Long-Tailed Unconventional Class I Myosins in Health and Disease


Abstract
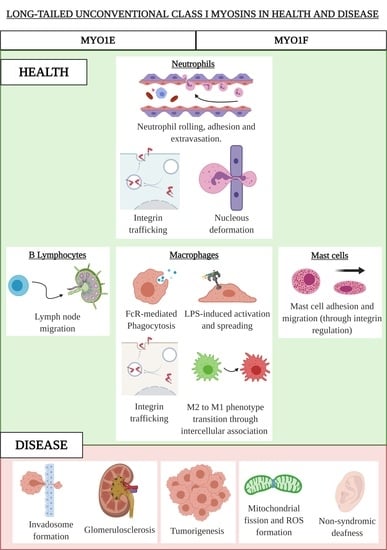
:
{kind=link}
{kind=link}
{kind=link}
1. Unconventional Myosins: General Introduction
2. Class I Unconventional Myosins
3. The Long-Tailed Unconventional Class I Myosins
3.1. MYO1E and MYO1F in Neutrophils
3.2. MYO1E and MYO1F in Macrophages
3.3. MYO1F in Mast Cells
3.4. MYO1E in B Lymphocytes
4. Class I Unconventional Long-Tailed Myosins in Diseases
4.1. MYO1E and MYO1F in Cancer
4.1.1. MYO1E
4.1.2. MYO1F
4.2. MYO1E in Kidney Diseases
4.3. MYO1F in Hearing Loss
5. Conclusions and Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Foth, B.J.; Goedecke, M.C.; Soldati, D. New insights into myosin evolution and classification. Proc. Natl. Acad. Sci. USA 2006, 103, 3681–3686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langelaan, D.N.; Liburd, J.; Yang, Y.; Miller, E.; Chitayat, S.; Crawley, S.W.; Côté, G.P.; Smith, S.P. Structure of the Single-lobe Myosin Light Chain C in Complex with the Light Chain-binding Domains of Myosin-1C Provides Insights into Divergent IQ Motif Recognition. J. Biol. Chem. 2016, 291, 19607–19617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Houdusse, A.; Cohen, C. Target sequence recognition by the calmodulin superfamily: Implications from light chain binding to the regulatory domain of scallop myosin. Proc. Natl. Acad. Sci. USA 1995, 92, 10644–10647. [Google Scholar] [CrossRef] [Green Version]
- Bähler, M.; Rhoads, A. Calmodulin signaling via the IQ motif. FEBS Lett. 2002, 513, 107–113. [Google Scholar] [CrossRef]
- Forkey, J.N.; Quinlan, M.E.; Shaw, M.A.; Corrie, J.E.T.; Goldman, Y.E. Three-dimensional structural dynamics of myosin V by single-molecule fluorescence polarization. Nature 2003, 422, 399–404. [Google Scholar] [CrossRef] [PubMed]
- Veigel, C.; Schmitz, S.; Wang, F.; Sellers, J.R. Load-dependent kinetics of myosin-V can explain its high processivity. Nat. Cell Biol. 2005, 7, 861–869. [Google Scholar] [CrossRef]
- Krendel, M.; Mooseker, M.S. Myosins: Tails (and Heads) of Functional Diversity. Physiology 2005, 20, 239–251. [Google Scholar] [CrossRef] [Green Version]
- Woolner, S.; Bement, W.M. Unconventional myosins acting unconventionally. Trends Cell Biol. 2009, 19, 245–252. [Google Scholar] [CrossRef] [Green Version]
- Langford, G.M. Actin- and microtubule-dependent organelle motors: Interrelationships between the two motility systems. Curr. Opin. Cell Biol. 1995, 7, 82–88. [Google Scholar] [CrossRef]
- Batters, C.; Veigel, C. Mechanics and Activation of Unconventional Myosins. Traffic 2016, 17, 860–871. [Google Scholar] [CrossRef] [Green Version]
- Fili, N.; Toseland, C.P. Unconventional Myosins: How Regulation Meets Function. Int. J. Mol. Sci. 2019, 21, 67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thompson, R.F.; Langford, G.M. Myosin superfamily evolutionary history. Anat. Rec. 2002, 268, 276–289. [Google Scholar] [CrossRef] [PubMed]
- Nambiar, R.; McConnell, R.E.; Tyska, M.J. Control of cell membrane tension by myosin-I. Proc. Natl. Acad. Sci. USA 2009, 106, 11972–11977. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berg, J.S.; Powell, B.C.; Cheney, R.E. A millennial myosin census. Mol. Biol. Cell 2001, 12, 780–794. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.V.; Mehal, W.Z.; Dong, X.; Heinrich, V.; Pypaert, M.; Mellman, I.; Dembo, M.; Mooseker, M.S.; Wu, D.; Flavell, R.A. Modulation of Cell Adhesion and Motility in the Immune System by Myo1f. Science 2006, 314, 136–139. [Google Scholar] [CrossRef] [Green Version]
