Mechanism of Secondary Ganglioside and Lipid Accumulation in Lysosomal Disease


Abstract
:1. Introduction
2. Primary Storage Compounds in Gangliosidoses and Historical Aspects
2.1. GM2 Gangliosidoses
2.1.1. Tay–Sachs Disease (B Variant) and B1 Variant
2.1.2. Sandhoff Disease (SD) (0 Variant)
2.1.3. GM2 Activator Protein Deficiency (AB Variant)
2.2. GM1 Gangliosidosis
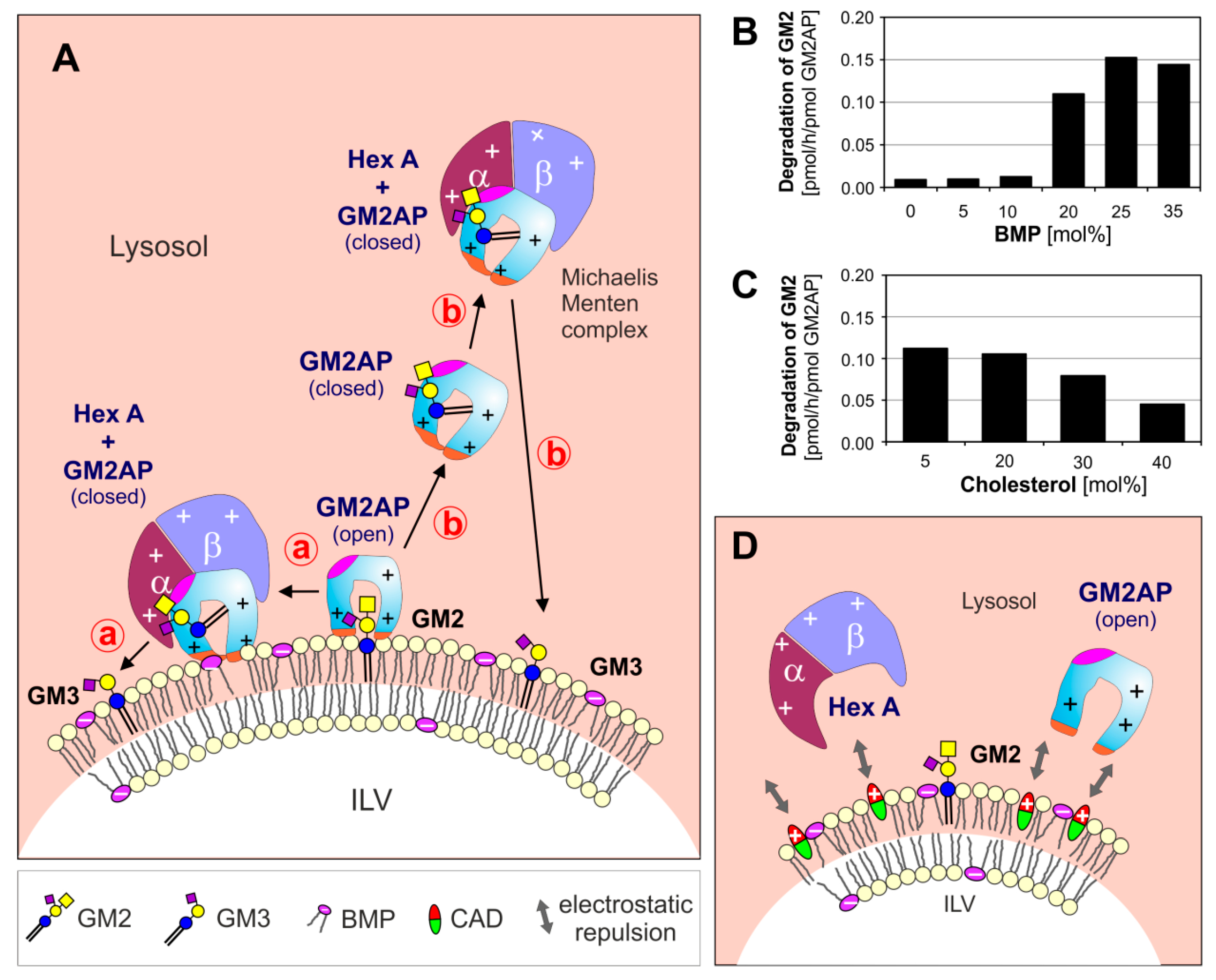
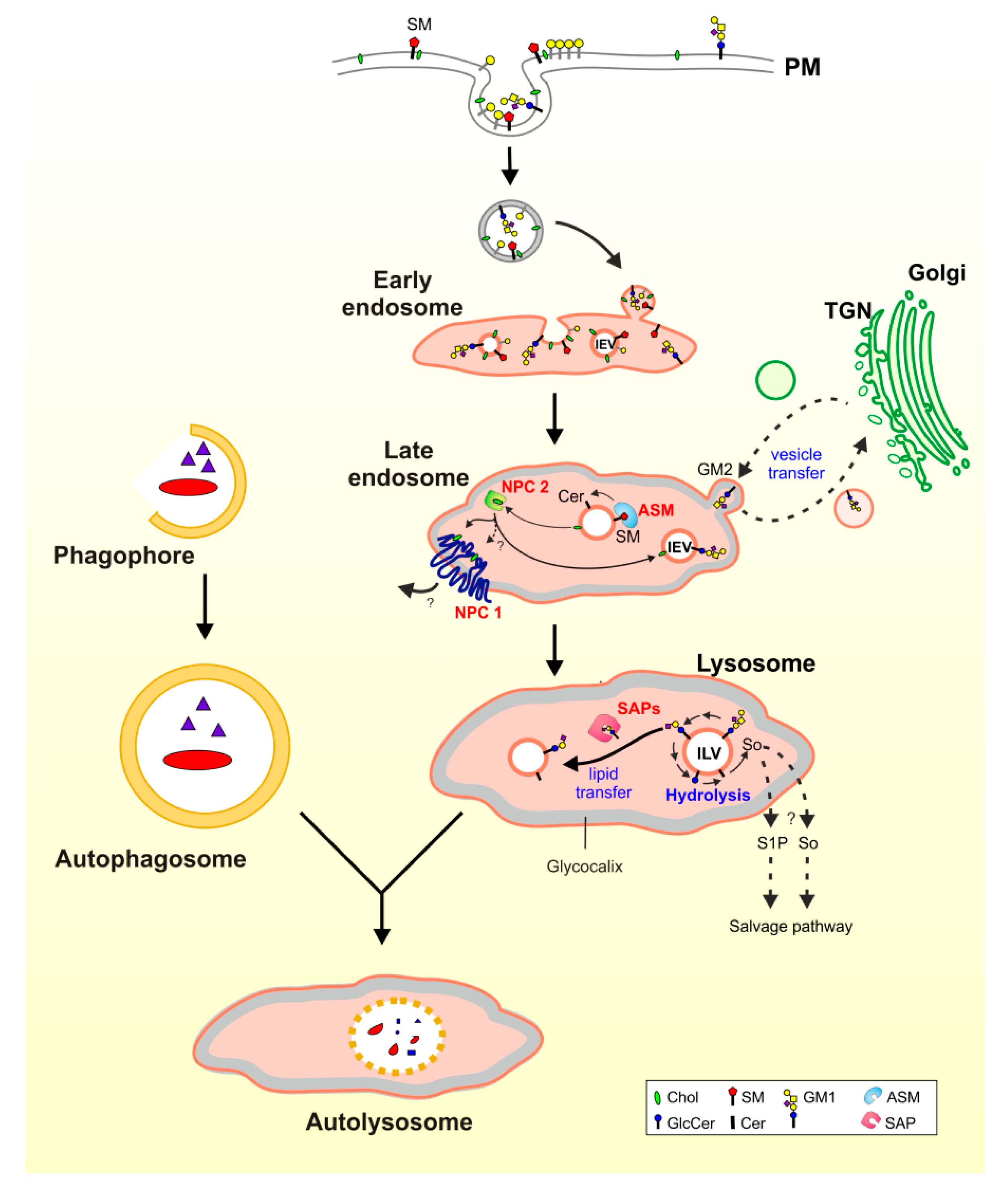
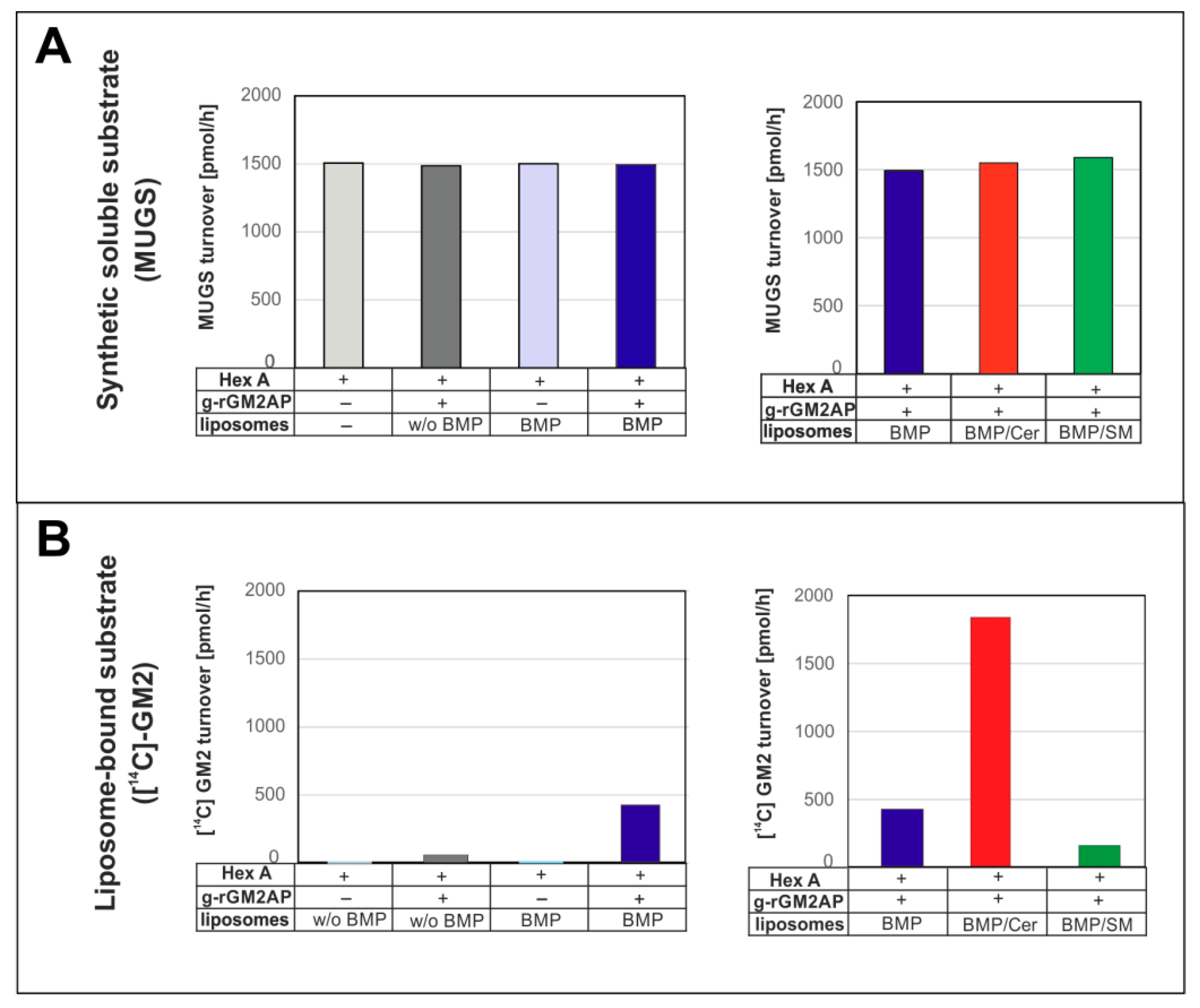
3. Lysosomal Catabolism of GGs
Maturation of ILVs and Regulation of GG Catabolism at ILV Surfaces
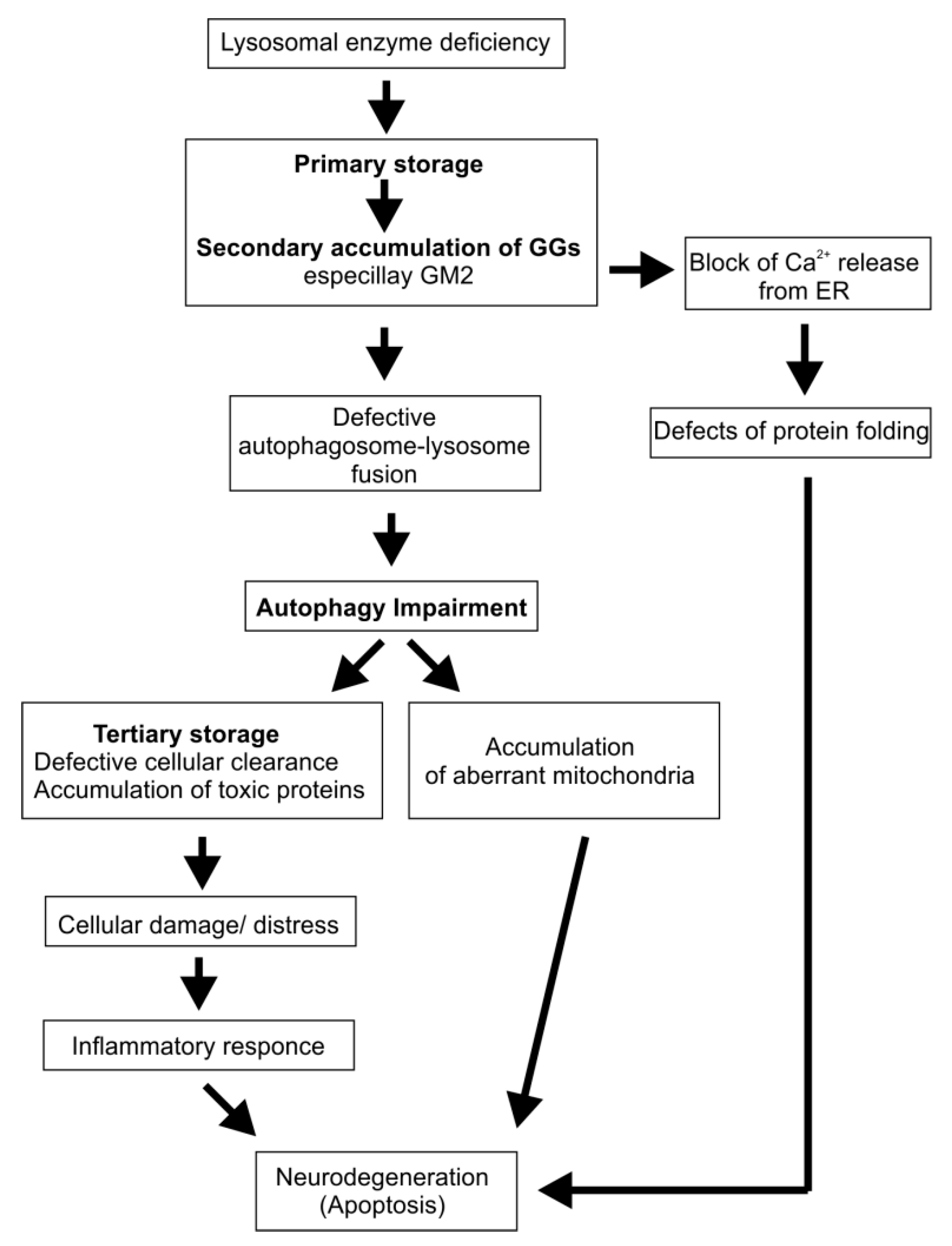
4. Cascading Errors in LSDs
5. Lysosomal Storage Disorders with Secondary Ganglioside Accumulation
5.1. Sphingolipidoses
5.1.1. Niemann–Pick Disease Type A and B
5.1.2. Niemann–Pick Disease Type C
5.1.3. Gaucher Disease
5.1.4. Krabbe Disease
5.1.5. Metachromatic Leukodystrophy (MLD)
5.1.6. Farber Disease
5.1.7. Prosaposin Deficiency
5.2. Mucopolysaccharidoses (MPSs)
5.2.1. MPS I (Hurler Syndrome)
5.2.2. MPS II (Hunter Syndrome)
5.2.3. MPS III (Sanfilippo Syndrome)
5.2.4. MPS VI (Maroteaux–Lamy Syndrome)
5.2.5. MPS VII (Sly Syndrome)
5.3. Mucolipidoses (MLs)
5.3.1. Mucolipidosis ΙΙ (I-cell Disease) and Mucolipidosis ΙIΙ (Pseudo-Hurler Polydystrophy)
5.3.2. Mucolipidosis ΙV (Mucolipidin 1 Deficiency)
5.4. Glycoproteinoses
5.4.1. Galactosialidosis
5.4.2. α-Mannosidosis
5.4.3. Sialidosis
5.5. Neuronal Ceroid Lipofuscinoses (NCLs)
5.5.1. NCL 3 (Batten Disease)
5.5.2. NCL 6
5.5.3. NCL 10 (Congenital Cathepsin D deficiency)
5.6. Hereditary Spastic Paraplegia (HSP)
Hereditary Spastic Paraplegia Caused by Mutations in the AP 5/SPG11/SPG15 Complex
5.7. TgCRND8—An Alzheimer’s Disease Mouse Model
6. Drug-Induced GG Accumulation
7. Conclusions and Perspectives
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ASM | Acid sphingomyelinase |
| BMP | Bis(monoacylglycero)phosphate |
| CAD | Cationic amphiphilic drug |
| GBA1 | Glucosylceramide-β-glucosidase |
| GG | Ganglioside |
| GSL | Glycosphingolipid |
| GM2AP | GM2 activator protein |
| Hex A | β-Hexosaminidase A |
| Hex B | β-Hexosaminidase B |
| HSP | Hereditary spastic paraplegia |
| IVL | Intralysosomal luminal vesicle |
| LSD | Lysosomal storage disorders |
| MLD | Metachromatic leukodystrophy |
| MPS | Mucopolysaccharidoses |
| NCL | Neuronal ceroid lipofuscinoses |
| NEU | Neuraminidase |
| NPC1 | Niemann–Pick disease protein C type 1 |
| NPC2 | Niemann–Pick disease protein C type 2 |
| SAP | Sphingolipid activator protein |
| Sap | Saposin |
| SD | Sandhoff disease |
| TSD | Tay–Sachs disease |
References
- Lehovský, M. Clinical pictures in patients with thesaurismosis of gangliosides. Rev. Czech. Med. 1972, 18, 145–148. [Google Scholar] [PubMed]
- Van Bogaert, L.; Klenk, E. Clinical, histopathological and chemical aspects of phosphatide thesaurismosis. G Psichiatr. Neuropatol. 1953, 81, 879–880. [Google Scholar] [PubMed]
- Schuchman, E.H.; Desnick, R.J. Niemann–Pick disease types A and B: Sphingomyelinase deficiencies. In The Metabolic and Molecular Bases of Inherited Disease, 8th ed.; Scriver, C.R., Beaudet, A.L., Sly, W.S., Valle, D., Eds.; McGraw-Hill: New York, NY, USA, 2001; pp. 3589–3610. [Google Scholar]
- Patterson, M.C.; Vanier, M.T.; Suzuki, K.; Morris, J.A.; Eugene, C.; Neufeld, E.B.; Blanchette-Mackie Joan, E.; Pentchev, P.G. Niemann–Pick disease type C: A lipid trafficking disorders. In The Metabolic and Molecular Bases of Inherited Disease, 8th ed.; Scriver, C.R., Beaudet, A.L., Sly, W.S., Valle, D., Eds.; McGraw-Hill: New York, NY, USA, 2001; pp. 3611–3633. [Google Scholar]
- Neufeld, E.; Muenzer, J. The mucopolysaccharidoses. In The Metabolic and Molecular Bases of Inherited Disease, 8th ed.; Scriver, C.R., Beaudet, A.L., Sly, W.S., Valle, D., Eds.; McGraw-Hill: New York, NY, USA, 2001; pp. 3421–3452. [Google Scholar]
- Anheuser, S.; Breiden, B.; Sandhoff, K. Membrane lipids and their degradation compounds control GM2 catabolism at intralysosomal luminal vesicles. J. Lipid Res. 2019, 60, 1099–1111. [Google Scholar] [CrossRef]
- Anheuser, S.; Breiden, B.; Sandhoff, K. Ganglioside GM2 catabolism is inhibited by storage compounds of mucopolysaccharidoses and by cationic amphiphilic drugs. Mol. Genet. Metab 2019, 128, 75–83. [Google Scholar] [CrossRef] [PubMed]
- Breiden, B.; Sandhoff, K. Lysosomal glycosphingolipid storage diseases. Annu. Rev. Biochem. 2019, 88, 461–485. [Google Scholar] [CrossRef] [PubMed]
- Sandhoff, R.; Sandhoff, K. Emerging concepts of ganglioside metabolism. FEBS Lett. 2018, 592, 3835–3864. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oninla, V.O.; Breiden, B.; Babalola, J.O.; Sandhoff, K. Acid sphingomyelinase activity is regulated by membrane lipids and facilitates cholesterol transfer by NPC2. J. Lipid Res. 2014, 55, 2606–2619. [Google Scholar] [CrossRef] [Green Version]
- Sachs, B. On arrested cerebral development with speciel reference to its cortical pathology. J. Neur. Ment. Dis. 1887, 14, 541–553. [Google Scholar] [CrossRef]
- Sachs, B. A family form of idiocy, generally fatal, associated with early blindness (amaurotic family idiocy). J. Nerv. Ment. Dis. 1896, 21, 475–479. [Google Scholar]
- Klenk, E. Niemann–Pick’sche Krankheit und Amaurotische Idiotie. Hoppe-Seyler’s Z Physiol. Chem. 1939, 262, 128–143. [Google Scholar] [CrossRef]
- Sandhoff, K.; Harzer, K. Gangliosides and gangliosidoses: Principles of molecular and metabolic pathogenesis. J. Neurosci. 2013, 33, 10195–10208. [Google Scholar] [CrossRef] [PubMed]
- Tay, W. Symmetrical changes in the region of the yellow spot in each eye of an infant. Trans. Ophthalmol. Soc. 1881, 1, 55–57. [Google Scholar] [CrossRef]
- Jatzkewitz, H.; Sandhoff, K. On a biochemically special form of infantile amaturotic idiocy. Biochim. Biophys. Acta. 1963, 70, 354–356. [Google Scholar] [CrossRef]
- Sandhoff, K. Die Amaurotische Idiotie des Menschen als Störung im Glykosphingolipidstoffwechsel. Doctoral Thesis, University of Munich, Munich, Germany, 1965. [Google Scholar]
- Kuhn, R.; Wiegandt, H. Die Konstitution der Ganglio-N-tetraose und des Gangliosids GI. Chem. Ber. 1963, 96, 866–880. [Google Scholar] [CrossRef]
- Sandhoff, K. The GM2-gangliosidoses and the elucidation of the beta-hexosaminidase system. Adv. Genet. 2001, 44, 67–91. [Google Scholar]
- Sandhoff, K. Variation of beta-N-acetylhexosaminidase-pattern in Tay–Sachs disease. FEBS Lett. 1969, 4, 351–354. [Google Scholar] [CrossRef] [Green Version]
