Winnie-APCMin/+ Mice: A Spontaneous Model of Colitis-Associated Colorectal Cancer Combining Genetics and Inflammation

 ,
,  ,
,  , , ,
, , , 
 ,
,  , ,
, , 
Abstract
:
1. Introduction
2. Results
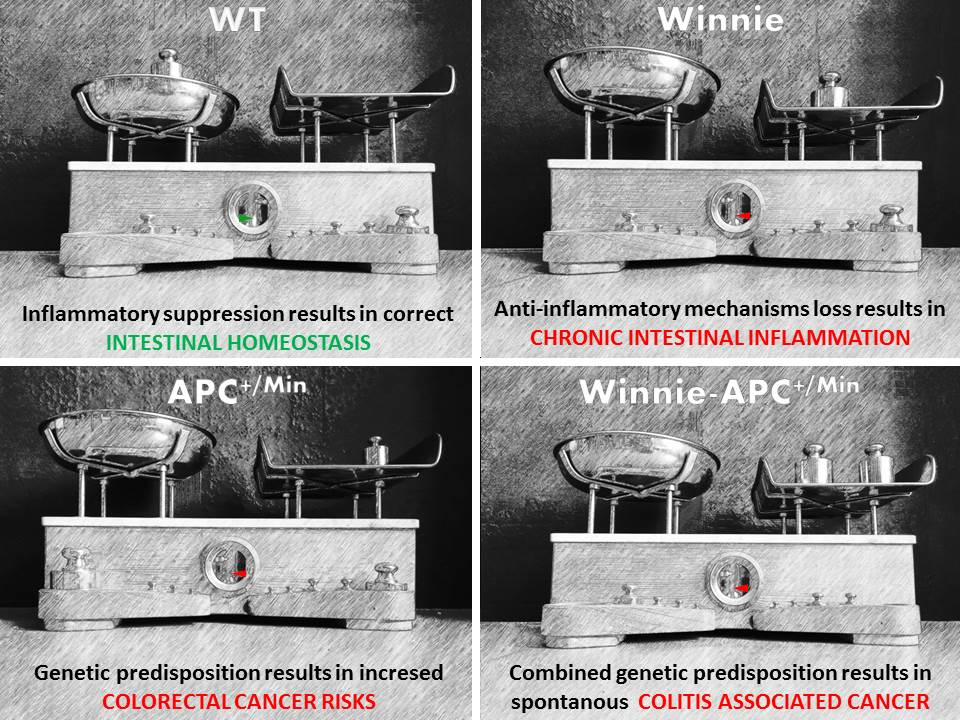
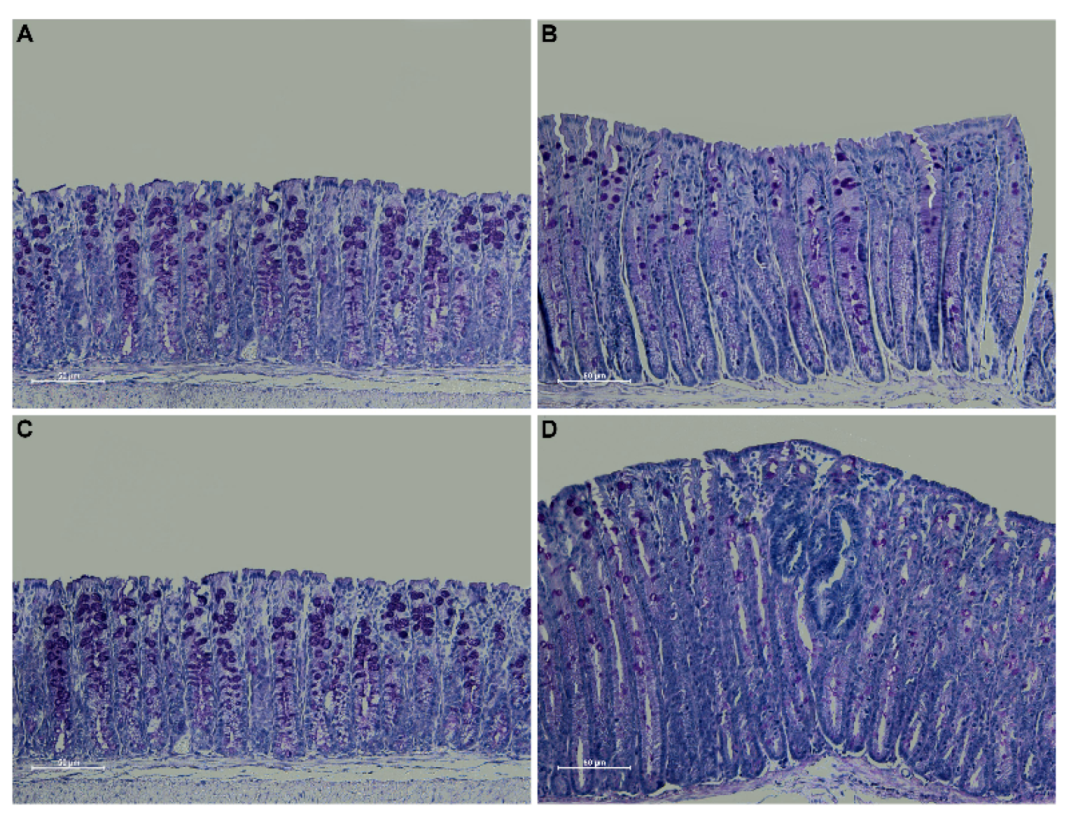
2.1. Creation and Characterization of the New Spontaneous Model of Colitis-Associated Colorectal Cancer: The Winnie-APCMin/+ Model
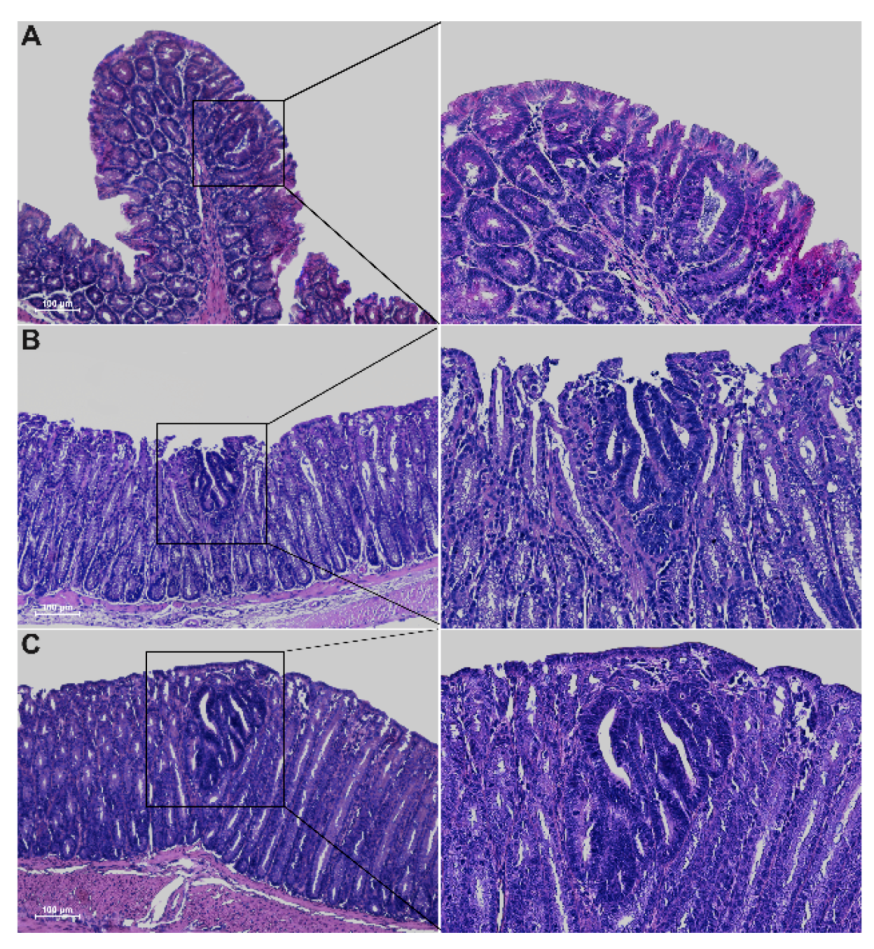
2.2. The Novel Winnie-ApcMin/+ -Mouse Strain Develops Colitis-Associated CRC Uniquely in the Colon with Early Appearance of Aggressive Dysplastic Lesions Compared to Parental Controls
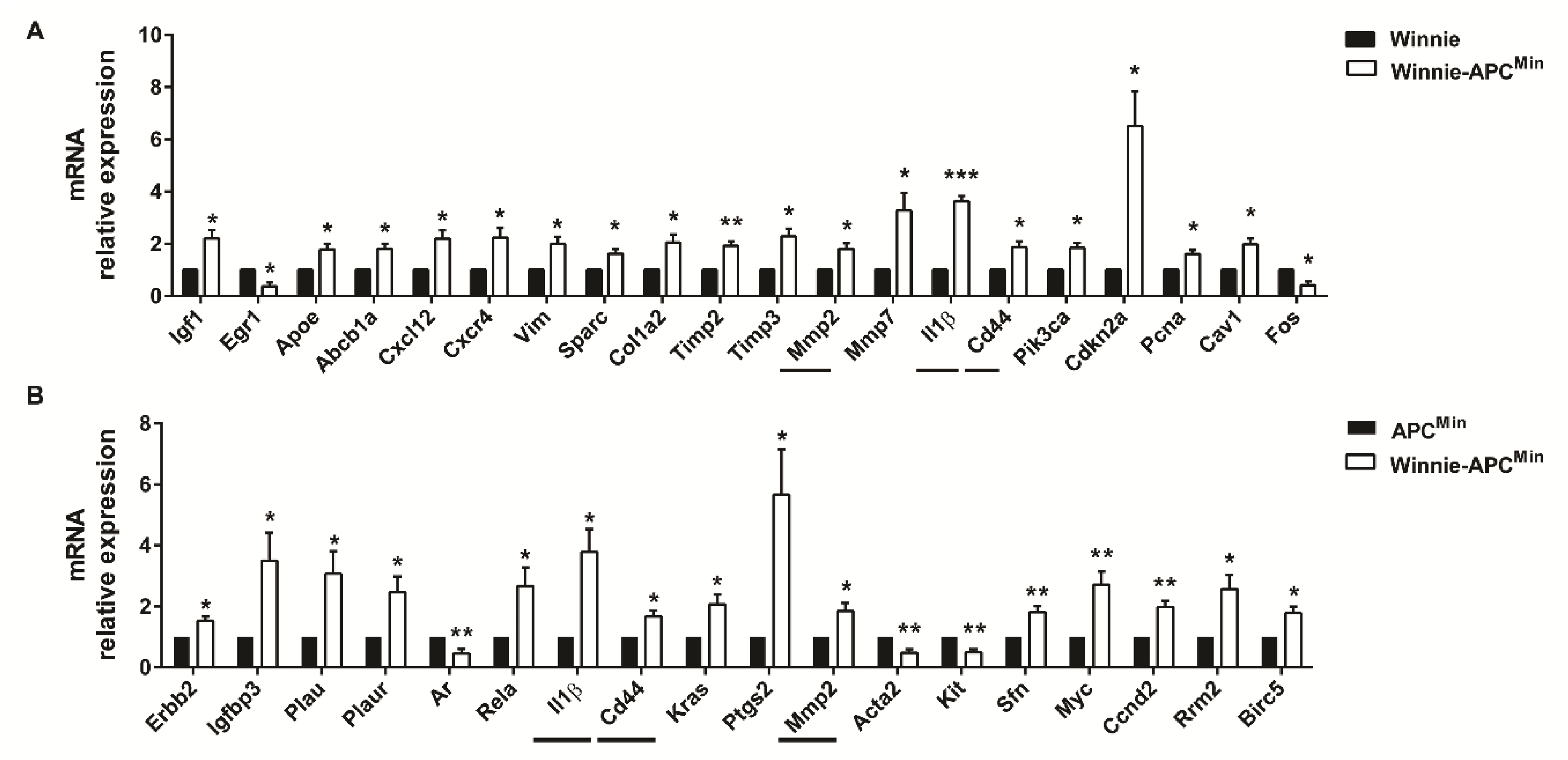
2.3. Gene Expression Profile from 5-Week-Old Winnie-ApcMin/+ Mice Shows Reduced Apoptosis and Increased Cellular Proliferation and Cytoskeletal Remodeling Patterns
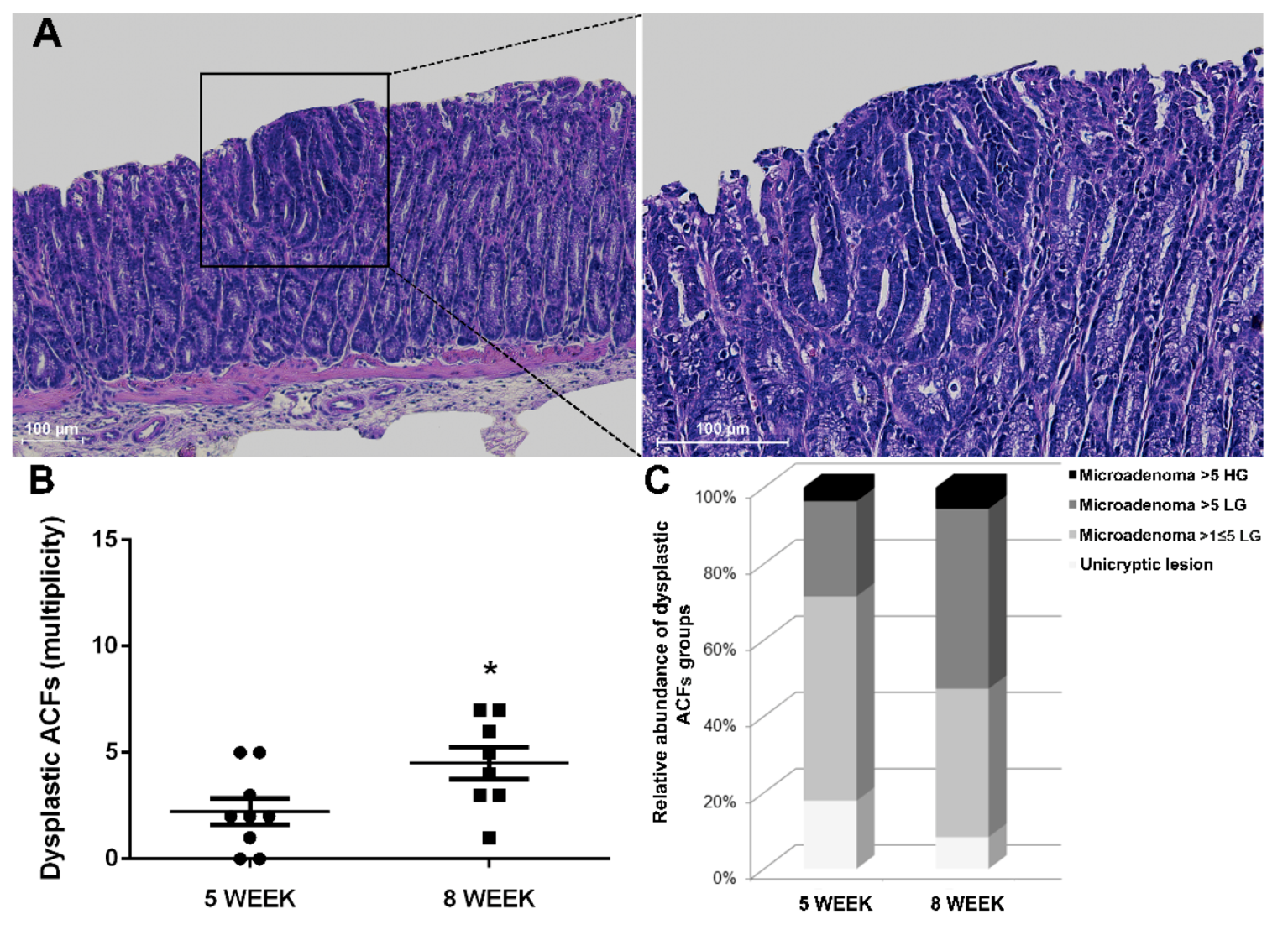
2.4. Winnie-ApcMin/+ Model Confirms the Multi-Step Nature of CRC Increasing the Multiplicity and Tumor Grading of Dysplastic ACFs in Older 8-Week-Old Mice
3. Discussion
4. Material and Methods
4.1. Mice
4.2. Histology
4.3. RNA Extraction from Colon Tissue and qPCR Analysis
4.4. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ACFs | Aberrant crypt foci |
| AOM | Azoxymethane |
| CAC | Colitis-associated cancer |
| CRC | Colorectal cancer |
| FAP | Familial adenomatous polyposis |
| HG | High-grade |
| IBD | Inflammatory bowel diseases |
| LG | Low-grade |
| PAS | Periodic acid Schiff |
| UC | Ulcerative colitis |
| WT | Wild-type |
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [Green Version]