- Patino-Lopez, G.; Aravind, L.; Dong, X.; Kruhlak, M.J.; Ostap, E.M.; Shaw, S. Myosin 1G Is an Abundant Class I Myosin in Lymphocytes Whose Localization at the Plasma Membrane Depends on Its Ancient Divergent Pleckstrin Homology (PH) Domain (Myo1PH). J. Biol. Chem. 2010, 285, 8675–8686. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, E.; Kalli, A.C.; Yasuoka, K.; Sansom, M.S.P. Interactions of Pleckstrin Homology Domains with Membranes: Adding Back the Bilayer via High-Throughput Molecular Dynamics. Structure 2016, 24, 1421–1431. [Google Scholar] [CrossRef] [Green Version]
- Yu, H.Y.E.; Bement, W.M. Multiple myosins are required to coordinate actin assembly with coat compression during compensatory endocytosis. Mol. Biol. Cell 2007, 18, 4096–4105. [Google Scholar] [CrossRef]
- Feeser, E.A.; Ignacio, C.M.G.; Krendel, M.; Ostap, E.M. Myo1e binds anionic phospholipids with high affinity. Biochemistry 2010, 49, 9353–9360. [Google Scholar] [CrossRef] [Green Version]
- Kurochkina, N.; Guha, U. SH3 domains: Modules of protein–protein interactions. Biophys. Rev. 2013, 5, 29–39. [Google Scholar] [CrossRef] [Green Version]
- Navinés-Ferrer, A.; Ainsua-Enrich, E.; Serrano-Candelas, E.; Sayós, J.; Martin, M. Myo1f, an Unconventional Long-Tailed Myosin, Is a New Partner for the Adaptor 3BP2 Involved in Mast Cell Migration. Front. Immunol. 2019, 10. [Google Scholar] [CrossRef] [PubMed]
- Nourshargh, S.; Alon, R. Leukocyte Migration into Inflamed Tissues. Immunity 2014, 41, 694–707. [Google Scholar] [CrossRef] [Green Version]
- Hind, L.E.; Vincent, W.J.B.; Huttenlocher, A. Leading from the Back: The Role of the Uropod in Neutrophil Polarization and Migration. Dev. Cell 2016, 38, 161–169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jorch, S.K.; Kubes, P. An emerging role for neutrophil extracellular traps in noninfectious disease. Nat. Med. 2017, 23, 279–287. [Google Scholar] [CrossRef] [PubMed]
- Vestweber, D. How leukocytes cross the vascular endothelium. Nat. Rev. Immunol. 2015, 15, 692–704. [Google Scholar] [CrossRef]
- Schnoor, M.; García Ponce, A.; Vadillo, E.; Pelayo, R.; Rossaint, J.; Zarbock, A. Actin dynamics in the regulation of endothelial barrier functions and neutrophil recruitment during endotoxemia and sepsis. Cell. Mol. Life Sci. 2017, 74, 1985–1997. [Google Scholar] [CrossRef]
- Vadillo, E.; Chánez-Paredes, S.; Vargas-Robles, H.; Guerrero-Fonseca, I.M.; Castellanos-Martínez, R.; García-Ponce, A.; Nava, P.; Girón-Pérez, D.A.; Santos-Argumedo, L.; Schnoor, M. Intermittent rolling is a defect of the extravasation cascade caused by Myosin1e-deficiency in neutrophils. Proc. Natl. Acad. Sci. USA 2019, 116, 26752–26758. [Google Scholar] [CrossRef]
- Chen, G.; Dimitriou, I.; Milne, L.; Lang, K.S.; Lang, P.A.; Fine, N.; Ohashi, P.S.; Kubes, P.; Rottapel, R. The 3BP2 Adapter Protein Is Required for Chemoattractant-Mediated Neutrophil Activation. J. Immunol. 2012, 189, 2138–2150. [Google Scholar] [CrossRef] [Green Version]
- Salvermoser, M.; Pick, R.; Weckbach, L.T.; Zehrer, A.; Löhr, P.; Drechsler, M.; Sperandio, M.; Soehnlein, O.; Walzog, B. Myosin 1f is specifically required for neutrophil migration in 3D environments during acute inflammation. Blood 2018, 131, 1887–1898. [Google Scholar] [CrossRef]
