1. Introduction
Primary malignant brain tumors remain a devastating tumor entity characterized by poor prognoses despite extensive research and gains in knowledge concerning its genesis and progression [
1]. Microglia, the innate immune cells of the brain and infiltrating monocytes/macrophages play a pivotal role in glioma promotion and growth and contribute to the tumor tissue by up to 30% of the cells [
2]. Glioma induces the polarization of glioma associated microglia/macrophages (GAMs) into a glioma-supporting phenotype through complex interactions involving several signaling mechanisms [
3]. One major mechanism that contributes to glioma invasiveness is the degradation of the surrounding extracellular matrix by matrix metalloproteases (MMPs). MMPs are a group of enzymes that proteolytically cleave diverse proteoglycans, collagens and gelatin [
4]. We discovered that microglial cells express MMP14 as a membrane bound protein which serves to activate glioma released MMP2. While microglia in the healthy brain do not express MMP14, it is upregulated in glioma associated GAMs. This upregulation is induced by versican released from glioma cells which signals via toll like receptor 2 (TLR2). This interaction between glioma and GAMs is an important contributor to microglia/macrophage-induced glioma invasiveness und progression [
5,
6]. TLRs are membrane-bound receptors that recognize a broad spectrum of pathogen-associated molecular patterns. In addition, TLRs can be activated by endogenous ligands such as versican and tenascin C [
7]. In the glioma context, TLR4 and in particular TLR2 are mediators of glioma-GAM interactions. The expression of TLR2 and MMPs in glioma tissue was found to be inversely correlated with patients’ median survival according to TCGA analysis. In our previous study, we demonstrated that the downregulation of TLR2 in microglia by minocycline treatment correlates with the downregulation of MMP14 and increased survival of brain tumor bearing mice [
5]. Recently, Mistry et al. [
8] identified a loop within the intracellular adapter domain of TLR2 that can be blocked by O-Vanillin, and thus identified O-Vanillin as an TLR2 inhibitor. O-Vanillin is an isomer of the well-known food supplement vanillin and was studied in different malignancies including colon, ovarian and lung cancer and melanoma [
9,
10,
11]. So far, the use of O-Vanillin as a potential drug in gliomas has not yet been explored.
In the present study, we tested whether O-Vanillin can be used to inhibit glioma-induced TLR2 signaling in murine microglia. O-Vanillin was able to attenuate the glioma-induced increase in mmp14 and mmp9 in cultured microglia as well as in microglia/macrophages isolated from human glioma samples. Moreover, O-Vanillin also attenuated the growth of gliomas injected into murine brain slice cultures and this effect was dependent on the presence of microglia.
3. Discussion
Glioma cells polarize GAMs into a tumor promoting phenotype and these cells acquire an expression profile which is distinct from M1 or M2 profile [
15]. Their tumor promoting activity is mediated by an increased production and activation of matrix metalloproteinases, in particular MMP9 and MMP14 [
12,
13]. Thus, glioma cells exploit GAMs to promote glioma invasiveness, immune evasion and survival [
3]. TLRs induce the glioma-associated phenotype of GAMs. We have shown that the endogenous ligand versican activates TLR2 in GAMs and is an important pathway for glioma-GAM communication [
12]. The TLR2 pathway has been established as a tumor promoting component in different malignancies like ovarian carcinomas, melanomas, or gliomas [
9,
16]. The expression of TLR2 in glioma samples, which consists of up to 30% of microglia/macrophages, is inversely correlated with patients’ overall survival [
6,
17]. We also found a negative correlation between
MMP14 and
MMP9 expression on overall survival [
18]. Thus, TLR2 has emerged as a novel target for impairing glioma growth and we could demonstrate in a mouse glioma model that interfering with TLR2 signaling by using TLR2—specific antibodies attenuated glioma growth [
6].