- Sandhoff, K.; Harzer, K.; Wässle, W.; Jatzkewitz, H. Enzyme alterations and lipid storage in three variants of Tay–Sachs disease. J. Neurochem. 1971, 18, 2469–2489. [Google Scholar] [CrossRef]
- Conzelmann, E.; Sandhoff, K. AB variant of infantile GM2 gangliosidosis: Deficiency of a factor necessary for stimulation of hexosaminidase A-catalyzed degradation of ganglioside GM2 and glycolipid GA2. Proc. Natl. Acad. Sci. USA 1978, 75, 3979–3983. [Google Scholar] [CrossRef] [Green Version]
- Okada, S.; O’Brien, J.S. Tay–Sachs disease: Generalized absence of a beta-D-N-acetylhexosaminidase component. Science 1969, 165, 698–700. [Google Scholar] [CrossRef]
- Sandhoff, K. The hydrolysis of Tay–Sachs ganglioside (TSG) by human N-acetyl-beta-D-hexosaminidase A. FEBS Lett. 1970, 11, 342–344. [Google Scholar] [CrossRef] [Green Version]
- Sandhoff, R.; Schulze, H.; Sandhoff, K. Ganglioside metabolism in health and disease. Prog. Mol. Biol. Transl. Sci. 2018, 156, 1–62. [Google Scholar] [CrossRef] [PubMed]
- Sandhoff, K.; Jatzkewitz, H.; Peters, G. Die infantile amaurotische Idiotie und verwandte Formen als Gangliosid-Speicherkrankheiten. Naturwissenschaften 1969, 56, 356–362. [Google Scholar] [CrossRef] [PubMed]
- Hepbildikler, S.T.; Sandhoff, R.; Kölzer, M.; Proia, R.L.; Sandhoff, K. Physiological substrates for human lysosomal beta -hexosaminidase S. J. Biol. Chem. 2002, 277, 2562–2572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gravel, R.; Kaback, M.M.; Proia, R.L.; Sandhoff, K.; Suzuki, K.; Suzuki, K. The GM2 gangliosidoses. In The Metabolic and Molecular Bases of Inherited Disease, 8th ed.; Scriver, C.R., Beaudet, A.L., Sly, W.S., Valle, D., Eds.; McGraw-Hill: New York, NY, USA, 2001; pp. 3827–3876. [Google Scholar]
- Sandhoff, K.; Andreae, U.; Jatzkewitz, H. Deficient hexosaminidase activity in an exceptional case of Tay–Sachs disease with additional storage of kidney globoside in visceral organs. Pathol Eur. 1968, 3, 278–285. [Google Scholar] [CrossRef]
- Pilz, H.; Müller, D.; Sandhoff, K.; ter Meulen, V. Tay–Sachssche Krankheit mit Hexosaminidase-Defekt. Klinische, morphologische und biochemische Befunde bei einem Fall mit viszeraler Speicherung von Nierenglobosid. Dtsch Med. Wochenschr 1968, 93, 1833–1839. [Google Scholar] [CrossRef]
- Suzuki, K.; Chen, G.C. GM1-gangliosidosis (generalized gangliosidosis). Morphology and chemical pathology. Pathol Eur. 1968, 3, 389–408. [Google Scholar]
- Siegel, D.A.; Walkley, S.U. Growth of ectopic dendrites on cortical pyramidal neurons in neuronal storage diseases correlates with abnormal accumulation of GM2 ganglioside. J. Neurochem. 1994, 62, 1852–1862. [Google Scholar] [CrossRef]
- Kytzia, H.J.; Hinrichs, U.; Maire, I.; Suzuki, K.; Sandhoff, K. Variant of GM2-gangliosidosis with hexosaminidase A having a severely changed substrate specificity. Embo J. 1983, 2, 1201–1205. [Google Scholar] [CrossRef]
- Kytzia, H.J.; Sandhoff, K. Evidence for two different active sites on human beta-hexosaminidase A. Interaction of GM2 activator protein with beta-hexosaminidase A. J. Biol. Chem. 1985, 260, 7568–7572. [Google Scholar]
- Sango, K.; Yamanaka, S.; Hoffmann, A.; Okuda, Y.; Grinberg, A.; Westphal, H.; McDonald, M.P.; Crawley, J.N.; Sandhoff, K.; Suzuki, K.; et al. Mouse models of Tay–Sachs and Sandhoff diseases differ in neurologic phenotype and ganglioside metabolism. Nat. Genet. 1995, 11, 170–176. [Google Scholar] [CrossRef]
- Jeyakumar, M.; Smith, D.; Eliott-Smith, E.; Cortina-Borja, M.; Reinkensmeier, G.; Butters, T.D.; Lemm, T.; Sandhoff, K.; Perry, V.H.; Dwek, R.A.; et al. An inducible mouse model of late onset Tay–Sachs disease. Neurobiol. Dis. 2002, 10, 201–210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sango, K.; McDonald, M.P.; Crawley, J.N.; Mack, M.L.; Tifft, C.J.; Skop, E.; Starr, C.M.; Hoffmann, A.; Sandhoff, K.; Suzuki, K.; et al. Mice lacking both subunits of lysosomal [beta]-hexosaminidase display gangliosidosis and mucopolysaccharidosis. Nat. Genet. 1996, 14, 348–352. [Google Scholar]
- Anheuser, S.; Breiden, B.; Schwarzmann, G.; Sandhoff, K. Membrane lipids regulate ganglioside GM2 catabolism and GM2 activator protein activity. J. Lipid Res. 2015, 56, 1747–1761. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Brien, J.S.; Stern, M.B.; Landing, B.H.; O’Brien, J.K.; Donnell, G.N. Generalized gangliosidosis: Another inborn error of ganglioside metabolism? Am. J. Dis. Child. 1965, 109, 338–346. [Google Scholar] [CrossRef]
- Lawrence, R.; Van Vleet, J.L.; Mangini, L.; Harris, A.; Martin, N.; Clark, W.; Chandriani, S.; LeBowitz, J.H.; Giugliani, R.; d’Azzo, A.; et al. Characterization of glycan substrates accumulating in GM1 gangliosidosis. Mol. Genet. Metab. Rep. 2019, 21, 100524. [Google Scholar] [CrossRef]
- Wilkening, G.; Linke, T.; Uhlhorn-Dierks, G.; Sandhoff, K. Degradation of membrane-bound ganglioside GM1. Stimulation by bis(monoacylglycero)phosphate and the activator proteins SAP-B and GM2-AP. J. Biol. Chem. 2000, 275, 35814–35819. [Google Scholar] [CrossRef] [Green Version]
- Miyagi, T.; Takahashi, K.; Yamamoto, K.; Shiozaki, K.; Yamaguchi, K. Biological and pathological roles of ganglioside sialidases. Prog. Mol. Biol. Transl. Sci. 2018, 156, 121–150. [Google Scholar]
- Timur, Z.K.; Akyildiz Demir, S.; Marsching, C.; Sandhoff, R.; Seyrantepe, V. Neuraminidase-1 contributes significantly to the degradation of neuronal B-series gangliosides but not to the bypass of the catabolic block in Tay–Sachs mouse models. Mol. Genet. Metab. Rep. 2015, 4, 72–82. [Google Scholar] [CrossRef]
- Smutova, V.; Albohy, A.; Pan, X.; Korchagina, E.; Miyagi, T.; Bovin, N.; Cairo, C.W.; Pshezhetsky, A.V. Structural basis for substrate specificity of mammalian neuraminidases. PLoS ONE 2014, 9, e106320. [Google Scholar] [CrossRef] [Green Version]
- Monti, E.; Bonten, E.; D’Azzo, A.; Bresciani, R.; Venerando, B.; Borsani, G.; Schauer, R.; Tettamanti, G. Sialidases in vertebrates: A family of enzymes tailored for several cell functions. Adv. Carbohydr. Chem. Biochem. 2010, 64, 403–479. [Google Scholar]