- Balkwill, F.; Mantovani, A. Inflammation and cancer: Back to Virchow? Lancet 2001, 357, 539–545. [Google Scholar] [CrossRef]
- Coussens, L.M.; Werb, Z. Inflammation and cancer. Nature 2002, 420, 860–867. [Google Scholar] [CrossRef]
- Cuzick, J.; Otto, F.; Baron, J.A.; Brown, P.H.; Burn, J.; Greenwald, P.; Jankowski, J.; La Vecchia, C.; Meyskens, F.; Senn, H.J.; et al. Aspirin and non-steroidal anti-inflammatory drugs for cancer prevention: An international consensus statement. Lancet. Oncol. 2009, 10, 501–507. [Google Scholar] [CrossRef] [Green Version]
- Abraham, C.; Cho, J.H. Inflammatory bowel disease. N. Engl. J. Med. 2009, 361, 2066–2078. [Google Scholar] [CrossRef]
- Costa Cdos, S.; Rohden, F.; Hammes, T.O.; Margis, R.; Bortolotto, J.W.; Padoin, A.V.; Mottin, C.C.; Guaragna, R.M. Resveratrol upregulated SIRT1, FOXO1, and adiponectin and downregulated PPARγ1-3 mRNA expression in human visceral adipocytes. Obes. Surg. 2011, 21, 356–361. [Google Scholar] [CrossRef]
- Crohn, B.R.H. The sigmoidoscopic picture of chronic ulcerative colitis (non-specific). Am. J. Med. Sci. 1925, 170, 220–228. [Google Scholar] [CrossRef]
- Eaden, J.A.; Abrams, K.R.; Mayberry, J.F. The risk of colorectal cancer in ulcerative colitis: A meta-analysis. Gut 2001, 48, 526–535. [Google Scholar] [CrossRef] [Green Version]
- Jess, T.; Rungoe, C.; Peyrin-Biroulet, L. Risk of colorectal cancer in patients with ulcerative colitis: A meta-analysis of population-based cohort studies. Clin. Gastroenterol. Hepatol. Off. Clin. Pract. J. Am. Gastroenterol. Assoc. 2012, 10, 639–645. [Google Scholar] [CrossRef]