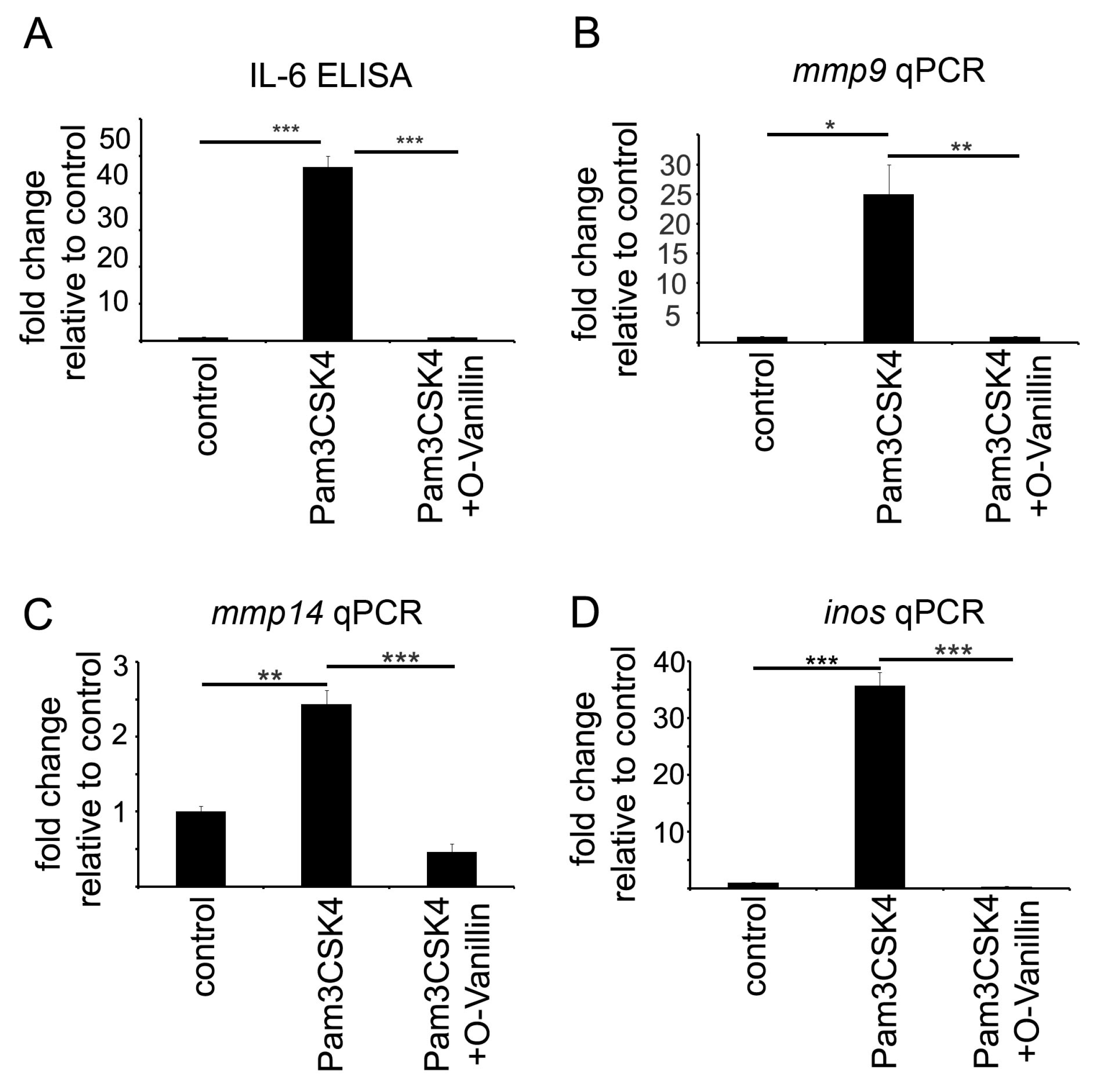
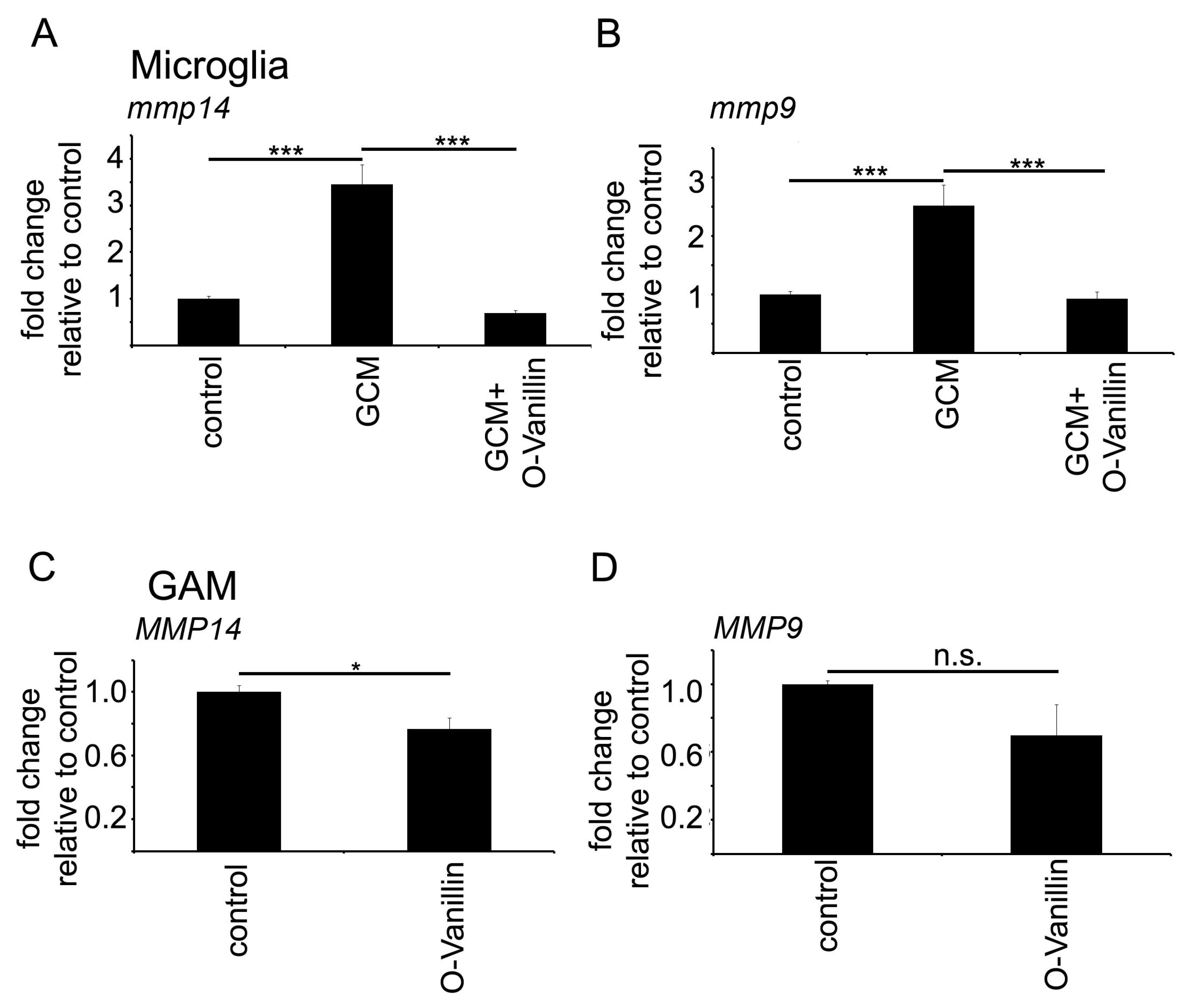
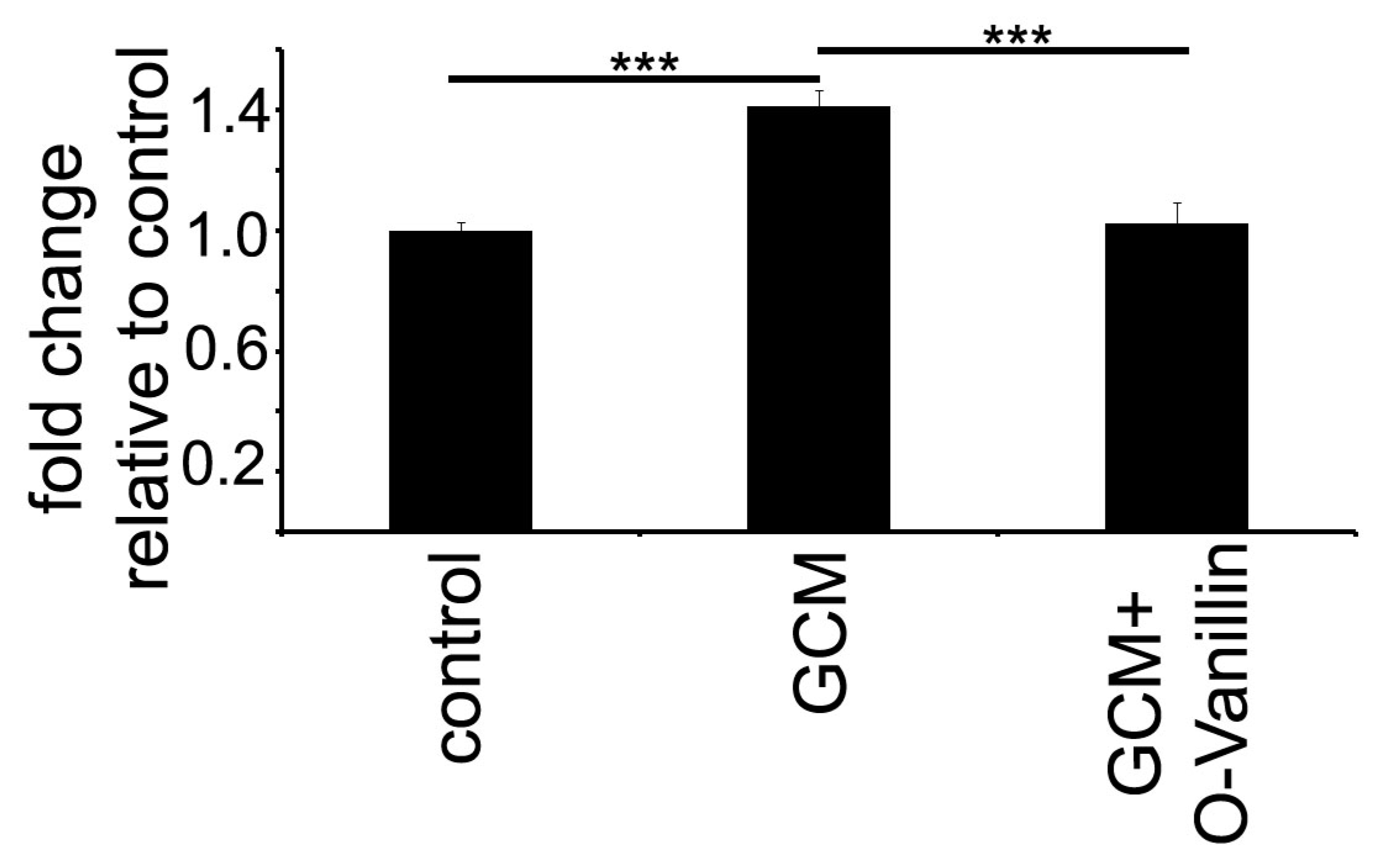
Mistry et al. identified O-Vanillin using computer-aided structure analysis as potential inhibitor of TLR2 signaling by blocking its heterodimerization. They were able to inhibit the TLR2-mediated increase in
IL8 RNA in peripheral macrophages using O-Vanillin. We adapted these finding to the context of GAMs. We showed that O-Vanillin is a specific TLR2 inhibitor in primary microglia (
Figure 1). We found that the TLR2 agonist Pam3CSK4 induced an increased
mmp9,
mmp14, and
inos expression [
19,
20,
21]. A quantity of 100 µM O-Vanillin was able to inhibit this induction. Moreover, the increase in
MMP9 and
MMP14 expression by GCM was also blocked by O-Vanillin. We therefore conclude that O-Vanillin works as an TLR2 inhibitor in microglia.