- D’Azzo, A.; Machado, E.; Annunziata, I. Pathogenesis, emerging therapeutic targets and treatment in sialidosis. Expert Opin. Orphan. Drugs. 2015, 3, 491–504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonten, E.; van der Spoel, A.; Fornerod, M.; Grosveld, G.; d’Azzo, A. Characterization of human lysosomal neuraminidase defines the molecular basis of the metabolic storage disorder sialidosis. Genes Dev. 1996, 10, 3156–3169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Azzo, A.; Bonten, E. Molecular mechanisms of pathogenesis in a glycosphingolipid and a glycoprotein storage disease. Biochem Soc. Trans. 2010, 38, 1453–1457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fingerhut, R.; van der Horst, G.T.J.; Verheijen, F.W.; Conzelmann, E. Degradation of gangliosides by the lysosomal sialidase requires an activator protein. Eur J. Biochem. 1992, 208, 623–629. [Google Scholar] [CrossRef] [PubMed]
- Abdul-Hammed, M.; Breiden, B.; Schwarzmann, G.; Sandhoff, K. Lipids regulate the hydrolysis of membrane bound glucosylceramide by lysosomal β-glucocerebrosidase. J. Lipid Res. 2017, 58, 563–577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akiyama, H.; Kobayashi, S.; Hirabayashi, Y.; Murakami-Murofushi, K. Cholesterol glucosylation is catalyzed by transglucosylation reaction of ß-glucosidase 1. Biochem. Biophys. Res. Com. 2013, 441, 838–843. [Google Scholar] [CrossRef] [Green Version]
- Eskelinen, E.-L.; Tanaka, Y.; Saftig, P. At the acidic edge: Emerging functions for lysosomal membrane proteins. Trends Cell. Biol. 2003, 13, 137–145. [Google Scholar] [CrossRef]
- Kobayashi, T.; Beuchat, M.H.; Lindsay, M.; Frias, S.; Palmiter, R.D.; Sakuraba, H.; Parton, R.G.; Gruenberg, J. Late endosomal membranes rich in lysobisphosphatidic acid regulate cholesterol transport. Nat. Cell Biol. 1999, 1, 113–118. [Google Scholar] [CrossRef]
- Möbius, W.; van Donselaar, E.; Ohno-Iwashita, Y.; Shimada, Y.; Heijnen, H.F.; Slot, J.W.; Geuze, H.J. Recycling compartments and the internal vesicles of multivesicular bodies harbor most of the cholesterol found in the endocytic pathway. Traffic (Copenhagen, Denmark) 2003, 4, 222–231. [Google Scholar] [CrossRef]
- Graf, C.G.F.; Schulz, C.; Schmälzlein, M.; Heinlein, C.; Mönnich, M.; Perkams, L.; Püttner, M.; Boos, I.; Hessefort, M.; Lombana Sanchez, J.N.; et al. Synthetic glycoforms reveal carbohydrate-dependent bioactivity of human saposin D. Angew Chem. Int. Ed. Engl. 2017, 56, 5252–5257. [Google Scholar] [CrossRef]
- Kölzer, M.; Werth, N.; Sandhoff, K. Interactions of acid sphingomyelinase and lipid bilayers in the presence of the tricyclic antidepressant desipramine. FEBS Lett. 2004, 559, 96–98. [Google Scholar] [CrossRef] [Green Version]
- Yu, C.; Sun, Q.; Zhou, H. Enzymatic screening and diagnosis of lysosomal storage diseases. N. Am. J. Med. Sci.. 2013, 6, 186–193. [Google Scholar] [CrossRef] [PubMed]
- Galjaard, H. Genetic metabolic diseases: Early diagnosis and prenatal analysis; Elsevier-Noth Holland Biomedical Press: Amsterdam, The Netherlands, 1980. [Google Scholar]
- Kolter, T.; Proia, R.L.; Sandhoff, K. Combinatorial ganglioside biosynthesis. J. Biol. Chem. 2002, 277, 25859–25862. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Meer, G.; Voelker, D.R.; Feigenson, G.W. Membrane lipids: Where they are and how they behave. Nat. Rev. Mol. Cell Biol. 2008, 9, 112–124. [Google Scholar] [CrossRef]
- Abdul-Hammed, M.; Breiden, B.; Adebayo, M.A.; Babalola, J.O.; Schwarzmann, G.; Sandhoff, K. Role of endosomal membrane lipids and NPC2 in cholesterol transfer and membrane fusion. J. Lipid Res. 2010, 51, 1747–1760. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.L.; Motamed, M.; Infante, R.E.; Abi-Mosleh, L.; Kwon, H.J.; Brown, M.S.; Goldstein, J.L. Identification of surface residues on Niemann–Pick C2 essential for hydrophobic handoff of cholesterol to NPC1 in lysosomes. Cell Metab. 2010, 12, 166–173. [Google Scholar] [CrossRef] [Green Version]
- Vanier, M.T. Biochemical studies in Niemann–Pick disease I. Major sphingolipids of liver and spleen. Biochim. Biophys Acta. 1983, 750, 178–184. [Google Scholar] [CrossRef]
- Jatzkewitz, H.; Pilz, H.; Sandhoff, K. Quantitative Bestimmungen von Gangliosiden und ihren Neuraminsäurefreien Derivaten bei infantilen, juvenilen und adulten Formen der amaurotischen Idiotie und einer spätinfantilen biochemischen Sonderform. J. Neurochem. 1965, 12, 135–144. [Google Scholar] [CrossRef]
- Remmel, N.; Locatelli-Hoops, S.; Breiden, B.; Schwarzmann, G.; Sandhoff, K. Saposin B mobilizes lipids from cholesterol-poor and bis(monoacylglycero)phosphate-rich membranes at acidic pH. Unglycosylated patient variant saposin B lacks lipid-extraction capacity. FEBS J. 2007, 274, 3405–3420. [Google Scholar] [CrossRef]
- Locatelli-Hoops, S.; Remmel, N.; Klingenstein, R.; Breiden, B.; Rossocha, M.; Schoeniger, M.; Koenigs, C.; Saenger, W.; Sandhoff, K. Saposin A mobilizes lipids from low cholesterol and high bis(monoacylglycerol)phosphate-containing membranes: Patient variant Saposin A lacks lipid extraction capacity. J. Biol. Chem. 2006, 281, 32451–32460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walkley, S.U. Pathogenic cascades in lysosomal disease-Why so complex? J. Inherit. Metab. Dis. 2009, 32, 181–189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Breiden, B.; Sandhoff, K. Emerging mechanisms of drug-induced phospholipidosis. Biol. Chem. 2020, 401, 31–46. [Google Scholar] [CrossRef] [PubMed]
- Sarmientos, F.; Schwarzmann, G.; Sandhoff, K. Specificity of human glucosylceramide beta-glucosidase towards synthetic glucosylsphingolipids inserted into liposomes. Kinetic studies in a detergent-free assay system. Eur J. Biochem. 1986, 160, 527–535. [Google Scholar] [CrossRef] [PubMed]
- Walkley, S.U.; Vanier, M.T. Secondary lipid accumulation in lysosomal disease. Biochim. Biophys. Acta. 2009, 1793, 726–736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tessitore, A.; del, P.M.M.; Sano, R.; Ma, Y.; Mann, L.; Ingrassia, A.; Laywell, E.D.; Steindler, D.A.; Hendershot, L.M.; d’Azzo, A. GM1-ganglioside-mediated activation of the unfolded protein response causes neuronal death in a neurodegenerative gangliosidosis. Mol. Cell. 2004, 15, 753–766. [Google Scholar] [CrossRef]
- Jmoudiak, M.; Futerman, A.H. Gaucher disease: Pathological mechanisms and modern management. Br. J. Haematol. 2005, 129, 178–188. [Google Scholar] [CrossRef]
- Schulze, H.; Sandhoff, K. Lysosomal lipid storage diseases. Cold Spring Harb Perspect Biol 2011, 3, 287–305. [Google Scholar] [CrossRef]
- Toulmay, A.; Prinz, W.A. Lipid transfer and signaling at organelle contact sites: The tip of the iceberg. Curr. Opin. Cell Biol. 2011, 23, 458–463. [Google Scholar] [CrossRef] [Green Version]