- Jess, T.; Simonsen, J.; Jorgensen, K.T.; Pedersen, B.V.; Nielsen, N.M.; Frisch, M. Decreasing risk of colorectal cancer in patients with inflammatory bowel disease over 30 years. Gastroenterology 2012, 143, 375–381.e371, quiz e313-374. [Google Scholar] [CrossRef]
- Peneau, A.; Savoye, G.; Turck, D.; Dauchet, L.; Fumery, M.; Salleron, J.; Lerebours, E.; Ligier, K.; Vasseur, F.; Dupas, J.L.; et al. Mortality and cancer in pediatric-onset inflammatory bowel disease: A population-based study. Am. J. Gastroenterol. 2013, 108, 1647–1653. [Google Scholar] [CrossRef] [PubMed]
- Su, L.K.; Kinzler, K.W.; Vogelstein, B.; Preisinger, A.C.; Moser, A.R.; Luongo, C.; Gould, K.A.; Dove, W.F. Multiple intestinal neoplasia caused by a mutation in the murine homolog of the APC gene. Science 1992, 256, 668–670. [Google Scholar] [CrossRef] [PubMed]
- Heazlewood, C.K.; Cook, M.C.; Eri, R.; Price, G.R.; Tauro, S.B.; Taupin, D.; Thornton, D.J.; Png, C.W.; Crockford, T.L.; Cornall, R.J.; et al. Aberrant mucin assembly in mice causes endoplasmic reticulum stress and spontaneous inflammation resembling ulcerative colitis. Plos Med. 2008, 5, e54. [Google Scholar] [CrossRef] [Green Version]
- Velcich, A.; Yang, W.; Heyer, J.; Fragale, A.; Nicholas, C.; Viani, S.; Kucherlapati, R.; Lipkin, M.; Yang, K.; Augenlicht, L. Colorectal cancer in mice genetically deficient in the mucin Muc2. Science 2002, 295, 1726–1729. [Google Scholar] [CrossRef]
- Eri, R.D.; Adams, R.J.; Tran, T.V.; Tong, H.; Das, I.; Roche, D.K.; Oancea, I.; Png, C.W.; Jeffery, P.L.; Radford-Smith, G.L.; et al. An intestinal epithelial defect conferring ER stress results in inflammation involving both innate and adaptive immunity. Mucosal Immunol. 2011, 4, 354–364. [Google Scholar] [CrossRef]