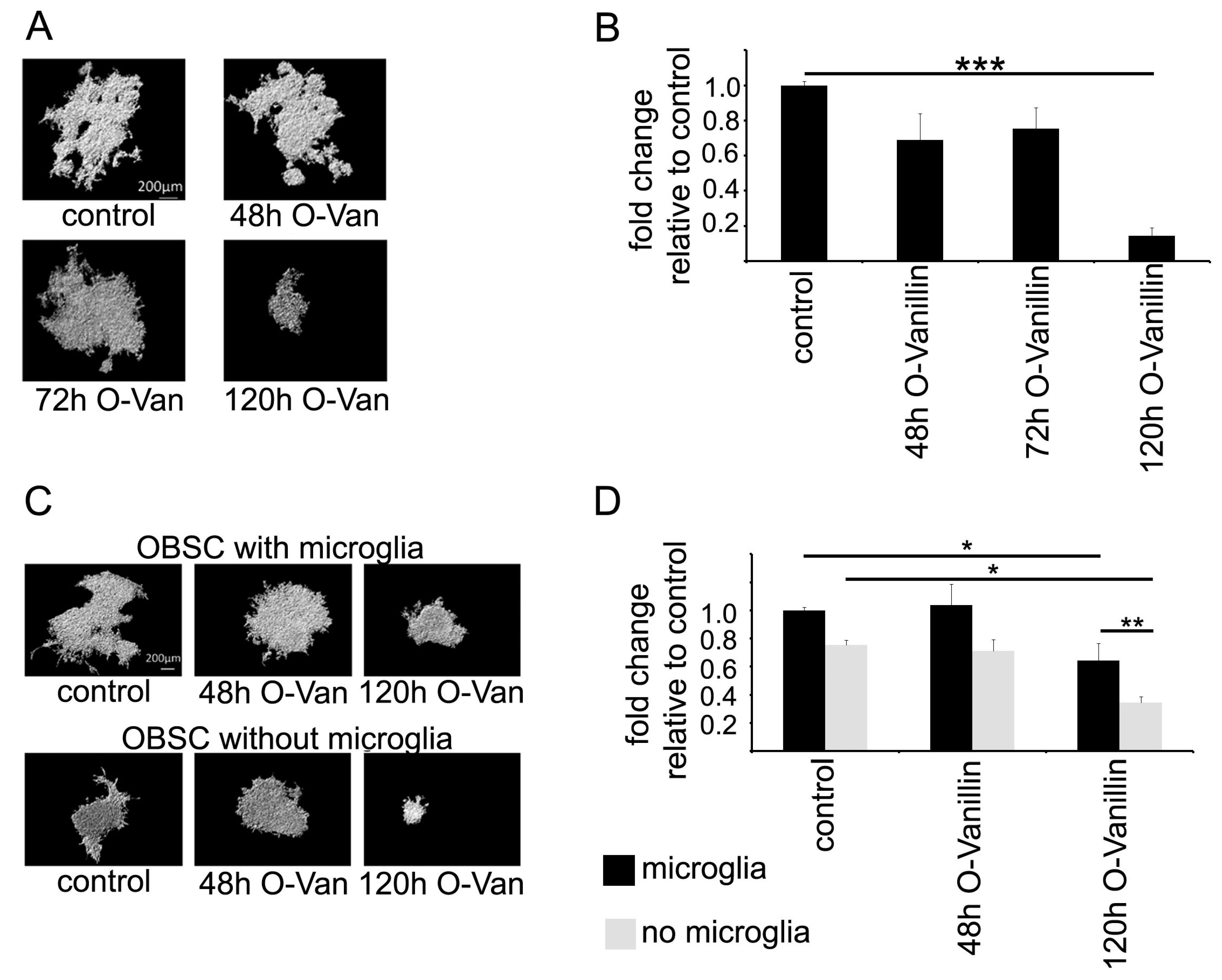
To test the impact of O-Vanillin on glioma growth, we used an organotypic slice model in which we could quantify glioma growth. Treatment of the slices with 100 µM O-Vanillin led to a significant reduction in tumor volume by 85%. We also tested whether the impact of O-Vanillin was mediated by microglia by depleting these cells from the slices. As previously described, there was a reduction in glioma growth through the depletion of microglia, but treatment with O-Vanillin led even to a further decrease in glioma growth. This can be explained by our observation that O-Vanillin directly inhibited glioma growth. As a further mechanism of O-Vanillin action, we observed an inhibitory effect on microglia proliferation induced by GCM. It is, however, not yet fully explored to what extent and by which mechanism glioma induce the proliferation of microglia. In contrast to our observation in mice, conditioned medium from the rat glioma cell line C6 did not alter rat microglial proliferation [
21]. Li and Graeber (2012) speculate that gliomas secrete monocyte colony stimulating factor (M-CSF) which triggers the proliferation of GAM via M-CSF receptor (M-CSFR) [
22].