- Hanada, K. Organelle contacts: Sub-organelle zones to facilitate rapid and accurate inter-organelle trafficking of lipids. Traffic. 2020, 21, 189–196. [Google Scholar] [CrossRef]
- Liu, E.A.; Lieberman, A.P. The intersection of lysosomal and endoplasmic reticulum calcium with autophagy defects in lysosomal diseases. Neurosci. Lett. 2019, 697, 10–16. [Google Scholar] [CrossRef]
- Ballabio, A.; Gieselmann, V. Lysosomal disorders: From storage to cellular damage. Biochim. Biophys. Acta. 2009, 1793, 684–696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pelled, D.; Riebeling, C.; van Echten-Deckert, G.; Sandhoff, K.; Futerman, A.H. Reduced rates of axonal and dendritic growth in embryonic hippocampal neurones cultured from a mouse model of Sandhoff disease. Neuropathol. Appl. Neurobiol. 2003, 29, 341–349. [Google Scholar] [CrossRef] [PubMed]
- Virgolini, M.J.; Feliziani, C.; Cambiasso, M.J.; Lopez, P.H.; Bollo, M. Neurite atrophy and apoptosis mediated by PERK signaling after accumulation of GM2-ganglioside. Biochim. Biophys. Acta. 2019, 1866, 225–239. [Google Scholar] [CrossRef] [PubMed]
- Takamura, A.; Higaki, K.; Kajimaki, K.; Otsuka, S.; Ninomiya, H.; Matsuda, J.; Ohno, K.; Suzuki, Y.; Nanba, E. Enhanced autophagy and mitochondrial aberrations in murine G(M1)-gangliosidosis. Biochem. Biophys. Res. Commun. 2008, 367, 616–622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niemann, A. Un unbekanntes Krankheitsbild. Jahrb. Kinderheilkd. 1914, 79, 1–3. [Google Scholar]
- Klenk, E. Über die Natur der Phosphatide der Milz bei der Niemann–Pickschen Krankheit. Hoppe Seylers Z Physiol. Chem. 1934, 229, 151. [Google Scholar] [CrossRef]
- Brady, R.O.; Kanfer, J.; Mock, M.; Fredrickson, D. The metabolism of sphingomyelin. Evidence of an enzymatic deficiency in Niemann–Pick disease. Proc. Natl. Acad. Sci. USA 1966, 55, 367–370. [Google Scholar] [CrossRef] [Green Version]
- Quintern, L.E.; Weitz, G.; Nehrkorn, H.; Tager, J.M.; Schram, A.W.; Sandhoff, K. Acid sphingomyelinase from human urine: Purification and characterization. Biochim. Biophys. Acta. 1987, 922, 323–336. [Google Scholar] [CrossRef]
- Ferlinz, K.; Hurwitz, R.; Moczall, H.; Lansmann, S.; Schuchman, E.H.; Sandhoff, K. Functional characterization of the N-glycosylation sites of human acid sphingomyelinase by site-directed mutagenesis. Eur. J. Biochem. 1997, 243, 511–517x. [Google Scholar] [CrossRef]
- Hurwitz, R.; Ferlinz, K.; Vielhaber, G.; Moczall, H.; Sandhoff, K. Processing of human acid sphingomyelinase in normal and I-cell fibroblasts. J. Biol. Chem. 1994, 269, 5440–5445. [Google Scholar]
- Quintern, L.E.; Schuchman, E.H.; Levran, O.; Suchi, M.; Ferlinz, K.; Reinke, H.; Sandhoff, K.; Desnick, R.J. Isolation of cDNA clones encoding human acid sphingomyelinase: Occurrence of alternatively processed transcripts. Embo J. 1989, 8, 2469–2473. [Google Scholar] [CrossRef] [PubMed]
- Ferlinz, K.; Hurwitz, R.; Sandhoff, K. Molecular basis of acid sphingomyelinase deficiency in a patient with Niemann–Pick disease type A. Biochem. Biophys. Res. Commun. 1991, 179, 1187–1191. [Google Scholar] [CrossRef]
- Levran, O.; Desnick, R.; Schuchman, E. Niemann–Pick disease: A frequent missense mutation in the acid sphingomyelinase gene of Ashkenazi Jewish type A and B patiens. Proc. Natl. Acad. Sci. USA 1991, 88, 3748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greenbaum, M.; Hoffman, L.M.; Schneck, L. Ceramide hexosides in Niemann–Pick disease brain. J. Neurol. 1976, 213, 251–255. [Google Scholar] [CrossRef] [PubMed]
- Vellodi, A.; Hobbs, J.R.; O’Donnell, N.M.; Coulter, B.S.; Hugh-Jones, K. Treatment of Niemann–Pick disease type B by allogeneic bone marrow transplantation. Br. Med. J. 1987, 295, 1375–1376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schuchman, E.H.; Desnick, R.J. Types A and B Niemann–Pick disease. Mol. Genet. Metab. 2017, 120, 27–33. [Google Scholar] [CrossRef] [Green Version]
- Samaranch, L.; Perez-Canamas, A.; Soto-Huelin, B.; Sudhakar, V.; Jurado-Arjona, J.; Hadaczek, P.; Avila, J.; Bringas, J.R.; Casas, J.; Chen, H.; et al. Adeno-associated viral vector serotype 9-based gene therapy for Niemann–Pick disease type A. Sci. Transl. Med. 2019, 11. [Google Scholar] [CrossRef]
- Vanier, M.T. Complex lipid trafficking in Niemann–Pick disease type C. J. Inherit. Metab. Dis. 2015, 38, 187–199. [Google Scholar] [CrossRef]
- Sleat, D.; Wiseman, J.; El Banna, M.; Price, S.; Verot, L.; Shen, M.; Tint, G.; Vanier, M.; Walkley, S.; Lobel, P. Genetic evidence for nonredundant functional cooperativity between NPC1 and NPC2 in lipid transport. Proc. Natl Acad Sci USA 2004, 101, 5886–5891. [Google Scholar] [CrossRef] [Green Version]
- Carstea, E.D.; Morris, J.A.; Coleman, K.G.; Loftus, S.K.; Zhang, D.; Cummings, C.; Gu, J.; Rosenfeld, M.A.; Pavan, W.J.; Krizman, D.B.; et al. Niemann–Pick C1 disease gene: Homology to mediators of cholesterol homeostasis. Science 1997, 277, 228–231. [Google Scholar] [CrossRef] [Green Version]
- Naureckiene, S.; Sleat, D.E.; Lackland, H.; Fensom, A.; Vanier, M.T.; Wattiaux, R.; Jadot, M.; Lobel, P. Identification of HE1 as the second gene of Niemann–Pick C disease. Science 2000, 290, 2298–2301. [Google Scholar] [CrossRef] [PubMed]
- Lloyd-Evans, E.; Morgan, A.J.; He, X.; Smith, D.A.; Elliot-Smith, E.; Sillence, D.J.; Churchill, G.C.; Schuchman, E.H.; Galione, A.; Platt, F.M. Niemann–Pick disease type C1 is a sphingosine storage disease that causes deregulation of lysosomal calcium. Nat. Med. 2008, 14, 1247–1255. [Google Scholar] [CrossRef] [PubMed]
- Zschoche, A.; Fürst, W.; Schwarzmann, G.; Sandhoff, K. Hydrolysis of lactosylceramide by human galactosylceramidase and GM1-beta-galactosidase in a detergent-free system and its stimulation by sphingolipid activator proteins, sap-B and sap-C. Activator proteins stimulate lactosylceramide hydrolysis. Eur. J. Biochem. 1994, 222, 83–90. [Google Scholar] [CrossRef] [PubMed]
- Hurwitz, R.; Ferlinz, K.; Sandhoff, K. The tricyclic antidepressant desipramine causes proteolytic degradation of lysosomal sphingomyelinase in human fibroblasts. Biol. Chem. Hoppe Seyler 1994, 375, 447–450. [Google Scholar] [CrossRef]
- Bhuvaneswaran, C.; Venkatesan, S.; Mitropoulos, K.A. Lysosomal accumulation of cholesterol and sphingomyelin: Evidence for inhibition of acid sphingomyelinase. Eur. J. Cell Biol. 1985, 37, 98–106. [Google Scholar]