- Femia, A.P.; Giannini, A.; Fazi, M.; Tarquini, E.; Salvadori, M.; Roncucci, L.; Tonelli, F.; Dolara, P.; Caderni, G. Identification of mucin depleted foci in the human colon. Cancer Prev. Res. (Phila. Pa.) 2008, 1, 562–567. [Google Scholar] [CrossRef] [Green Version]
- Higashi, D.; Futami, K.; Ishibashi, Y.; Egawa, Y.; Maekawa, T.; Matsui, T.; Iwashita, A.; Kuroki, M. Clinical course of colorectal cancer in patients with ulcerative colitis. Anticancer Res. 2011, 31, 2499–2504. [Google Scholar]
- Nasiri, S.; Kuenzig, M.E.; Benchimol, E.I. Long-term outcomes of pediatric inflammatory bowel disease. Semin. Pediatric Surg. 2017, 26, 398–404. [Google Scholar] [CrossRef]
- Rosenberg, D.W.; Giardina, C.; Tanaka, T. Mouse models for the study of colon carcinogenesis. Carcinogenesis 2009, 30, 183–196. [Google Scholar] [CrossRef] [Green Version]
- Okayasu, I.; Ohkusa, T.; Kajiura, K.; Kanno, J.; Sakamoto, S. Promotion of colorectal neoplasia in experimental murine ulcerative colitis. Gut 1996, 39, 87–92. [Google Scholar] [CrossRef] [Green Version]
- Jackstadt, R.; Sansom, O.J. Mouse models of intestinal cancer. J. Pathol. 2016, 238, 141–151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Santis, S.; Kunde, D.; Galleggiante, V.; Liso, M.; Scandiffio, L.; Serino, G.; Pinto, A.; Campiglia, P.; Sorrentino, R.; Cavalcanti, E.; et al. TNFα deficiency results in increased IL-1β in an early onset of spontaneous murine colitis. Cell Death Dis. 2017, 8, e2993. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dicarlo, M.; Teti, G.; Verna, G.; Liso, M.; Cavalcanti, E.; Sila, A.; Raveenthiraraj, S.; Mastronardi, M.; Santino, A.; Serino, G.; et al. Quercetin Exposure Suppresses the Inflammatory Pathway in Intestinal Organoids from Winnie Mice. Int. J. Mol. Sci. 2019, 20, 5771. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liso, M.; De Santis, S.; Verna, G.; Dicarlo, M.; Calasso, M.; Santino, A.; Gigante, I.; Eri, R.; Raveenthiraraj, S.; Sobolewski, A.; et al. A Specific Mutation in Muc2 Determines Early Dysbiosis in Colitis-Prone Winnie Mice. Inflamm. Bowel Dis. 2019, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Wilson, R.; Gundamaraju, R.; Vemuri, R.; Angelucci, C.; Geraghty, D.; Gueven, N.; Eri, R.D. Identification of Key Pro-Survival Proteins in Isolated Colonic Goblet Cells of Winnie, a Murine Model of Spontaneous Colitis. Inflamm Bowel Dis 2020, 26, 80–92. [Google Scholar] [CrossRef]
- Gundamaraju, R.; Vemuri, R.; Chong, W.C.; Myers, S.; Norouzi, S.; Shastri, M.D.; Eri, R. Interplay between Endoplasmic Reticular Stress and Survivin in Colonic Epithelial Cells. Cells 2018, 7, 171. [Google Scholar] [CrossRef] [Green Version]
- Witkiewicz, A.K.; Knudsen, K.E.; Dicker, A.P.; Knudsen, E.S. The meaning of p16(ink4a) expression in tumors: Functional significance, clinical associations and future developments. Cell Cycle (Georget. Tex.) 2011, 10, 2497–2503. [Google Scholar] [CrossRef] [Green Version]
