Arctigenin Enhances the Cytotoxic Effect of Doxorubicin in MDA-MB-231 Breast Cancer Cells


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
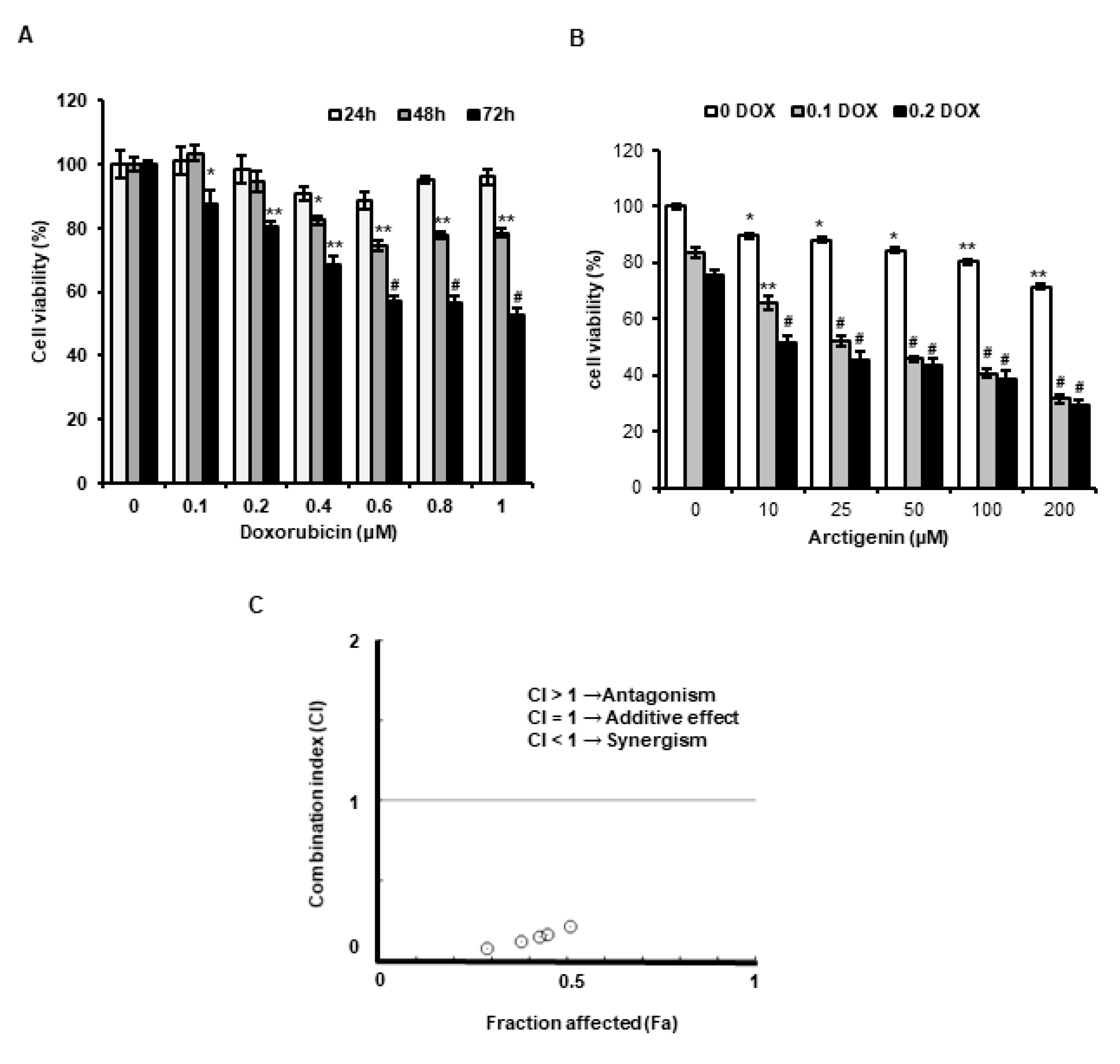
2.1. ATG Enhanced DOX-Induced MDA-MB-231 Cell Death
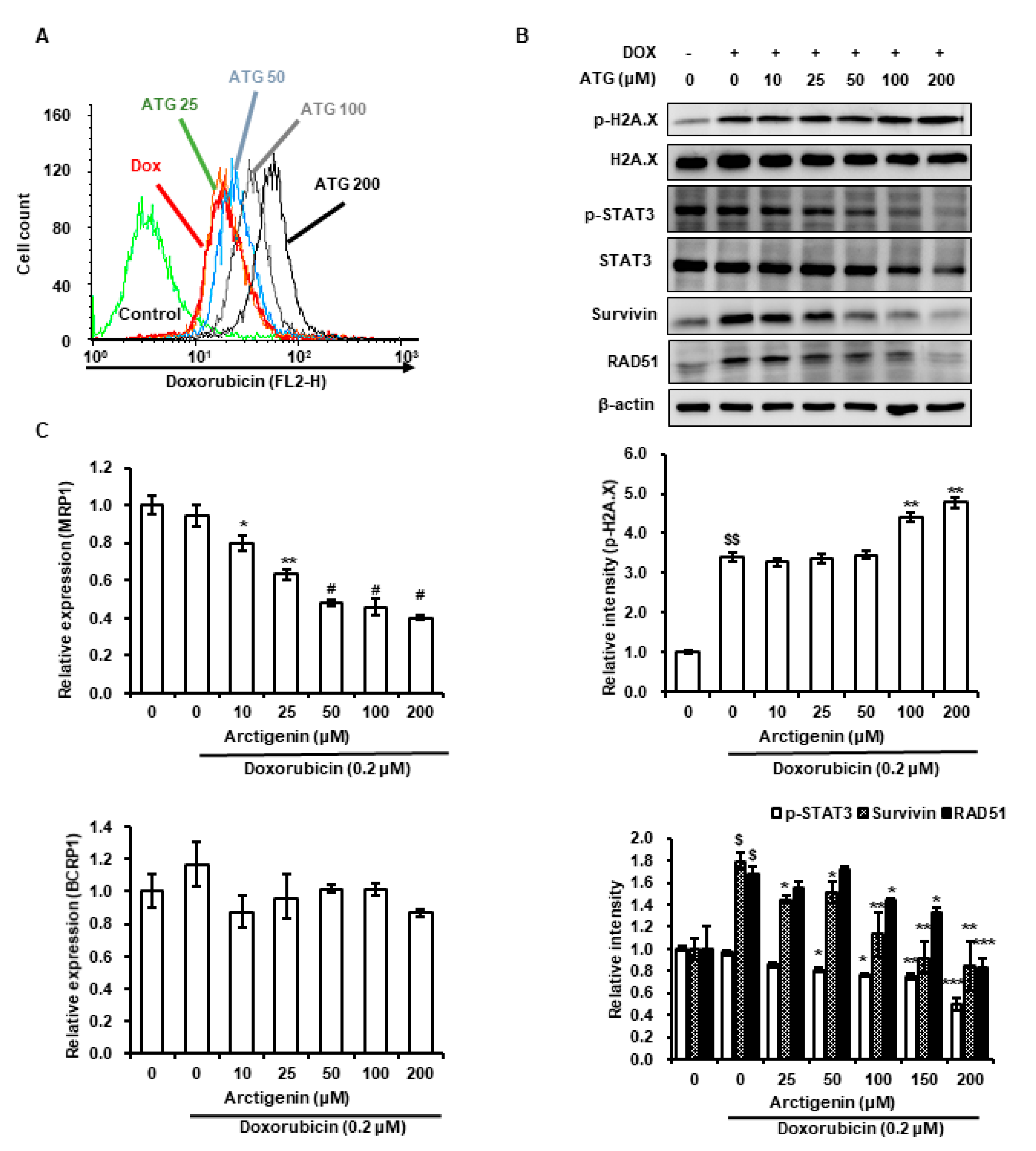
2.2. DOX Uptake by MDA-MB-231 Cells Was Increased by ATG
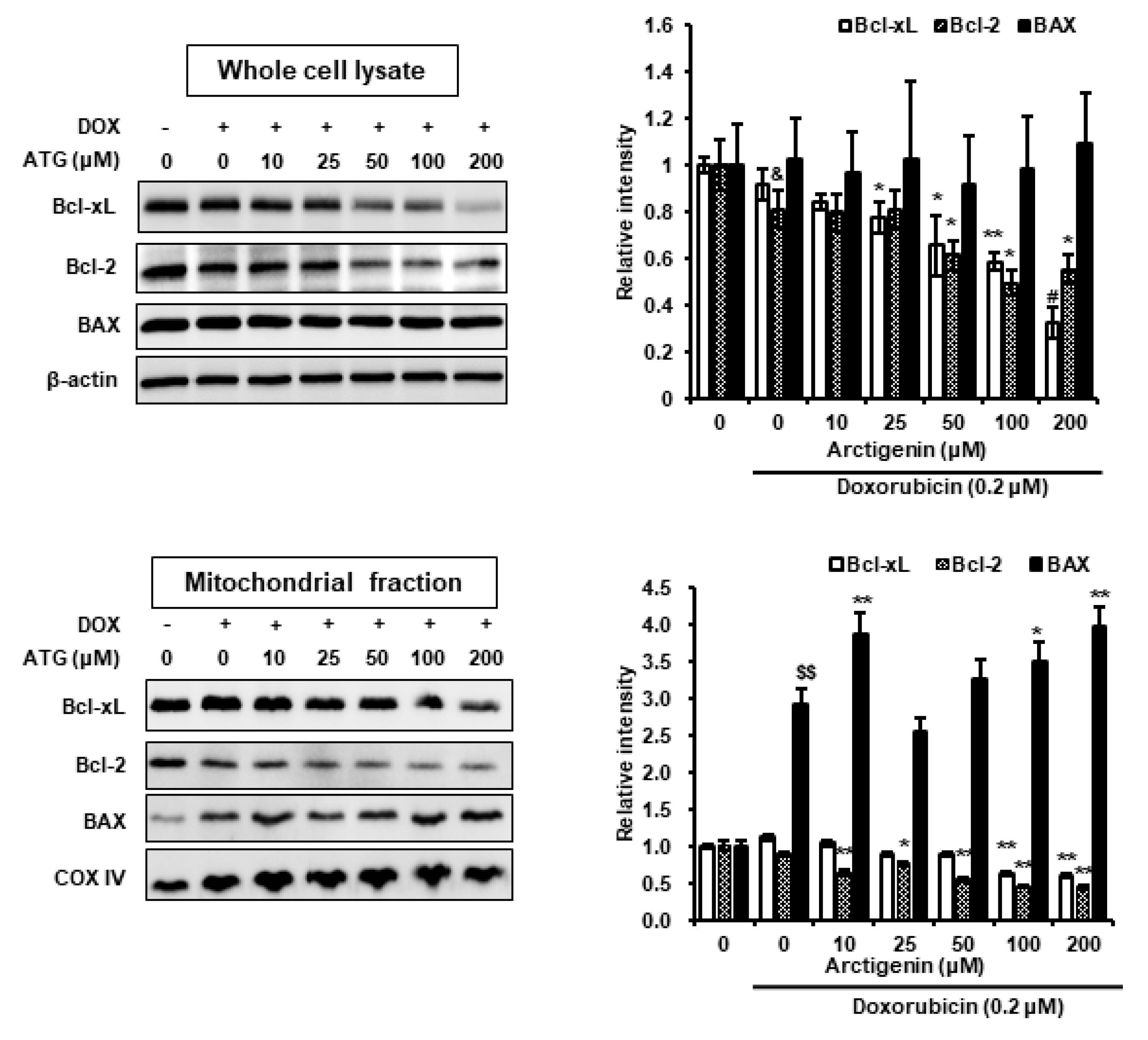
2.3. Cell Death by ATG/DOX Co-Treatment Was Associated with