- Shen, D.; Wang, X.; Li, X.; Zhang, X.; Yao, Z.; Dibble, S.; Dong, X.P.; Yu, T.; Lieberman, A.P.; Showalter, H.D.; et al. Lipid storage disorders block lysosomal trafficking by inhibiting a TRP channel and lysosomal calcium release. Nat. Commun. 2012, 3, 731. [Google Scholar] [CrossRef] [Green Version]
- Sitarska, D.; Ługowska, A. Laboratory diagnosis of the Niemann–Pick type C disease: An inherited neurodegenerative disorder of cholesterol metabolism. Metab. Brain Dis. 2019, 34, 1253–1260. [Google Scholar] [CrossRef] [Green Version]
- Deodato, F.; Boenzi, S.; Taurisano, R.; Semeraro, M.; Sacchetti, E.; Carrozzo, R.; Dionisi-Vici, C. The impact of biomarkers analysis in the diagnosis of Niemann–Pick C disease and acid sphingomyelinase deficiency. Clinica. Chimica. Acta. 2018, 486, 387–394. [Google Scholar] [CrossRef]
- Hammerschmidt, T.G.; de Oliveira Schmitt Ribas, G.; Saraiva-Pereira, M.L.; Bonatto, M.P.; Kessler, R.G.; Souza, F.T.S.; Trapp, F.; Michelin-Tirelli, K.; Burin, M.G.; Giugliani, R.; et al. Molecular and biochemical biomarkers for diagnosis and therapy monitorization of Niemann–Pick type C patients. Int. J. Dev. Neurosci. Off. J. Int. Soc. Dev. Neurosci. 2018, 66, 18–23. [Google Scholar] [CrossRef]
- Nilsson, O.; Svennerholm, L. Accumulation of glucosylceramide and glucosylsphingosine (Psychosine) in gerebrum and gerebellum in infantile and juvenile Gaucher disease. J. Neurochem. 1982, 39, 709–718. [Google Scholar] [CrossRef]
- Brady, R.O.; Kanfer, J.N.; Shapiro, D. Metabolism of glucocerebrosides. Evidence of an enzymatic deficiency in Gaucher’s disease. Biochem. Biophys. Res. Com. 1965, 18, 221–225. [Google Scholar] [CrossRef]
- Barton, N.W.; Brady, R.O.; Dambrosia, J.M.; Di Bisceglie, A.M.; Doppelt, S.H.; Hill, S.C.; Mankin, H.J.; Murray, G.J.; Parker, R.I.; Argoff, C.E.; et al. Replacement therapy for inherited enzyme deficiency — Macrophage-targeted glucocerebrosidase for Gaucher’s disease. N Engl. J. Med. 1991, 324, 1464–1470. [Google Scholar] [CrossRef] [PubMed]
- Doering, T.; Proia, R.L.; Sandhoff, K. Accumulation of protein-bound epidermal glucosylceramides in beta-glucocerebrosidase deficient type 2 Gaucher mice. FEBS Lett. 1999, 447, 167–170. [Google Scholar] [CrossRef] [Green Version]
- Breiden, B.; Sandhoff, K. The role of sphingolipid metabolism in cutaneous permeability barrier formation. Biochim. Biophys. Acta. 2014, 1841, 441–452. [Google Scholar] [CrossRef]
- Ghauharali-van der Vlugt, K.; Langeveld, M.; Poppema, A.; Kuiper, S.; Hollak, C.E.; Aerts, J.M.; Groener, J.E. Prominent increase in plasma ganglioside GM3 is associated with clinical manifestations of type I Gaucher disease. Clin. Chim. Acta. 2008, 389, 109–113. [Google Scholar] [CrossRef]
- Gornati, R.; Berra, B.; Montorfano, G.; Martini, C.; Ciana, G.; Ferrari, P.; Romano, M.; Bembi, B. Glycolipid analysis of different tissues and cerebrospinal fluid in type II Gaucher disease. J. Inherit. Metab. Dis. 2002, 25, 47–55. [Google Scholar] [CrossRef]
- Rolfs, A.; Giese, A.K.; Grittner, U.; Mascher, D.; Elstein, D.; Zimran, A.; Bottcher, T.; Lukas, J.; Hubner, R.; Golnitz, U.; et al. Glucosylsphingosine is a highly sensitive and specific biomarker for primary diagnostic and follow-up monitoring in Gaucher disease in a non-Jewish, Caucasian cohort of Gaucher disease patients. PLoS ONE 2013, 8, e79732. [Google Scholar] [CrossRef]
- Hurvitz, N.; Dinur, T.; Becker-Cohen, M.; Cozma, C.; Hovakimyan, M.; Oppermann, S.; Demuth, L.; Rolfs, A.; Abramov, A.; Zimran, A.; et al. Glucosylsphingosine (lyso-Gb1) as a Biomarker for Monitoring Treated and Untreated Children with Gaucher Disease. Int. J. Mol. Sci. 2019, 20, 3033. [Google Scholar] [CrossRef] [Green Version]
- Vanier, M.T.; Svennerholm, L. Chemical pathology of Krabbe’s disease. Ceramide-hexosides and gangliosides of brain. Acta Paediatr Scand. 1975, 64, 641–648. [Google Scholar] [CrossRef]
- Escolar, M.L.; Kiely, B.T.; Shawgo, E.; Hong, X.; Gelb, M.H.; Orsini, J.J.; Matern, D.; Poe, M.D. Psychosine, a marker of Krabbe phenotype and treatment effect. Mol. Genet. Metab. 2017, 121, 271–278. [Google Scholar] [CrossRef]
- Mikulka, C.R.; Sands, M.S. Treatment for Krabbe’s disease: Finding the combination. J. Neurosci. Res. 2016, 94, 1126–1137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Von Figura, K.; Gieselmann, V.; Jaeken, J. Metachroimatic leukodysrtophy. In The Metabolic and Molecular Bases of Inherited Disease, 8th ed.; Scriver, C.R., Beaudet, A.L., Sly, W.S., Valle, D., Eds.; McGraw-Hill: New York, NY, USA, 2001; pp. 3695–3724. [Google Scholar]
- Suzuki, K. Ganglioside patterns of normal and pathological brains. In Inborn Disorders of Sphingolipid Metabolism; Pergamon: Oxford, UK, 1967; pp. 215–230. [Google Scholar]
- Sevin, C.; Verot, L.; Benraiss, A.; Van Dam, D.; Bonnin, D.; Nagels, G.; Fouquet, F.; Gieselmann, V.; Vanier, M.T.; De Deyn, P.P.; et al. Partial cure of established disease in an animal model of metachromatic leukodystrophy after intracerebral adeno-associated virus-mediated gene transfer. Gene Therapy 2007, 14, 405–414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beerepoot, S.; Nierkens, S.; Boelens, J.J.; Lindemans, C.; Bugiani, M.; Wolf, N.I. Peripheral neuropathy in metachromatic leukodystrophy: Current status and future perspective. Orphanet. J. Rare Dis. 2019, 14, 240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klein, A.; Henseler, M.; Klein, C.; Suzuki, K.; Harzer, K.; Sandhoff, K. Sphingolipid activator protein D (sap-D) stimulates the lysosomal degradation of ceramide in vivo. Biochem. Biophys. Res. Commun. 1994, 200, 1440–1448. [Google Scholar] [CrossRef]
- Ferreira, C.R.; Gahl, W.A. Lysosomal storage diseases. Transl. Sci. Rare Dis. 2017, 2, 1–71. [Google Scholar] [CrossRef] [Green Version]
- Antonarakis, S.E.; Valle, D.; Moser, H.W.; Moser, A.; Qualman, S.J.; Zinkham, W.H. Phenotypic variability in siblings with Farber disease. J. Pediatr. 1984, 104, 406–409. [Google Scholar] [CrossRef]
- Cozma, C.; Iurașcu, M.-I.; Eichler, S.; Hovakimyan, M.; Brandau, O.; Zielke, S.; Böttcher, T.; Giese, A.-K.; Lukas, J.; Rolfs, A. C26-Ceramide as highly sensitive biomarker for the diagnosis of Farber Disease. Sci. Rep. 2017, 7, 6149. [Google Scholar] [CrossRef] [Green Version]