- Gavert, N.; Shvab, A.; Sheffer, M.; Ben-Shmuel, A.; Haase, G.; Bakos, E.; Domany, E.; Ben-Ze’ev, A. c-Kit is suppressed in human colon cancer tissue and contributes to L1-mediated metastasis. Cancer Res. 2013, 73, 5754–5763. [Google Scholar] [CrossRef] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 5-WEEK | Non-Dysplastic ACFs | Dysplastic ACFs | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| All Groups | Unicryptic Lesion | Microadenoma >1 ≤ 5-LG | Microadenoma >5-LG | Microadenoma >5-HG | ||||||||
| Genotype (nr. mice) | Incidence (%) | Multiplicity (mean ± SEM) | Incidence (%) | Multiplicity (mean ± SEM) | Incidence (%) | Multiplicity (mean ± SEM) | Incidence (%) | Multiplicity (mean ± SEM) | Incidence (%) | Multiplicity (mean ± SEM) | Incidence (%) | Multiplicity (mean ± SEM) |
| Proximal Colon | ||||||||||||
| WT (8) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Winnie (8) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| APCMin/+ (10) | 20 | 0.2 ± 0.13 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Winnie-APCMin/+ (9) | 0 | 0 | 11.1 | 0.1 ± 0.1 | 100 | 0.1 ± 0.1 | 0 | 0 | 0 | 0 | 0 | 0 |
| Medial Colon | ||||||||||||
| WT (8) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Winnie (8) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| APCMin/+ (10) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Winnie-APCMin/+ (9) | 10 | 0.2 ± 0.2 | 66.7 | 1.1 ± 0.4 | 10 | 0.1 ± 0.1 | 70 | 0.78 ± 0.28 | 20 | 0.22 ± 0.15 | 0 | 0 |
| Distal Colon | ||||||||||||
| WT (8) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Winnie (8) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| APCMin/+ (10) | 0 | 0 | 30 | 0.3 ± 0.15 | 33.3 | 0.1 ± 0.1 | 66.7 | 0.2 ± 0.13 | 0 | 0 | 0 | 0 |