Down-Regulations in Bcl-2 and Bcl-xL and Increases in BAX Levels in Mitochondria
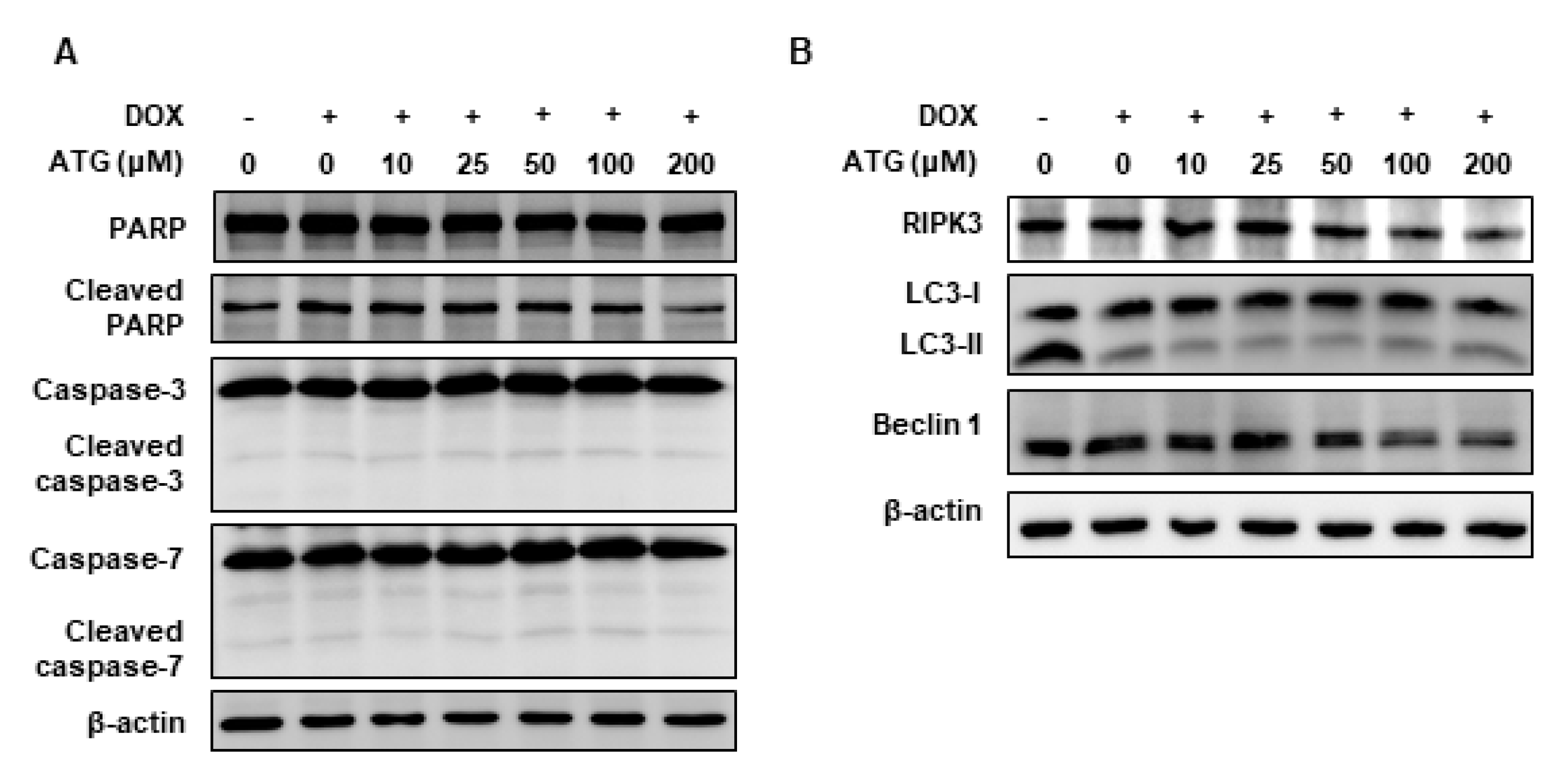
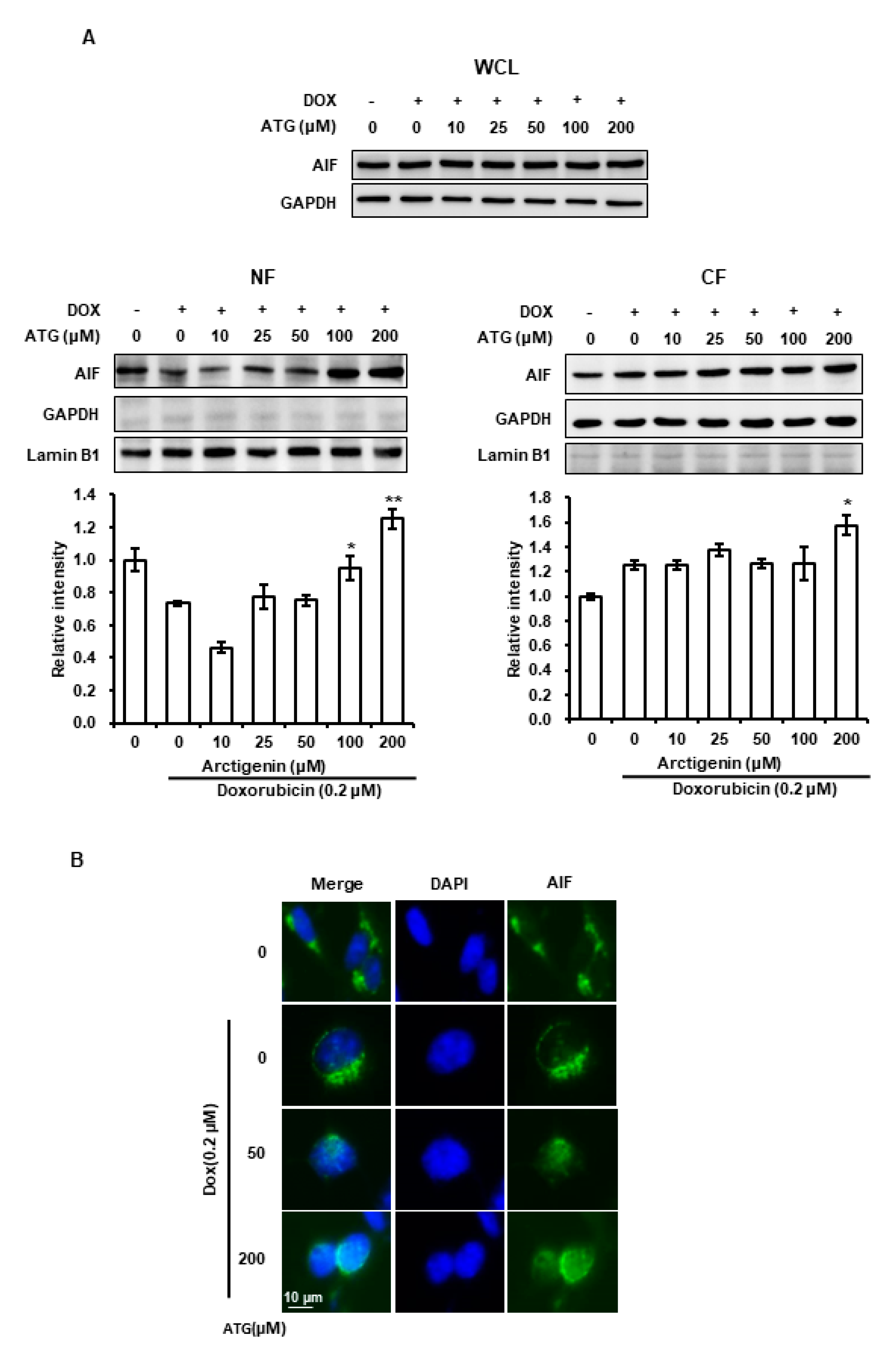
2.4. MDA-MB-231 Cell Death by ATG/DOX Co-Treatment Was Induced AIF-Dependently
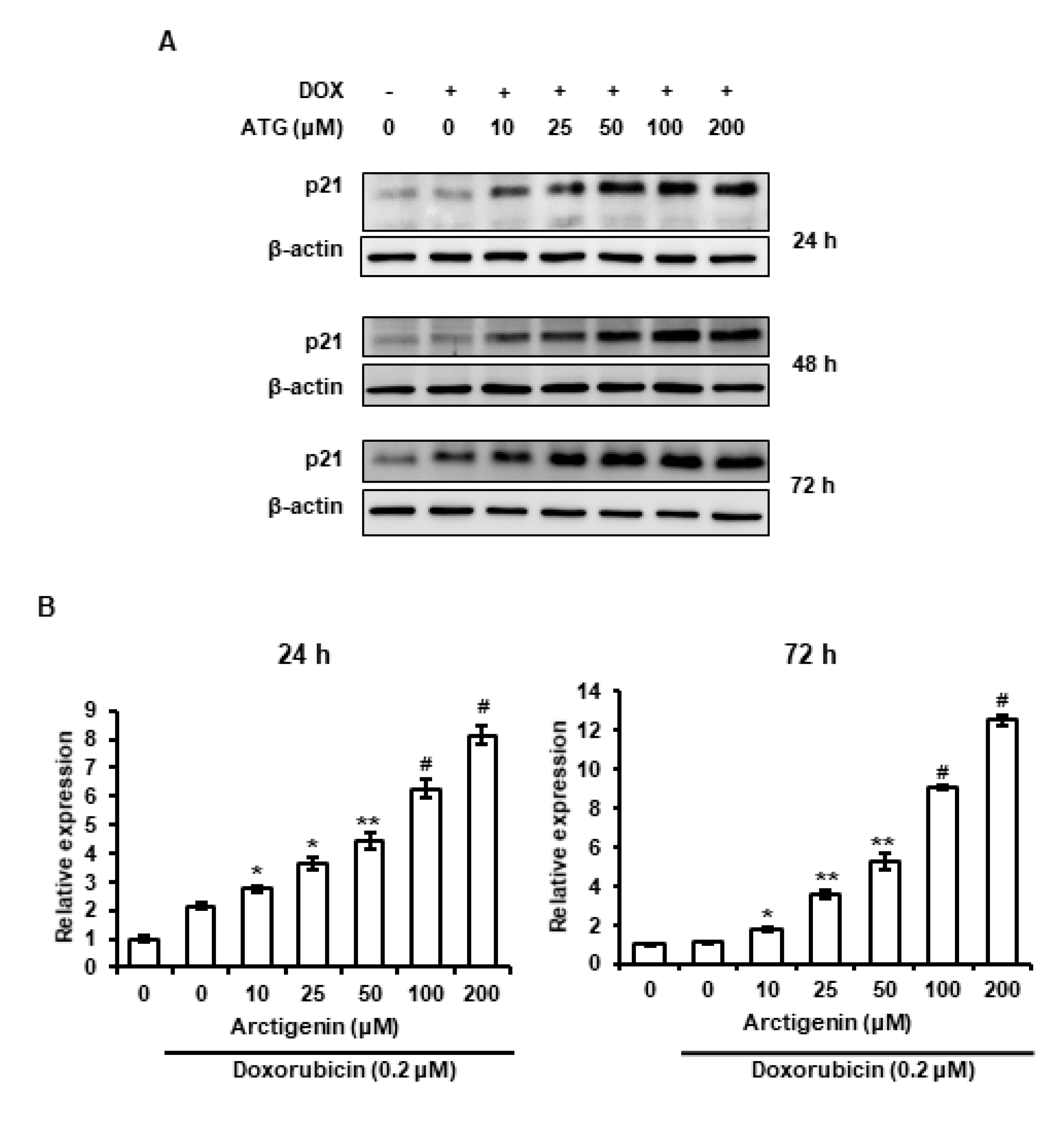
2.5. Sustained Increase of p21 Was Associated with ATG/DOX Co-Treatment-Induced Cell Death
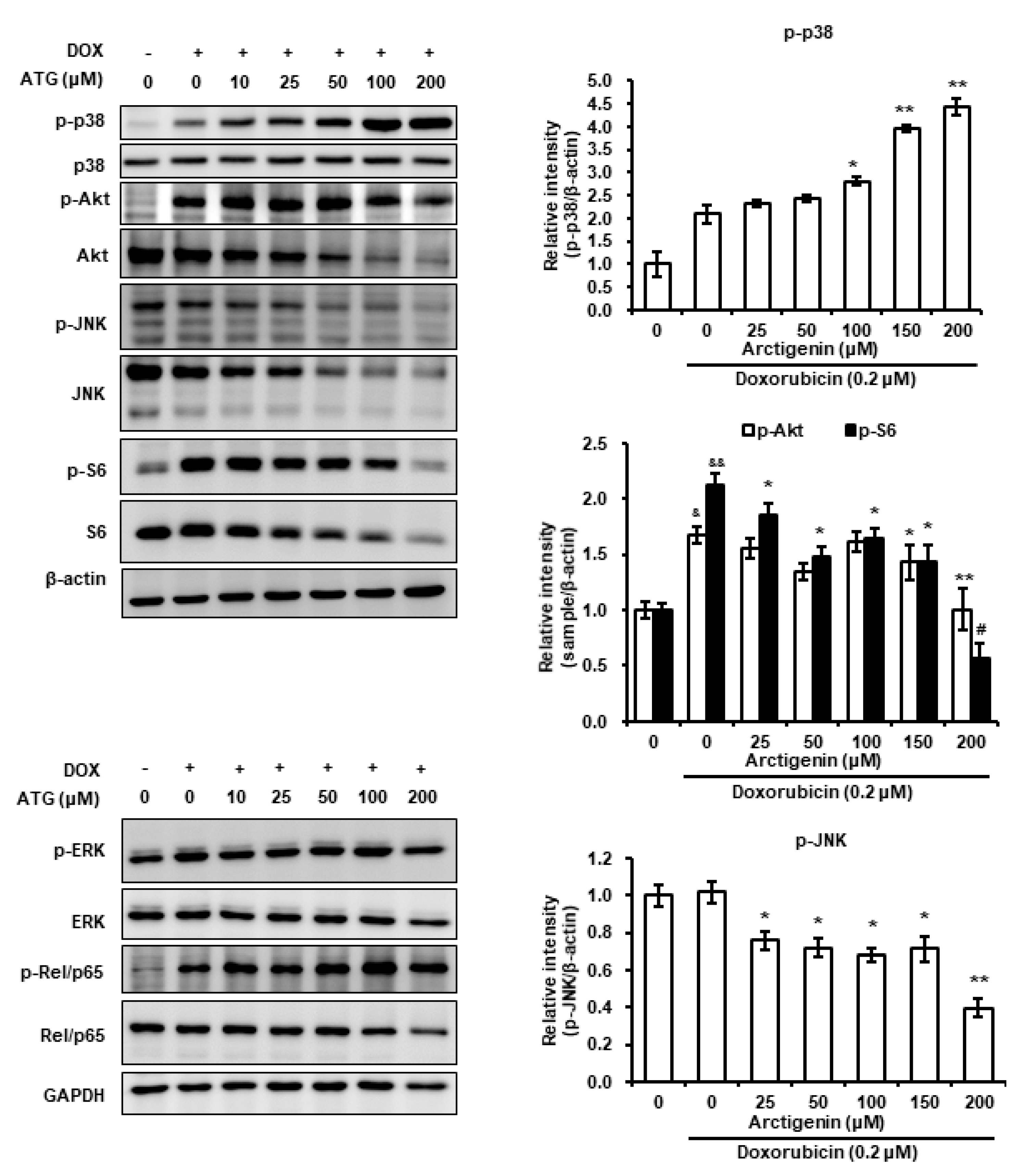