- He, X.; Dworski, S.; Zhu, C.; DeAngelis, V.; Solyom, A.; Medin, J.A.; Simonaro, C.M.; Schuchman, E.H. Enzyme replacement therapy for Farber disease: Proof-of-concept studies in cells and mice. BBA Clin. 2017, 7, 85–96. [Google Scholar] [CrossRef]
- Fujita, N.; Suzuki, K.; Vanier, M.T.; Popko, B.; Maeda, N.; Klein, A.; Henseler, M.; Sandhoff, K.; Nakayasu, H. Targeted disruption of the mouse sphingolipid activator protein gene: A complex phenotype, including severe leukodystrophy and wide-spread storage of multiple sphingolipids. Hum. Mol. Genet. 1996, 5, 711–725. [Google Scholar] [CrossRef]
- Constantopoulos, G.; Dekaban, A.S. Neurochemistry of the mucopolysaccharidoses: Brain lipids and lysosomal enzymes in patients with four types of mucopolysaccharidosis and in normal controls. J. Neurochem. 1978, 30, 965–973. [Google Scholar] [CrossRef]
- Stapleton, M.; Arunkumar, N.; Kubaski, F.; Mason, R.W.; Tadao, O.; Tomatsu, S. Clinical presentation and diagnosis of mucopolysaccharidoses. Mol. Genet. Metab 2018, 125, 4–17. [Google Scholar] [CrossRef] [PubMed]
- Donati, M.A.; Pasquini, E.; Spada, M.; Polo, G.; Burlina, A. Newborn screening in mucopolysaccharidoses. Ital. J. Pediatrics 2018, 44, 126. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Wood, T.; Young, S.P.; Millington, D.S. A straightforward, quantitative ultra-performance liquid chromatography-tandem mass spectrometric method for heparan sulfate, dermatan sulfate and chondroitin sulfate in urine: An improved clinical screening test for the mucopolysaccharidoses. Mol. Genet. Metab. 2015, 114, 123–128. [Google Scholar] [CrossRef]
- Kiely, B.T.; Kohler, J.L.; Coletti, H.Y.; Poe, M.D.; Escolar, M.L. Early disease progression of Hurler syndrome. Orphanet. J. Rare Dis. 2017, 12, 32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Constantopoulos, G.; Iqbal, K.; Dekaban, A.S. Mucopolysaccharidosis types IH, IS, II and IIIA: Glycosaminoglycans and lipids of isolated brain cells and other fractions from autopsied tissues. J. Neurochem. 1980, 34, 1399–1411. [Google Scholar] [CrossRef]
- Wraith, J.E.; Scarpa, M.; Beck, M.; Bodamer, O.A.; De Meirleir, L.; Guffon, N.; Meldgaard Lund, A.; Malm, G.; Van der Ploeg, A.T.; Zeman, J. Mucopolysaccharidosis type II (Hunter syndrome): A clinical review and recommendations for treatment in the era of enzyme replacement therapy. Eur. J. Pediatrics 2008, 167, 267–277. [Google Scholar] [CrossRef] [Green Version]
- Saville, J.T.; Flanigan, K.M.; Truxal, K.V.; McBride, K.L.; Fuller, M. Evaluation of biomarkers for Sanfilippo syndrome. Mol. Genet. Metab. 2019, 128, 68–74. [Google Scholar] [CrossRef]
- Liour, S.S.; Jones, M.Z.; Suzuki, M.; Bieberich, E.; Yu, R.K. Metabolic studies of glycosphingolipid accumulation in mucopolysaccharidosis IIID. Mol. Genet. Metab. 2001, 72, 239–247. [Google Scholar] [CrossRef]
- Gaffke, L.; Pierzynowska, K.; Piotrowska, E.; Węgrzyn, G. How close are we to therapies for Sanfilippo disease? Metab. Brain Dis. 2018, 33, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Lieberman, A.P.; Puertollano, R.; Raben, N.; Slaugenhaupt, S.; Walkley, S.U.; Ballabio, A. Autophagy in lysosomal storage disorders. Autophagy 2012, 8, 719–730. [Google Scholar] [CrossRef] [Green Version]
- Tessitore, A.; Pirozzi, M.; Auricchio, A. Abnormal autophagy, ubiquitination, inflammation and apoptosis are dependent upon lysosomal storage and are useful biomarkers of mucopolysaccharidosis VI. Pathogenetics. 2009, 2, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walkley, S.U. Secondary accumulation of gangliosides in lysosomal storage disorders. Semin Cell Dev. Biol 2004, 15, 433–444. [Google Scholar] [CrossRef] [PubMed]
- Harmatz, P.; Whitley, C.B.; Waber, L.; Pais, R.; Steiner, R.; Plecko, B.; Kaplan, P.; Simon, J.; Butensky, E.; Hopwood, J.J. Enzyme replacement therapy in mucopolysaccharidosis VI (Maroteaux–Lamy syndrome). J. Pediatrics 2004, 144, 574–580. [Google Scholar] [CrossRef] [PubMed]
- McGlynn, R.; Dobrenis, K.; Walkley, S.U. Differential subcellular localization of cholesterol, gangliosides, and glycosaminoglycans in murine models of mucopolysaccharide storage disorders. J. Comp. Neurol. 2004, 480, 415–426. [Google Scholar] [CrossRef] [PubMed]
- Dacremont, G.; Kint, J.A.; Cocquyt, G. Brain sphingolipids in I cell disease (mucolipidosis II). J. Neurochem. 1974, 22, 599–602. [Google Scholar] [CrossRef]
- LaPlante, J.M.; Falardeau, J.; Sun, M.; Kanazirska, M.; Brown, E.M.; Slaugenhaupt, S.A.; Vassilev, P.M. Identification and characterization of the single channel function of human mucolipin-1 implicated in mucolipidosis type IV, a disorder affecting the lysosomal pathway. FEBS Lett. 2002, 532, 183–187. [Google Scholar] [CrossRef] [Green Version]
- Boudewyn, L.C.; Walkley, S.U. Current concepts in the neuropathogenesis of mucolipidosis type IV. J. Neurochem. 2019, 148, 669–689. [Google Scholar] [CrossRef] [Green Version]
- Cam, L.; Tettamanti, G.; Berra, B.; Sale, F.O.; Borrone, C.; Gatti, R.; Durand, P.; Martin, J.J. Mucolipidosis IV, a sialolipidosis due to ganglioside sialidase deficiency. J. Inherit. Metab. Dis. 1982, 5, 218–224. [Google Scholar] [CrossRef]
- Tellez-Nagel, I.; Rapin, I.; Iwamoto, T.; Johnson, A.B.; Norton, W.T.; Nitowsky, H. Mucolipidosis IV. Clinical, ultrastructural, histochemical, and chemical studies of a case, including a brain biopsy. Arch. Neurol. 1976, 33, 828–835. [Google Scholar] [CrossRef]
- D’Azzo, A.; Andria, G.; Strisciuglio, P.; Galjaard, H. Galactosialidosis. In The Metabolic and Molecular Bases of Inherited Disease; Scriver, C., Beaudet, A., Sly, W., Valle, D., Eds.; McGraw-Hill: New York, NY, USA, 2001; pp. 3811–3826. [Google Scholar]
- Yoshino, H.; Miyashita, K.; Miyatani, N.; Ariga, T.; Hashimoto, Y.; Tsuji, S.; Oyanagi, K.; Ohama, E.; Ikuta, F.; Suzuki, A.; et al. Abnormal glycosphingolipid metabolism in the nervous system of galactosialidosis. J. Neurol. Sci. 1990, 97, 53–65. [Google Scholar] [CrossRef]
- Miyatake, T.; Atsumi, T.; Obayashi, T.; Mizuno, Y.; Ando, S.; Ariga, T.; Matsui-Nakamura, K.; Yamada, T. Adult type neuronal storage disease with neuraminidase deficiency. Ann. Neurol. 1979, 6, 232–244. [Google Scholar] [CrossRef] [PubMed]
- Thomas, G.H. Disorders of glycoprotein degradation: α-mannosidosis, β-mannosidosis, fucosidosis, and sialidosis. In The Metabolic and Molecular Bases of Inherited Disease; Scriver, C.R., Beaudet, A.L., Sly, W.S., Valle, D., Eds.; McGraw-Hill: New York, NY, USA, 2001; pp. 3507–3633. [Google Scholar]