| Winnie-APCMin/+ (9) | 0 | 0 | 77.8 | 2.2 ± 0.62 | 20 | 0.44 ± 0.24 | 50 | 1.1 ± 0.6 | 25 | 0.6 ± 0.24 | 5 | 0.1 ± 0.1 |
| 8-WEEK | Non-Dysplastic ACFs | Dysplastic ACFs | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| All Groups | Unicryptic Lesion | Microadenoma >1 ≤ 5-LG | Microadenoma >5-LG | Microadenoma >5-HG | ||||||||
| Genotype (nr. mice) | Incidence (%) | Multiplicity (mean ± SEM) | Incidence (%) | Multiplicity (mean ± SEM) | Incidence (%) | Multiplicity (mean ± SEM) | Incidence (%) | Multiplicity (mean ± SEM) | Incidence (%) | Multiplicity (mean ± SEM) | Incidence (%) | Multiplicity (mean ± SEM) |
| Proximal Colon | ||||||||||||
| WT (8) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Winnie (8) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| APCMin/+ (8) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Winnie-APCMin/+ (8) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Medial Colon | ||||||||||||
| WT (8) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Winnie (8) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| APCMin/+ (8) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Winnie-APCMin/+ (8) | 0 | 0 | 70 | 1.4 ± 0.47 | 13.3 | 0.2 ± 0.13 | 40 | 0.55 ± 0.26 | 26.7 | 0.4 ± 0.2 | 20 | 0.3 ± 0.2 |
| Distal Colon | ||||||||||||
| WT (8) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Winnie (8) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| APCMin/+ (8) | 0 | 0 | 37.5 | 0.38 ± 0.18 | 0 | 0 | 33.4 | 0.13 ± 0.13 | 33.3 | 0.13 ± 0.13 | 33.3 | 0.13 ± 0.13 |
| Winnie-APCMin/+ (8) | 0 | 0 | 100 | 4.8 ± 0.77 | 10.4 | 0.5 ± 0.2 | 43.8 | 2.1 ± 0.4 | 41.7 | 2 ± 0.6 | 4.2 | 0.2 ± 0.13 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