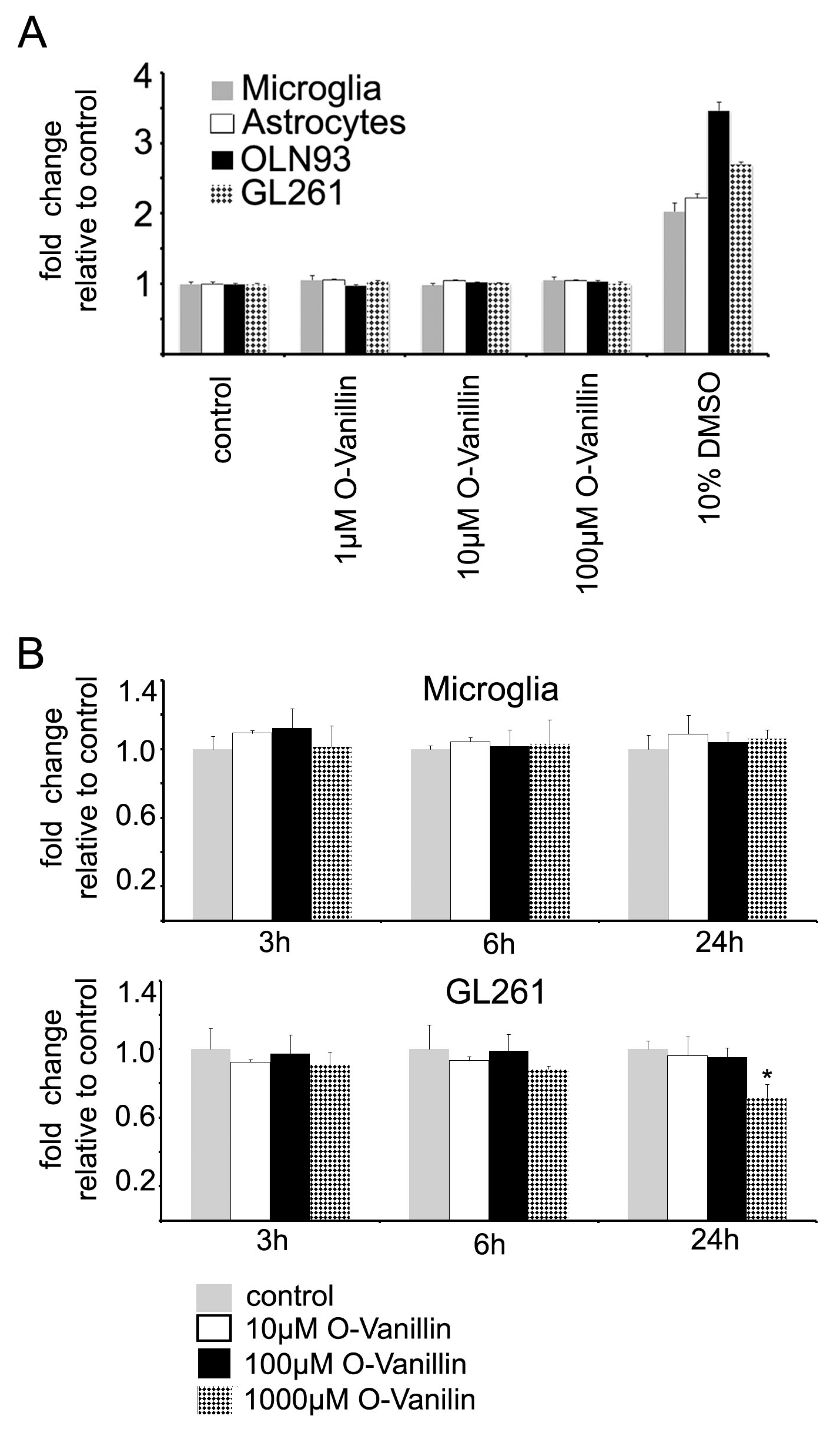
O-Vanillin demonstrated a good tolerability at even high concentrations up to 2 mM on a cellular level and 100 mg/kg (660 µM) over one month in vivo in rodents without evidence for macroscopic anomalies or cell death [
11,
23]. We also tested the effect of O-Vanillin on the survival of oligodendrocytes using a cell line as a model, primary cultured astrocytes and microglia, and found no impact on cell viability using the propidium iodide and Alamar Blue assays. At higher concentrations and longer exposure times, O-Vanillin decreased glioma cell survival while having no impact on the survival of glial cells. Marton et al. reported that O-Vanillin reduced the growth of a human Melanoma cell line [
10]. We thus identified O-Vanillin as a potential drug to attenuate glioma growth. Since O-Vanillin was shown to penetrate the blood-brain barrier [
24], there should be further studies conducted to clarify the therapeutic effect of O-Vanillin in vivo.
4. Materials and Methods
4.1. Animals
We made primary microglial cell cultures and organotypic brain slice cultures (OBSC) from C57/Bl6 wild-type mice (Charles River Laboratories, Wilmington, MA, USA) for all in vitro experiments. The mice were bred and maintained in the animal housing facility of the Max Delbrück Center as per ethical rules and approval of the local governmental institutions (01 January 2018; X9005/18; §9 and §11 of German animal protection low). The animals lived according to a 12/12 dark-light rhythm and had access to water and food ad libitum.