- Wang, Y.; Jin, H.; Wang, W.; Wang, F.; Zhao, H. Myosin1f-mediated neutrophil migration contributes to acute neuroinflammation and brain injury after stroke in mice. J. Neuroinflammation 2019, 16, 77. [Google Scholar] [CrossRef] [Green Version]
- Freeman, S.A.; Grinstein, S. Phagocytosis: Receptors, signal integration, and the cytoskeleton. Immunol. Rev. 2014, 262, 193–215. [Google Scholar] [CrossRef] [PubMed]
- Blander, J.M.; Medzhitov, R. Regulation of Phagosome Maturation by Signals from Toll-Like Receptors. Science 2004, 304, 1014–1018. [Google Scholar] [CrossRef] [PubMed]
- Wenzel, J.; Held, C.; Palmisano, R.; Teufel, S.; David, J.-P.; Wittenberg, T.; Lang, R. Measurement of TLR-Induced Macrophage Spreading by Automated Image Analysis: Differential Role of Myd88 and MAPK in Early and Late Responses. Front. Physiol. 2011, 2, 71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weintz, G.; Olsen, J.V.; Frühauf, K.; Niedzielska, M.; Amit, I.; Jantsch, J.; Mages, J.; Frech, C.; Dölken, L.; Mann, M.; et al. The phosphoproteome of toll-like receptor-activated macrophages. Mol. Syst. Biol. 2010, 6, 371. [Google Scholar] [CrossRef] [PubMed]
- Wenzel, J.; Ouderkirk, J.L.; Krendel, M.; Lang, R. Class I myosin Myo1e regulates TLR4-triggered macrophage spreading, chemokine release, and antigen presentation via MHC class II. Eur. J. Immunol. 2015, 45, 225–237. [Google Scholar] [CrossRef] [Green Version]
- Paul, P.; van den Hoorn, T.; Jongsma, M.L.M.; Bakker, M.J.; Hengeveld, R.; Janssen, L.; Cresswell, P.; Egan, D.A.; van Ham, M.; ten Brinke, A.; et al. A Genome-wide Multidimensional RNAi Screen Reveals Pathways Controlling MHC Class II Antigen Presentation. Cell 2011, 145, 268–283. [Google Scholar] [CrossRef] [Green Version]
- Piedra-Quintero, Z.L.; Serrano, C.; Villegas-Sepúlveda, N.; Maravillas-Montero, J.L.; Romero-Ramírez, S.; Shibayama, M.; Medina-Contreras, O.; Nava, P.; Santos-Argumedo, L. Myosin 1F Regulates M1-Polarization by Stimulating Intercellular Adhesion in Macrophages. Front. Immunol. 2019, 9, 3118. [Google Scholar] [CrossRef]
- Barger, S.R.; Reilly, N.S.; Shutova, M.S.; Li, Q.; Maiuri, P.; Heddleston, J.M.; Mooseker, M.S.; Flavell, R.A.; Svitkina, T.; Oakes, P.W.; et al. Membrane-cytoskeletal crosstalk mediated by myosin-I regulates adhesion turnover during phagocytosis. Nat. Commun. 2019, 10, 1249. [Google Scholar] [CrossRef] [Green Version]
- García-García, E.; Rosales, C. Signal transduction during Fc receptor-mediated phagocytosis. J. Leukoc. Biol. 2002, 107, 357–362. [Google Scholar]
- Masters, T.A.; Sheetz, M.P.; Gauthier, N.C. F-actin waves, actin cortex disassembly and focal exocytosis driven by actin-phosphoinositide positive feedback. Cytoskeleton 2016, 73, 180–196. [Google Scholar] [CrossRef]
- Galli, S.J.; Kalesnikoff, J.; Grimbaldeston, M.A.; Piliponsky, A.M.; Williams, C.M.M.; Tsai, M. MAST CELLS AS “TUNABLE” EFFECTOR AND IMMUNOREGULATORY CELLS: Recent Advances. Annu. Rev. Immunol. 2005, 23, 749–786. [Google Scholar] [CrossRef] [PubMed]
- Dahlin, J.S.; Hallgren, J. Mast cell progenitors: Origin, development and migration to tissues. Mol. Immunol. 2015, 63, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Gilfillan, A.M.; Beaven, M.A. Regulation of mast cell responses in health and disease. Crit. Rev. Immunol. 2011, 31, 475–529. [Google Scholar] [CrossRef] [PubMed]