De Santis, S.; Verna, G.; Serino, G.; Armentano, R.; Cavalcanti, E.; Liso, M.; Dicarlo, M.; Coletta, S.; Mastronardi, M.; Lippolis, A.; et al. Winnie-APCMin/+ Mice: A Spontaneous Model of Colitis-Associated Colorectal Cancer Combining Genetics and Inflammation. Int. J. Mol. Sci. 2020, 21, 2972. https://doi.org/10.3390/ijms21082972
De Santis S, Verna G, Serino G, Armentano R, Cavalcanti E, Liso M, Dicarlo M, Coletta S, Mastronardi M, Lippolis A, et al. Winnie-APCMin/+ Mice: A Spontaneous Model of Colitis-Associated Colorectal Cancer Combining Genetics and Inflammation. International Journal of Molecular Sciences. 2020; 21(8):2972. https://doi.org/10.3390/ijms21082972
Chicago/Turabian StyleDe Santis, Stefania, Giulio Verna, Grazia Serino, Raffaele Armentano, Elisabetta Cavalcanti, Marina Liso, Manuela Dicarlo, Sergio Coletta, Mauro Mastronardi, Antonio Lippolis, and et al. 2020. "Winnie-APCMin/+ Mice: A Spontaneous Model of Colitis-Associated Colorectal Cancer Combining Genetics and Inflammation" International Journal of Molecular Sciences 21, no. 8: 2972. https://doi.org/10.3390/ijms21082972
APA StyleDe Santis, S., Verna, G., Serino, G., Armentano, R., Cavalcanti, E., Liso, M., Dicarlo, M., Coletta, S., Mastronardi, M., Lippolis, A., Tafaro, A., Santino, A., Pinto, A., Campiglia, P., Huang, A. Y., Cominelli, F., Pizarro, T. T., & Chieppa, M. (2020). Winnie-APCMin/+ Mice: A Spontaneous Model of Colitis-Associated Colorectal Cancer Combining Genetics and Inflammation. International Journal of Molecular Sciences, 21(8), 2972. https://doi.org/10.3390/ijms21082972