4.2. Cell Culture
Primary microglial cells were cultivated as described previously (Markovic et al., 2005) [
14]. Briefly, the cerebral cortices of newborn C57Bl/6 mice were carefully cleaned of blood vessels and meninges, then the tissue was enzymatically digested with Trypsin/DNase (Gibco/Life Technologies, Carlsbad, CA, USA) and mechanically dissociated with a fire-polished glass Pasteur pipette. Cultures were incubated at 37 °C, 5% CO
2 humidified atmosphere in complete growth medium (10% fetal calf serum (FCS)/Dulbecco´s modified Eagles Medium (DMEM with 200 mM glutamine, 100 U/mL penicillin and 100 µg/mL streptomycin all from Gibco/Life Technologies) and the next day adherent cells were washed 5 times with phosphate buffered solution (PBS)(Gibco/Life Technologies). After culturing for one week followed by culturing with L929 fibroblast cell line (RRID:CVCL_AR58; obtained from American Type Culture Collection, Manassas, VA, USA) conditioned medium for two days the microglia are now seen as floating cells or as semi-adherent cells on top of an astrocytic monolayer. These were then harvested by shaking the culture flask.
4.3. Astrocytes
To generate C57/Bl6 wild type astrocytes, the animals were processed as described above. After the third microglial shake off, the confluent flasks were washed thoroughly with PBS. Trypsin/EDTA was used to detach the cells. The reaction was stopped with complete growth medium and the cells were centrifuged at 500× g for 5 min at 4 °C. Then the cells were counted in a Neubauer chamber (Paul Marienfeld, Lauda-Koenigshoffen, Germany) according to the manufacturer´s instructions and plated as required for each experiment.
4.4. Glioma Cell Lines
The GL261 cells (RRID:CVCL_Y003)were obtained from the Jackson Laboratory (Bar Harbor, ME, USA). The cells were grown in DMEM with 10 % FCS, 200 mM glutamine, 100 U/mL penicillin and 100 µg/mL streptomycin (all from Gibco/Life Technologies). Red fluorescent mCherry expressing GL261 glioma cells were generated according to the protocol of Vinnakota et al. [
5]. First, we transfected OmicsLinknontargeted short hairpin RNA tagged with mCherry (GeneCopoeia, Rockville, MD, USA) according to the manufacturer’s instructions. Transfected GL261 cells were then treated with 5 µg/mL puromycin (Gibco/Life Technologies) for the selection. After the 15th passage, the cell line was discarded.
4.4.1. OLN93 Oligodendrocyte Cell Line
The immortalized oligodendrocyte progenitor cell line OLN93 (RRID:CVCL_5850; gift from C. Richter-Landsberg) was passaged in complete growth medium and split when confluent [
25,
26].
4.4.2. Human Glioma Samples
All human glioma samples were provided by the Department of Neurosurgery, University Hospital Schleswig-Holstein and Department of Neurosurgery HELIOS Hospital Berlin-Buch. After the Department for Pathology confirmed the diagnosis of a malignant glioma, the cells were further processed. All patients were informed and gave their permission for further scientific processing of the tissue samples. Analysis of resected human brain tumors was performed according to the rules by the Ethical Committee of University Hospital Kiel (UKSH D447/18) and Charité (EA4/098). Briefly, tumor tissue was taken during the surgery and was placed immediately in complete growth medium for cell isolation.
All experiments were performed with Mycoplasma-free cells.
4.4.3. Magnetic Cell Separation (MACS) of Human Brain TumorTissue
Glioma associated brain microglia/macrophages were isolated from the human tumor resected tissues. Fresh tissue was dissociated immediately after resection using a neural tissue dissociation kit (MiltenyiBiotec, BergischGladbach, Germany). Erythrocytes were lysed by adding 5 mL ammonium chloride solution. Thereafter, cells were resuspended in PBS containing 0.5% bovine serum albumin and 2 mM EDTA. Magnetic sorting for CD11b+ cells was then performed by using CD11b MicroBead kit (MiltenyiBiotec) following the manufacturer´s instruction. MACS into CD11b negative (Flow-through) and CD11b positive (CD11b+) enriched cell populations was done using several MACS columns in series. Both CD11b negative and CD11b+ fractions were collected. A purity check was performed after MACS by flow cytometry analysis of a small fraction of the sorted populations (BD FACS Aria, BD Biosciences, San Jose, CA, USA).
4.5. Production of Glioma Conditioned Medium (GCM)
GCM was prepared from the 80% confluent GL261 cultures by cultivation in complete culture medium overnight. The GCM was collected, briefly centrifuged and sterile-filtered through 0.2 µm filter mesh (Sartorius Stedim Biotech GmbH, Göttingen, Germany) and applied for further experiments.
Flow Cytometry
For all the flow cytometry analysis, after the treatment or isolation, cells were incubated for 30 min at 4 °C. Before fluorescence activated cell sorting (FACS), cells were stained with 2.5 µg/mL propidium iodide (Life Technologies) for 15 min. Flow cytometry was done by BD FACS Aria and data were analyzed using FlowJo software (Treestar, Ashland, OR, USA). Flow cytometry data were quantified by mean fluorescence intensity (MFI) and presented in histograms.