- Halova, I.; Draberova, L.; Draber, P. Mast Cell Chemotaxis – Chemoattractants and Signaling Pathways. Front. Immunol. 2012, 3, 119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ren, R.; Mayer, B.J.; Cicchetti, P.; Baltimore, D. Identification of a ten-amino acid proline-rich SH3 binding site. Science 1993, 259, 5098. [Google Scholar] [CrossRef] [PubMed]
- Ainsua-Enrich, E.; Álvarez-Errico, D.; Gilfillan, A.M.; Picado, C.; Sayós, J.; Rivera, J.; Martín, M. The Adaptor 3BP2 Is Required for Early and Late Events in FcεRI Signaling in Human Mast Cells. J. Immunol. 2012, 189, 2727–2734. [Google Scholar] [CrossRef] [Green Version]
- Ainsua-Enrich, E.; Serrano-Candelas, E.; Álvarez-Errico, D.; Picado, C.; Sayós, J.; Rivera, J.; Martín, M. The Adaptor 3BP2 Is Required for KIT Receptor Expression and Human Mast Cell Survival. J. Immunol. 2015, 194, 4309–4318. [Google Scholar] [CrossRef] [Green Version]
- Dráber, P.; Dráber, P. Membrane-Cytoskeleton Dynamics in the Course of Mast Cell Activation. Methods Mol. Biol. 2015, 1220, 219–237. [Google Scholar]
- Mesin, L.; Ersching, J.; Victora, G.D. Germinal Center B Cell Dynamics. Immunity 2016, 45, 471–482. [Google Scholar] [CrossRef] [Green Version]
- Girard, J.-P.; Moussion, C.; Förster, R. HEVs, lymphatics and homeostatic immune cell trafficking in lymph nodes. Nat. Rev. Immunol. 2012, 12, 762–773. [Google Scholar] [CrossRef]
- Mionnet, C.; Sanos, S.L.; Mondor, I.; Jorquera, A.; Laugier, J.-P.; Germain, R.N.; Bajénoff, M. High endothelial venules as traffic control points maintaining lymphocyte population homeostasis in lymph nodes. Blood 2011, 118, 6115–6122. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y.; Niederstrasser, H.; Edwards, M.; Jackson, C.E.; Cooper, J.A. Distinct Roles for CARMIL Isoforms in Cell Migration. Mol. Biol. Cell 2009, 20, 5290–5305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Girón-Pérez, D.A.; Vadillo, E.; Schnoor, M.; Santos-Argumedo, L. Myo1e modulates the recruitment of activated B cells to inguinal lymph nodes. J. Cell Sci. 2020, 133, jcs235275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parsons, J.T.; Martin, K.H.; Slack, J.K.; Taylor, J.M.; Weed, S.A. Focal Adhesion Kinase: A regulator of focal adhesion dynamics and cell movement. Oncogene 2000, 19, 5606–5613. [Google Scholar] [CrossRef] [Green Version]
- Ouderkirk, J.L.; Krendel, M. Non-muscle myosins in tumor progression, cancer cell invasion, and metastasis. Cytoskeleton 2014, 71, 447–463. [Google Scholar] [CrossRef] [Green Version]
- Heim, J.B.; Squirewell, E.J.; Neu, A.; Zocher, G.; Sominidi-Damodaran, S.; Wyles, S.P.; Nikolova, E.; Behrendt, N.; Saunte, D.M.; Lock-Andersen, J.; et al. Myosin-1E interacts with FAK proline-rich region 1 to induce fibronectin-type matrix. Proc. Natl. Acad. Sci. USA 2017, 114, 3933–3938. [Google Scholar] [CrossRef] [Green Version]
- Hoek, K.S.; Schlegel, N.C.; Brafford, P.; Sucker, A.; Ugurel, S.; Kumar, R.; Weber, B.L.; Nathanson, K.L.; Phillips, D.J.; Herlyn, M.; et al. Metastatic potential of melanomas defined by specific gene expression profiles with no BRAF signature. Pigment Cell Res. 2006, 19, 290–302. [Google Scholar] [CrossRef]
- Meves, A.; Nikolova, E.; Heim, J.B.; Squirewell, E.J.; Cappel, M.A.; Pittelkow, M.R.; Otley, C.C.; Behrendt, N.; Saunte, D.M.; Lock-Andersen, J.; et al. Tumor Cell Adhesion As a Risk Factor for Sentinel Lymph Node Metastasis in Primary Cutaneous Melanoma. J. Clin. Oncol. 2015, 33, 2509–2515. [Google Scholar] [CrossRef]