- Goodman, L.A.; Livingston, P.O.; Walkley, S.U. Ectopic dendrites occur only on cortical pyramidal cells containing elevated GM2 ganglioside in alpha-mannosidosis. Proc. Natl Acad Sci USA 1991, 88, 11330–11334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harmatz, P.; Cattaneo, F.; Ardigò, D.; Geraci, S.; Hennermann, J.B.; Guffon, N.; Lund, A.; Hendriksz, C.J.; Borgwardt, L. Enzyme replacement therapy with velmanase alfa (human recombinant alpha-mannosidase): Novel global treatment response model and outcomes in patients with alpha-mannosidosis. Mol. Genet. Metab. 2018, 124, 152–160. [Google Scholar] [CrossRef] [PubMed]
- Ceccarini, M.R.; Codini, M.; Conte, C.; Patria, F.; Cataldi, S.; Bertelli, M.; Albi, E.; Beccari, T. Alpha-Mannosidosis: Therapeutic Strategies. Int. J. Mol. Sci. 2018, 19, 1500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hofmann, S.L.; Peltonen, L. The neuronal ceroid lipofuscinosis. In The Metabolic and Molecular Bases of Inherited Disease, 8th ed.; Scriver, C.R., Beaudet, A.L., Sly, W.S., Valle, D., Eds.; McGraw-Hill: New York, NY, USA, 2001; pp. 3877–3894. [Google Scholar]
- Jabs, S.; Quitsch, A.; Käkelä, R.; Koch, B.; Tyynelä, J.; Brade, H.; Glatzel, M.; Walkley, S.; Saftig, P.; Vanier, M.T.; et al. Accumulation of bis(monoacylglycero)phosphate and gangliosides in mouse models of neuronal ceroid lipofuscinosis. J. Neurochem. 2008, 106, 1415–1425. [Google Scholar] [CrossRef] [PubMed]
- Walkley, S.U. Cellular pathology of lysosomal storage disorders. Brain Pathol. 1998, 8, 175–193. [Google Scholar] [CrossRef]
- Kyttälä, A.; Lahtinen, U.; Braulke, T.; Hofmann, S.L. Functional biology of the neuronal ceroid lipofuscinoses (NCL) proteins. Biochim. Biophys. Acta. 2006, 1762, 920–933. [Google Scholar] [CrossRef] [Green Version]
- Somogyi, A.; Petcherski, A.; Beckert, B.; Huebecker, M.; Priestman, D.A.; Banning, A.; Cotman, S.L.; Platt, F.M.; Ruonala, M.O.; Tikkanen, R. Altered expression of ganglioside metabolizing enzymes results in GM3 ganglioside accumulation in cerebellar cells of a mouse model of juvenile neuronal ceroid lipofuscinosis. Int. J. Mol. Sci. 2018, 19, 625. [Google Scholar] [CrossRef] [Green Version]
- Koike, M.; Shibata, M.; Waguri, S.; Yoshimura, K.; Tanida, I.; Kominami, E.; Gotow, T.; Peters, C.; von Figura, K.; Mizushima, N.; et al. Participation of autophagy in storage of lysosomes in neurons from mouse models of neuronal ceroid-lipofuscinoses (Batten disease). Am. J. Pathol. 2005, 167, 1713–1728. [Google Scholar] [CrossRef] [Green Version]
- Rakheja, D.; Bennett, M.J. Neuronal ceroid-lipofuscinoses. Transl. Sci. Rare Dis. 2018, 3, 83–95. [Google Scholar] [CrossRef] [Green Version]
- Harding, A.E. Classification of the hereditary ataxias and paraplegias. Lancet. 1983, 1, 1151–1155. [Google Scholar] [CrossRef]
- Hensiek, A.; Kirker, S.; Reid, E. Diagnosis, investigation and management of hereditary spastic paraplegias in the era of next-generation sequencing. J. Neurol. 2015, 262, 1601–1612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allison, R.; Edgar, J.R.; Pearson, G.; Rizo, T.; Newton, T.; Günther, S.; Berner, F.; Hague, J.; Connell, J.W.; Winkler, J.; et al. Defects in ER–endosome contacts impact lysosome function in hereditary spastic paraplegia. J. Cell Biol. 2017, 216, 1337–1355. [Google Scholar] [CrossRef] [PubMed]
- Robinson, M.S. Forty years of clathrin-coated vesicles. Traffic (Copenhagen, Denmark) 2015, 16, 1210–1238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirst, J.; Borner, G.H.; Edgar, J.; Hein, M.Y.; Mann, M.; Buchholz, F.; Antrobus, R.; Robinson, M.S. Interaction between AP-5 and the hereditary spastic paraplegia proteins SPG11 and SPG15. Mol. Biol. Cell. 2013, 24, 2558–2569. [Google Scholar] [CrossRef] [PubMed]
- Hirst, J.; Edgar, J.R.; Esteves, T.; Darios, F.; Madeo, M.; Chang, J.; Roda, R.H.; Dürr, A.; Anheim, M.; Gellera, C.; et al. Loss of AP-5 results in accumulation of aberrant endolysosomes: Defining a new type of lysosomal storage disease. Hum. Mol. Genet. 2015, 24, 4984–4996. [Google Scholar] [CrossRef] [Green Version]
- Khundadze, M.; Kollmann, K.; Koch, N.; Biskup, C.; Nietzsche, S.; Zimmer, G.; Hennings, J.C.; Huebner, A.K.; Symmank, J.; Jahic, A.; et al. A hereditary spastic paraplegia mouse model supports a role of ZFYVE26/SPASTIZIN for the endolysosomal system. PLoS Genetics 2013, 9, e1003988. [Google Scholar] [CrossRef] [Green Version]
- Renvoisé, B.; Chang, J.; Singh, R.; Yonekawa, S.; FitzGibbon, E.J.; Mankodi, A.; Vanderver, A.; Schindler, A.B.; Toro, C.; Gahl, W.A.; et al. Lysosomal abnormalities in hereditary spastic paraplegia types SPG15 and SPG11. Ann. Clin. Transl. Neurol. 2014, 1, 379–389. [Google Scholar] [CrossRef]
- Hirst, J.; Itzhak, D.N.; Antrobus, R.; Borner, G.H.H.; Robinson, M.S. Role of the AP-5 adaptor protein complex in late endosome-to-Golgi retrieval. PLoS Biology 2018, 16, e2004411. [Google Scholar] [CrossRef] [Green Version]
- Boutry, M.; Branchu, J.; Lustremant, C.; Pujol, C.; Pernelle, J.; Matusiak, R.; Seyer, A.; Poirel, M.; Chu-Van, E.; Pierga, A.; et al. Inhibition of lysosome membrane recycling causes accumulation of gangliosides that contribute to neurodegeneration. Cell Rep. 2018, 23, 3813–3826. [Google Scholar] [CrossRef]
- Branchu, J.; Boutry, M.; Sourd, L.; Depp, M.; Leone, C.; Corriger, A.; Vallucci, M.; Esteves, T.; Matusiak, R.; Dumont, M.; et al. Loss of spatacsin function alters lysosomal lipid clearance leading to upper and lower motor neuron degeneration. Neurobiol. Dis. 2017, 102, 21–37. [Google Scholar] [CrossRef]
- Ariga, T.; Yanagisawa, M.; Wakade, C.; Ando, S.; Buccafusco, J.J.; McDonald, M.P.; Yu, R.K. Ganglioside metabolism in a transgenic mouse model of Alzheimer’s disease: Expression of Chol-1alpha antigens in the brain. ASN Neuro. 2010, 2, e00044. [Google Scholar] [CrossRef] [PubMed]
- Molander-Melin, M.; Blennow, K.; Bogdanovic, N.; Dellheden, B.; Mansson, J.E.; Fredman, P. Structural membrane alterations in Alzheimer brains found to be associated with regional disease development; increased density of gangliosides GM1 and GM2 and loss of cholesterol in detergent-resistant membrane domains. J. Neurochem. 2005, 92, 171–182. [Google Scholar] [CrossRef] [PubMed]
- Pernber, Z.; Blennow, K.; Bogdanovic, N.; Månsson, J.E.; Blomqvist, M. Altered distribution of the gangliosides GM1 and GM2 in Alzheimer’s disease. Dement. Geriatr. Cogn. Disord. 2012, 33, 174–188. [Google Scholar] [CrossRef] [PubMed]