4.6. IL-6 ELISA
To investigate how the cytokine release was affected by the O-Vanillin treatment, 20,000 microglial cells were plated in a 96 well plate (Sarstedt, Nümbrecht, Germany) and maintained overnight. On the next day the cells were stimulated with Pam3CSK4 (InvivoGen, San Diego, CA, USA) and GCM for 6 h and treated with 100 µM O-Vanillin (Sigma-Aldrich, St. Louis, MO, USA). LPS (Enzo Life Sciences Inc., Farmingdale, NY, USA) was used as a positive control. After stimulation, the medium was discarded and fresh culture growth medium was added for 12 h. Then the medium was evacuated and stored at −20 °C. The IL-6 ELISA Assay was performed with ELISA Kit (R&D Systems, Minneapolis, MN, USA). For further experiments the medium was diluted 1:10 in an ELISA Buffer. Prior to the quantification of IL-6, the 96 well plates were precoated. The capture antibody was diluted in PBS according to manufacturer´s recommendations and pipetted into a 96-well microplate. The plates were sealed and incubated overnight. The next day the plates were washed 3 times with washing buffer, blocked for 1 h and washed again. Then, 100 µL of the diluted sample solution and standard solution were added to the prepared plates and incubated for 2 h at room temperature. Afterwards, the sample solutions were aspirated and the plates were washed again 3 times. The detection antibody was added and incubated for 2 h at room temperature with subsequent aspiration of the antibody and washing. After incubating the plates with 100 µL of the provided Streptavidin-HRP for 20 min and washing them again, the substrate solution was added and incubated for 20 min. To stop the reaction, we mixed 50 µL of Stop Solution into each well. The optical density of each well was analyzed using a plate reader at 450 nm (Infinite M200, Tecan, Männedorf, Switzerland).
4.7. Propidium Iodide (PI) Staining
A total of 20,000 neonatal C57/Bl6 microglial cells, 10,000 astrocytes and 5000 OLN93 oligodendrocytes were plated in different 96-well plates and were left to rest overnight. The next day, the cells were treated with 100 µM O-Vanillin for 24 or 48 h. Following the incubation period, the cells were washed with 200 µL PBS. Then, 100 µL complete growth medium containing 2.5 µg/mL PI (Life Technologies,) was added to stain the DNA of dead cells with permeable cell membranes. The cells were incubated for 15 min at room temperature and analyzed in a plate reader (Infinite M200, Tecan) at an excitation wavelength of 530 nm and a frequency of 25 flashes. The emission was analyzed at a wavelength of 645 nm. The fluorescence intensity of an untreated control group was defined as 100% living cells. As positive control 10 µL DMSO (Sigma-Aldrich) was added to every sample and the plate was analyzed again after 15 min. The assay was considered as working if the fluorescence intensity of incorporated PI increased at least twofold.
4.8. Proliferation Assay
To determine the number of proliferating cells, we analyzed the incorporation of blue fluorescent BrdU (Roche, Basel, Switzerland) into the DNA within 24 h. A total of 20,000 C57/Bl6 microglial cells were plated in a 96 well plate. Afterwards, the cells were treated with 100 µM O-Vanillin overnight. On the next morning, the medium was discarded and new complete growth medium containing fluorescent labeled BrdU and 100 µM O-Vanillin was added. The cells were incubated for another 24 h.
As a negative control we used dead cells after incubation in 10% DMSO for 24 h. As a positive control we used the well-known inductor of microglia proliferation, mMSCF (PeproTech, Rocky Hill, CT, USA) [
27]. The cells were analyzed in a plate reader (Infinite M200, Tecan). While mMCSF induced a significant increase in proliferation, the DMSO incubated cells showed no evidence for BrdU incorporation.
4.9. Alamar Blue Assay
The metabolism assay Alamar Blue (Alamar Blue Cell Viability Reagent, Thermo Fisher Scientific, Waltham, MA, USA) was applied for both primary murine neonatal microglia and murine GL 261 cells. Primary murine neonatal microglia were used directly after preparation from newborn C57BL/6 wildtype mice. Murine glioma GL261 cells were harvested from a 25 cm2 cell culture flask (Sarstedt). The cell number was adjusted to 10,000 cells/mL. Next, the cells were treated with different O-Vanillin concentrations and incubated for 3, 6 and 24 h. Fresh complete growth medium was added to the control wells. Finally, all media were replaced by 100 µL of fresh 10% Alamar Blue solution made by diluting Alamar Blue dye to fresh medium and the cells were incubated for further 3 h at 37 °C and 5% CO2. The Alamar Blue absorption spectrum in cells was measured using a plate reader (Infinite M200). The data was analysed according to the manufacturer´s protocol (Thermo Fisher Scientific, AlamarBlue Cell Viability Reagent).