- Serrels, A.; Lund, T.; Serrels, B.; Byron, A.; McPherson, R.C.; von Kriegsheim, A.; Gómez-Cuadrado, L.; Canel, M.; Muir, M.; Ring, J.E.; et al. Nuclear FAK Controls Chemokine Transcription, Tregs, and Evasion of Anti-tumor Immunity. Cell 2015, 163, 160–173. [Google Scholar] [CrossRef] [Green Version]
- Lahlou, H.; Sanguin-Gendreau, V.; Zuo, D.; Cardiff, R.D.; McLean, G.W.; Frame, M.C.; Muller, W.J. Mammary epithelial-specific disruption of the focal adhesion kinase blocks mammary tumor progression. Proc. Natl. Acad. Sci. USA 2007, 104, 20302–20307. [Google Scholar] [CrossRef] [Green Version]
- Ouderkirk-Pecone, J.L.; Goreczny, G.J.; Chase, S.E.; Tatum, A.H.; Turner, C.E.; Krendel, M. Myosin 1e promotes breast cancer malignancy by enhancing tumor cell proliferation and stimulating tumor cell de-differentiation. Oncotarget 2016, 7, 46419–46432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hallett, R.M.; Dvorkin-Gheva, A.; Bane, A.; Hassell, J.A. A Gene Signature for Predicting Outcome in Patients with Basal-like Breast Cancer. Sci. Rep. 2012, 2, 227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guy, C.T.; Cardiff, R.D.; Muller, W.J. Induction of mammary tumors by expression of polyomavirus middle T oncogene: A transgenic mouse model for metastatic disease. Mol. Cell. Biol. 1992, 12, 954–961. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ouderkirk, J.L.; Krendel, M. Myosin 1e is a component of the invadosome core that contributes to regulation of invadosome dynamics. Exp. Cell Res. 2014, 322, 265–276. [Google Scholar] [CrossRef] [Green Version]
- Bravo-Cordero, J.J.; Hodgson, L.; Condeelis, J. Directed cell invasion and migration during metastasis. Curr. Opin. Cell Biol. 2012, 24, 277–283. [Google Scholar] [CrossRef] [Green Version]
- Weaver, A.M. Invadopodia: Specialized Cell Structures for Cancer Invasion. Clin. Exp. Metastasis 2006, 23, 97–105. [Google Scholar] [CrossRef]
- Diquigiovanni, C.; Bergamini, C.; Evangelisti, C.; Isidori, F.; Vettori, A.; Tiso, N.; Argenton, F.; Costanzini, A.; Iommarini, L.; Anbunathan, H.; et al. Mutant MYO1F alters the mitochondrial network and induces tumor proliferation in thyroid cancer. Int. J. Cancer 2018, 143, 1706–1719. [Google Scholar] [CrossRef] [Green Version]
- Taki, T.; Akiyama, M.; Saito, S.; Ono, R.; Taniwaki, M.; Kato, Y.; Yuza, Y.; Eto, Y.; Hayashi, Y. The MYO1F, unconventional myosin type 1F, gene is fused to MLL in infant acute monocytic leukemia with a complex translocation involving chromosomes 7, 11, 19 and 22. Oncogene 2005, 24, 5191–5197. [Google Scholar] [CrossRef] [Green Version]
- Duhoux, F.P.; Ameye, G.; Libouton, J.-M.; Bahloula, K.; Iossifidis, S.; Chantrain, C.F.; Demoulin, J.-B.; Poirel, H.A. The t(11;19)(q23;p13) fusing MLL with MYO1F is recurrent in infant acute myeloid leukemias. Leuk. Res. 2011, 35, e171–e172. [Google Scholar] [CrossRef]
- Abate, F.; da Silva-Almeida, A.C.; Zairis, S.; Robles-Valero, J.; Couronne, L.; Khiabanian, H.; Quinn, S.A.; Kim, M.-Y.; Laginestra, M.A.; Kim, C.; et al. Activating mutations and translocations in the guanine exchange factor VAV1 in peripheral T-cell lymphomas. Proc. Natl. Acad. Sci. USA 2017, 114, 764–769. [Google Scholar] [CrossRef] [Green Version]