- Chan, R.B.; Oliveira, T.G.; Cortes, E.P.; Honig, L.S.; Duff, K.E.; Small, S.A.; Wenk, M.R.; Shui, G.; Di Paolo, G. Comparative lipidomic analysis of mouse and human brain with Alzheimer disease. J. Biol. Chem. 2012, 287, 2678–2688. [Google Scholar] [CrossRef] [Green Version]
- Yang, D.S.; Stavrides, P.; Mohan, P.S.; Kaushik, S.; Kumar, A.; Ohno, M.; Schmidt, S.D.; Wesson, D.; Bandyopadhyay, U.; Jiang, Y.; et al. Reversal of autophagy dysfunction in the TgCRND8 mouse model of Alzheimer’s disease ameliorates amyloid pathologies and memory deficits. Brain 2011, 134, 258–277. [Google Scholar] [CrossRef]
- Yang, D.-S.; Stavrides, P.; Saito, M.; Kumar, A.; Rodriguez-Navarro, J.A.; Pawlik, M.; Huo, C.; Walkley, S.U.; Saito, M.; Cuervo, A.M.; et al. Defective macroautophagic turnover of brain lipids in the TgCRND8 Alzheimer mouse model: Prevention by correcting lysosomal proteolytic deficits. Brain 2014, 137, 3300–3318. [Google Scholar] [CrossRef] [Green Version]
- Zhou, S.; Davidson, C.; McGlynn, R.; Stephney, G.; Dobrenis, K.; Vanier, M.T.; Walkley, S.U. Endosomal/lysosomal processing of gangliosides affects neuronal cholesterol sequestration in Niemann–Pick disease type C. Am. J. Pathol. 2011, 179, 890–902. [Google Scholar] [CrossRef]
- Beutler, E.; Grabowski, G.A. Gaucher disease. In The Metabolic and Molecular Bases of Inherited Disease, 8th ed.; Scriver, C.R., Beaudet, A.L., Sly, W.S., Valle, D., Eds.; McGraw-Hill: New York, NY, USA, 2001; pp. 3635–3668. [Google Scholar]
- Gilbert, E.F.; Dawson, G.; Zu Rhein, G.M.; Opitz, J.M.; Spranger, J.W. I-cell disease, mucolipidosis II. Pathological, histochemical, ultrastructural and biochemical observations in four cases. Z Kinderheilkd 1973, 114, 259–292. [Google Scholar] [CrossRef]
- Nilsson, O.; Fredman, P.; Klinghardt, G.W.; Dreyfus, H.; Svennerholm, L. Chloroquine-induced accumulation of gangliosides and phospholipids in skeletal muscles. Eur. J. Biochem. 1981, 116, 565–571. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Disease | Protein | Gene | Storage Compound | References |
|---|---|---|---|---|
| GM2 Gangliosidoses | ||||
| Tay–Sachs disease (B variant) | Hex A1, Hex S2 | HEXA | GM2, SM2a, lyso-GM2, GA2 | [28] |
| B1 variant | Hex A1 | HEXA | GM2 | [28] |
| Sandhoff disease | Hex A1, Hex B3 | HEXB | GM2, globoside, oligosaccharides, lyso-GM2 | [28,29,30] |
| GM2AP deficiency (AB variant) | GM2AP | GM2A | GM2 | [28] |
| GM1 Gangliosidosis | acid β-galactosidase | GBL1 | GM1, GA1, GM2, GM3, GA1a, lyso GM1 GlcCer Laccer, oligosaccharides, keratan sulfate | [31,32] |
| Disease | Protein | Gene | Major Storage Compound 1 | Accumulated Ganglioside | References |
|---|---|---|---|---|---|
| Sphingolipidoses | |||||
| Niemann–Pick disease type A, B | ASM | SMPD1 | SM1 | GM2, GM3 | [3,32,90] |
| Niemann–Pick disease type C | NPC1 | NPC1 | Chol 2 | GM2, GM3, GM1 | [4,32,179] |
| NPC2 | NPC2 | Chol 2 | GM2, GM3 | [4,95,179] | |
| Gaucher disease | β-glucosidase | GBA1 | GlcCer 3 | GM2, GM3, GM1, GD3 | [112,180] |
| Metachromatic leukodystrophy | Arylsulfatase A | ARSA | Sulfatide | GM2 | [120] |
| Krabbe disease | galactocerebrosidase | GALC | GalCer 4 | GD2, GD3, GM3 | [115] |
| Farber disease | acid ceramidase | ASAH1 | ceramide | Gangliosides | [123,124] |
| Mucopolysaccharidoses (MPS) | |||||
| MPS Ι (Hurler syndrome) | α-L iduronidase | IDUA | heparan sulfate, dermatan sulfate | GM2, GM3 | [32,128,133,142] |
| MPS ΙΙ (Hunter syndrome) | iduronate-2-sulfatase | IDS | heparan sulfate, dermatan sulfate | GM2, GM3 | [133] |
| MPS ΙΙΙA (Sanfilippo syndrome) | Heparin-N-sulfatase | SGSH | heparan sulfate | GM2, GM3, GD2 | [133,142] |
| MPS ΙΙΙB (Sanfilippo syndrome) | α-N-Acetylglucosaminidase | NAGLU | heparan sulfate | GM2, GM3, GD2 | [128] |
| MPS ΙΙΙC (Sanfilippo syndrome) | Acetyl-CoA: α-N-glucosaminide N-acetyltransferase | HGSNAT | heparan sulfate | GM2, GM3, GD2 | |
| MPS ΙΙΙD (Sanfilippo syndrome) | N-Acetylglucosamine-6-sulfatase | GNS | heparan sulfate | GM3, GM2, GD2 | [136] |
| MPS VΙ (Maroteaux–Lamy syndrome) | arylsulfatase B | ASRB | dermatan sulfate | GM2, GM3 | [140] |
| MPS VΙΙ (Sly syndrome) | β-glucuronidase | GUSB | heparan sulfate, dermatan sulfate, chondroitin sulfate | GM2, GM3 | [5,142] |
| Mucolipidoses | |||||
| Mucolipidosis ΙΙ (I-cell disease) Mucolipidosis ΙΙΙ (pseudo-Hurler polydystrophy) | N-acetylglucosamine-1-phosphotransferase | GNPTAB | GM1 | [143,181] | |
| Mucolipidosis ΙV (mucolipidin 1 deficiency) | TRPML1 | MCOLN1 | GM3, GD1a | [147] | |
| Glycoproteinoses | |||||
| Galactosialidosis | lysosomal protective protein–cathepsin A (PPCA) | CTSA | sialyloligosacchaides | GM2, GM3, GM1, GD1a | [149] |
| α-Mannosidosis | α-D-mannosidase | MAN2B1 | mannose-rich oligosaccharides | GM2, GM3 | [32,152] |
| Sialidosis | acid neuraminidase 1 | NEU1 | sialyloligosaccharides, sialoglycoproteins | GM3, GD3, GM4, LM1 | [151] |
| Neuronal ceroid lipofuscinoses (NCL) | |||||
| NCL 3 (Batten disease) | CLN3 | CLN3 | ATPase subunit c, lipofuscin | GM3 | [159] |
| NCL 6 | CLN 6 | NCLF | ATPase subunit c, lipofuscin | GM2, GM3 | [156] |
| NCL 10 (Congenital cathepsin D deficiency) | Cathapsin D | CTSD | ATPase subunit c, Sap A, Sap D, lipofuscin | GM2, GM3 | [156] |
| Hereditary spastic paraplegia (HSP) | |||||
| HSP type SPG 11 | spatacsin | SPG11 | p62 | GM2, GM3, GD2, GD3 | [171] |
| Alzheimer | |||||
| TgCRND8 (Alzheimer maus) | Aβ40, Aβ42 | GM1, GD1a, GD1b, GM2, GM3 | [178] | ||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Breiden, B.; Sandhoff, K. Mechanism of Secondary Ganglioside and Lipid Accumulation in Lysosomal Disease. Int. J. Mol. Sci. 2020, 21, 2566. https://doi.org/10.3390/ijms21072566
Breiden B, Sandhoff K. Mechanism of Secondary Ganglioside and Lipid Accumulation in Lysosomal Disease. International Journal of Molecular Sciences. 2020; 21(7):2566. https://doi.org/10.3390/ijms21072566
Chicago/Turabian StyleBreiden, Bernadette, and Konrad Sandhoff. 2020. "Mechanism of Secondary Ganglioside and Lipid Accumulation in Lysosomal Disease" International Journal of Molecular Sciences 21, no. 7: 2566. https://doi.org/10.3390/ijms21072566
APA StyleBreiden, B., & Sandhoff, K. (2020). Mechanism of Secondary Ganglioside and Lipid Accumulation in Lysosomal Disease. International Journal of Molecular Sciences, 21(7), 2566. https://doi.org/10.3390/ijms21072566