2.6. Involvement of p38 MAPK Phosphorylation in ATG/DOX Induced Cell Death
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Cell Culture
4.3. Cell Viability Analysis
4.4. Doxorubicin Uptake Assay
4.5. Determining Combined Drug Interactions
4.6. Quantitative Real-Time Polymerase Chain Reaction
4.7. Mitochondrial Fractionation
4.8. Nuclear Fractionation
4.9. Western Blotting
4.10. Fluorescence Immunocytochemistry
4.11. Statistical Analysis
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AIF | Apoptosis-inducing factor |
| ATG | Arctigenin |
| BAX | Bcl-2-associated X protein |
| Bcl-2 | B-cell lymphoma 2 |
| Bcl-xL | B-cell lymphoma-extra large |
| BCRP | Breast cancer resistance protein |
| CI-Fa | Combination index-Fraction affected |
| COX IV | Cytochrome c oxidase subunit 4 isoform 1 |
| Dox | Doxorubicin |
| ERK | Extracellular signal-regulated kinase 1/2 |
| GAPDH | Glyceraldehyde-3-phosphate dehydrogenase |
| H2A.X | H2A histone family member X |
| HER2 | Human epidermal growth factor receptor 2 |
| HR | Hormone receptor |
| JNK | C-Jun N-terminal kinase |
| LC3 | Microtubule-associated protein 1A/1B-light chain 3 |
| MRP1 | Multidrug resistance-associated protein 1 |
| mTOR | Mammalian target of rapamycin |
| NF-κB | Nuclear factor kappa-light-chain-enhancer of activated B cells |
| NSCLC | Non-small cell lung cancer |