- Boddicker, R.L.; Razidlo, G.L.; Dasari, S.; Zeng, Y.; Hu, G.; Knudson, R.A.; Greipp, P.T.; Davila, J.I.; Johnson, S.H.; Porcher, J.C.; et al. Integrated mate-pair and RNA sequencing identifies novel, targetable gene fusions in peripheral T-cell lymphoma. Blood 2016, 128, 1234–1245. [Google Scholar] [CrossRef] [PubMed]
- Pierchala, B.A.; Muñoz, M.R.; Tsui, C.C. Proteomic analysis of the slit diaphragm complex: CLIC5 is a protein critical for podocyte morphology and function. Kidney Int. 2010, 78, 868–882. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bi, J.; Chase, S.E.; Pellenz, C.D.; Kurihara, H.; Fanning, A.S.; Krendel, M. Myosin 1e is a component of the glomerular slit diaphragm complex that regulates actin reorganization during cell-cell contact formation in podocytes. Am. J. Physiol. Physiol. 2013, 305, F532–F544. [Google Scholar] [CrossRef] [Green Version]
- Fanning, A.S.; Van Itallie, C.M.; Anderson, J.M. Zonula occludens-1 and -2 regulate apical cell structure and the zonula adherens cytoskeleton in polarized epithelia. Mol. Biol. Cell 2012, 23, 577–590. [Google Scholar] [CrossRef] [PubMed]
- Krendel, M.; Kim, S.V.; Willinger, T.; Wang, T.; Kashgarian, M.; Flavell, R.A.; Mooseker, M.S. Disruption of Myosin 1e Promotes Podocyte Injury. J. Am. Soc. Nephrol. 2009, 20, 86–94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chase, S.E.; Encina, C.V.; Stolzenburg, L.R.; Tatum, A.H.; Holzman, L.B.; Krendel, M. Podocyte-specific knockout of myosin 1e disrupts glomerular filtration. Am. J. Physiol. Physiol. 2012, 303, F1099–F1106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mele, C.; Iatropoulos, P.; Donadelli, R.; Calabria, A.; Maranta, R.; Cassis, P.; Buelli, S.; Tomasoni, S.; Piras, R.; Krendel, M.; et al. MYO1E Mutations and Childhood Familial Focal Segmental Glomerulosclerosis. N. Engl. J. Med. 2011, 365, 295–306. [Google Scholar] [CrossRef] [Green Version]
- Nambiar, R.; McConnell, R.E.; Tyska, M.J. Myosin motor function: The ins and outs of actin-based membrane protrusions. Cell. Mol. Life Sci. 2010, 67, 1239–1254. [Google Scholar] [CrossRef] [Green Version]
- Chen, A.H.; Stephan, D.A.; Hasson, T.; Fukushima, K.; Nelissen, C.M.; Chen, A.F.; Jun, A.I.; Ramesh, A.; Van Camp, G.; Smith, R.J.H. MYO1F as a Candidate Gene for Nonsyndromic Deafness, DFNB15. Arch. Otolaryngol. Neck Surg. 2001, 127, 921. [Google Scholar] [CrossRef] [Green Version]
- Zadro, C.; Alemanno, M.S.; Bellacchio, E.; Ficarella, R.; Donaudy, F.; Melchionda, S.; Zelante, L.; Rabionet, R.; Hilgert, N.; Estivill, X.; et al. Are MYO1C and MYO1F associated with hearing loss? Biochim. Biophys. Acta Mol. Basis Dis. 2009, 1792, 27–32. [Google Scholar] [CrossRef] [Green Version]


© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Navinés-Ferrer, A.; Martín, M. Long-Tailed Unconventional Class I Myosins in Health and Disease. Int. J. Mol. Sci. 2020, 21, 2555. https://doi.org/10.3390/ijms21072555
Navinés-Ferrer A, Martín M. Long-Tailed Unconventional Class I Myosins in Health and Disease. International Journal of Molecular Sciences. 2020; 21(7):2555. https://doi.org/10.3390/ijms21072555
Chicago/Turabian StyleNavinés-Ferrer, A., and M. Martín. 2020. "Long-Tailed Unconventional Class I Myosins in Health and Disease" International Journal of Molecular Sciences 21, no. 7: 2555. https://doi.org/10.3390/ijms21072555
APA StyleNavinés-Ferrer, A., & Martín, M. (2020). Long-Tailed Unconventional Class I Myosins in Health and Disease. International Journal of Molecular Sciences, 21(7), 2555. https://doi.org/10.3390/ijms21072555