hoxha, C.; Angenendt, A.; Backes, C.S.; et al. A calcium optimum for cytotoxic T lymphocyte and natural killer cell cytotoxicity. J. Physiol. 2018, 596, 2681–2698. [Google Scholar] [CrossRef] [Green Version]
- Donnadieu, E.; Bismuth, G.; Trautmann, A. The intracellular Ca2+ concentration optimal for T cell activation is quite different after ionomycin or CD3 stimulation. Pflügers Arch. Eur. J. Physiol. 1995, 429, 546–554. [Google Scholar] [CrossRef]
- Shin, W.; Arpino, G.; Thiyagarajan, S.; Su, R.; Ge, L.; McDargh, Z.; Guo, X.; Wei, L.; Shupliakov, O.; Jin, A.; et al. Vesicle Shrinking and Enlargement Play Opposing Roles in the Release of Exocytotic Contents. Cell Rep. 2020, 30, 421–431.e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martina, J.A.; Wu, X.S.; Catalfamo, M.; Sakamoto, T.; Yi, C.; Hammer, J.A. Imaging of lytic granule exocytosis in CD8+ cytotoxic T lymphocytes reveals a modified form of full fusion. Cell. Immunol. 2011, 271, 267–279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, N.; A Martina, J.; Wu, X.S.; Iii, J.A.H.; Long, E.O. Two modes of lytic granule fusion during degranulation by natural killer cells. Immunol. Cell Boil. 2011, 89, 728–738. [Google Scholar] [CrossRef] [PubMed]
- Huppa, J.B.; Gleimer, M.; Sumen, C.; Davis, M.M. Continuous T cell receptor signaling required for synapse maintenance and full effector potential. Nat. Immunol. 2003, 4, 749–755. [Google Scholar] [CrossRef] [PubMed]
- Ou-Yang, C.-W.; Zhu, M.; Fuller, D.M.; Sullivan, S.A.; Chuck, M.I.; Ogden, S.; Li, Q.-J.; Zhang, W. Role of LAT in the Granule-Mediated Cytotoxicity of CD8 T Cells. Mol. Cell. Boil. 2012, 32, 2674–2684. [Google Scholar] [CrossRef] [Green Version]
- Myers, L.M.; Tal, M.C.; Torrez Dulgeroff, L.B.; Carmody, A.B.; Messer, R.J.; Gulati, G.; Yiu, Y.Y.; Staron, M.M.; Angel, C.L.; Sinha, R.; et al. A functional subset of CD8(+) T cells during chronic exhaustion is defined by SIRPalpha expression. Nat. Commun. 2019, 10, 794. [Google Scholar] [CrossRef] [Green Version]
- Hashimoto, M.; Kamphorst, A.O.; Im, S.J.; Kissick, H.; Pillai, R.; Ramalingam, S.R.; Araki, K.; Ahmed, R. CD8 T Cell Exhaustion in Chronic Infection and Cancer: Opportunities for Interventions. Annu. Rev. Med. 2018, 69, 301–318. [Google Scholar] [CrossRef]
- Halimani, M.; Pattu, V.; Marshall, M.; Chang, H.F.; Matti, U.; Jung, M.; Becherer, U.; Krause, E.; Hoth, M.; Schwarz, E.C.; et al. Syntaxin11 serves as a t- SNARE for the fusion of lytic granules in human cytotoxic T lymphocytes. Eur. J. Immunol. 2013, 44, 573–584. [Google Scholar] [CrossRef]
- Chitirala, P.; Ravichandran, K.; Galgano, D.; Sleiman, M.; Krause, E.; Bryceson, Y.T.; Rettig, J. Cytotoxic Granule Exocytosis From Human Cytotoxic T Lymphocytes Is Mediated by VAMP7. Front. Immunol. 2019, 10, 1855. [Google Scholar] [CrossRef] [Green Version]
- Chang, H.-F.; Mannebach, S.; Beck, A.; Ravichandran, K.; Krause, E.; Frohnweiler, K.; Fecher-Trost, C.; Schirra, C.; Pattu, V.; Flockerzi, V.; et al. Cytotoxic granule endocytosis depends on the Flower protein. J. Cell Boil. 2017, 217, 667–683. [Google Scholar] [CrossRef] [Green Version]




© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Estl, M.; Blatt, P.; Li, X.; Becherer, U.; Chang, H.-F.; Rettig, J.; Pattu, V. Various Stages of Immune Synapse Formation Are Differently Dependent on the Strength of the TCR Stimulus. Int. J. Mol. Sci. 2020, 21, 2475. https://doi.org/10.3390/ijms21072475
Estl M, Blatt P, Li X, Becherer U, Chang H-F, Rettig J, Pattu V. Various Stages of Immune Synapse Formation Are Differently Dependent on the Strength of the TCR Stimulus. International Journal of Molecular Sciences. 2020; 21(7):2475. https://doi.org/10.3390/ijms21072475
Chicago/Turabian StyleEstl, Michael, Pascal Blatt, Xuemei Li, Ute Becherer, Hsin-Fang Chang, Jens Rettig, and Varsha Pattu. 2020. "Various Stages of Immune Synapse Formation Are Differently Dependent on the Strength of the TCR Stimulus" International Journal of Molecular Sciences 21, no. 7: 2475. https://doi.org/10.3390/ijms21072475
APA StyleEstl, M., Blatt, P., Li, X., Becherer, U., Chang, H. -F., Rettig, J., & Pattu, V. (2020). Various Stages of Immune Synapse Formation Are Differently Dependent on the Strength of the TCR Stimulus. International Journal of Molecular Sciences, 21(7), 2475. https://doi.org/10.3390/ijms21072475