4.10. RNA Isolation/qPCR
For all RNA dependent experiments up to 300,000 cells were plated in a 12-well plate (Sarstedt). The standard treatment regimen was 100 µM O-Vanillin for 6 h. Isolated human CD11B positive cells were treated for 12 h. Cells were washed in cold PBS (Life technologies) to remove redundant complete growth medium. The isolation was carried out using a Promega RNA Isolation Kit (ReliaPrep RNA Cell Miniprep System, Promega, Fitchburg, MA, USA) following manufacturer´s instructions. The isolated RNA was dissolved in 15 µLRNAse free water (Sigma-Aldrich) and stored at −20 °C. The quality and total amount of RNA was analyzed using a Nano Drop (NanoDrop8000 Spectrophotometer, Thermo Fisher Scientific, Waltham, MA, USA). RNA was transcribed into cDNA using a TaKaRa cDNA Synthesis Kit (Takara Bio INC., Kyoto, Japan) following manufacturer´s instructions. The cDNA was stored at −20 °C. qPCR was performed in 96 well real time pPCR fast plates (7500 Fast Real-Time PCR System, Life Technologies). A master mix containing 10 µL SYBR Green (Life Technologies), 8 µLRNAse free water and 1 ng of cDNA was added to each well. The forward and reverse primers (
Table S1) were added at a concentration of 10 pg/mL. Then the plate was covered with an adhesive film (Applied Biosystems, Thermo Fisher Scientific) and centrifuged at 1000 rcf for 1 min at 4 °C (Centrifuge 5417 R Eppendorf, Wesseling-Berzdorf, Germany). TBP (Tata box binding protein) was used as house-keeping gene [
28]. We provide the raw qPCR values in data suppliment.
4.11. Organotypic Brain Slices
All working steps were carried out after all used material was disinfected with 70% ethylalcohol (EtOH) for at least 30 min. The brain slices were obtained from 13- to 16-day-old C57/Bl6 wild type mice. The mice were sacrificed by cervical dislocation and decapitated, brains were carefully removed and immediately transferred to ice cold PBS. The cerebellum and olfactory bulbs were cut off. The brain was glued (UHU Sekundenkleber, UHU GmbH and Co. KG, Bühl, Germany) in an upward position with its caudal end to the cutting table of the vibratome (MicrotomHM 650 V, Thermo Scientific) and its ventral end to an already fixed 5% Agar-Agar block (KobeI, Roth, Karlsruhe, Germany). The cutting table was fixed in a cooled cutting chamber and covered with ice cold PBS. A vibratome was used to cut the brain into 250 µm slices. The slices were directly transferred with a glass pipette to ice cold Hanks’ Balanced Salt Solution (HBSS) (Thermo Fisher Scientific). Up to 3 brain slices were plated on one semi-permeable membrane insert (Falcon cell culture inserts, 0.4 µm pore size, Corning Life Sciences, Amsterdam, The Netherlands) in a 6-well plate filled with 1 mL complete growth medium. Red fluorescent GL261 glioma cells were grown confluent and harvested. The cells were further concentrated up to 10,000/µL and 1 µL drawn up into a Hamilton 1 µL precision Syringe (Microliter Syringe Model Model 701 RN SYR, Hamiltion Company, Bonaduz, Switzerland) with micro needle. The syringe was attached to a customized micromanipulator. Twenty-four hours after preparing the murine brain slices, the culture inserts were transferred to a working 6-well plate equipped with a micromanipulator. All working steps were performed at 37 °C in complete growth medium. The precision syringe was adjusted 250 µm above the culture insert membrane and the brain slices were positioned. Then, 10,000 mCherry GL261 cells were injected into the region of basal ganglia in a depth of 150 µm by slowly pushing the plunger. During injection the syringe was slowly moved upwards. The mCherry GL261 cells were injected over a range of 50 µm. After injections of red fluorescent GL261, the culture inserts were transferred to a new plate filled with culture medium containing different concentrations of O-Vanillin. The brain slices were incubated for 5 days. The medium was renewed every two days.
4.12. Fixation of Brain Slices
Brain slices were fixed with 4% paraformaldehyde (Thermo Fisher Scientific, Paraformaldehyde, Methanol-free) for 1 h and the cell nuclei were stained with blue fluorescent dye Hoechst (Thermo Fisher Scientific, Hoechst 33,342 Solution) for 30 min at room temperature. After the brain slices were carefully cut out from the insert membrane and transferred on microscopy slides, mounted with Aqua-Poly/Mount (Polysciences, Inc., Aqua-Poly/Mount) and covered with coverslip.
4.13. Microscopy
All micrographs were taken using a Zeiss LSM 710 confocal microscope (Carl Zeiss, Oberkochen, Germany) with a 20× objective. Injected mCherry GL261 gliomas were visualized by excitation wavelength 550 nm and emission wavelength 645 nm and further analyzed at the depth of maximal surface area.
4.14. Tumor Size Measurements
The tumour area and volume were calculated using Imaris 8 software (Version 9.5.0, Bitplane Inc., St. Paul, MN, USA). Tumor volumes of high-resolution Zeiss LSM 710 confocal microscopy stacks were 3D rendered by application of 1 µm object detail, 15 threshold background and 1000 tridimensional pixels (voxels). The surface objects obtained were unified in one single object, and volume mean values were extracted.
4.15. Statistical Analysis
All analyses were performed using Prism Graphpad (Version 8.4.1, GraphPad Software, San Diego, CA, USA) and Microsoft Excel 2016. Statistically significant differences were determined with the Student´s t-test for parametric testing. One-way ANOVA was used to compare multiple groups with Bonferroni post-hoc test. Significance was defined at p values < 0.05 (*), <0.01 (**) and <0.001 (***).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}