| PARP | Poly (ADP-ribose) polymerase |
| RIPK3 | Receptor-interacting serine/threonine-protein kinase 3 |
| ROS | Reactive oxygen species |
| S6 | Ribosomal protein 6 |
| STAT3 | Signal transducer and activator of transcription 3 |
| TNBC | Triple-negative breast cancer |
References
- Prat, A.; Pineda, E.; Adamo, B.; Galvan, P.; Fernandez, A.; Gaba, L.; Diez, M.; Viladot, M.; Arance, A.; Munoz, M. Clinical implications of the intrinsic molecular subtypes of breast cancer. Breast 2015, 24, S26–S35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reddy, S.M.; Barcenas, C.H.; Sinha, A.K.; Hsu, L.; Moulder, S.L.; Tripathy, D.; Hortobagyi, G.N.; Valero, V. Long-term survival outcomes of triple-receptor negative breast cancer survivors who are disease free at 5 years and relationship with low hormone receptor positivity. Br. J. Cancer 2018, 118, 17–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liedtke, C.; Mazouni, C.; Hess, K.R.; Andre, F.; Tordai, A.; Mejia, J.A.; Symmans, W.F.; Gonzalez-Angulo, A.M.; Hennessy, B.; Green, M.; et al. Response to neoadjuvant therapy and long-term survival in patients with triple-negative breast cancer. J. Clin. Oncol. 2008, 26, 1275–1281. [Google Scholar] [CrossRef] [PubMed]
- Lin, N.U.; Vanderplas, A.; Hughes, M.E.; Theriault, R.L.; Edge, S.B.; Wong, Y.N.; Blayney, D.W.; Niland, J.C.; Winer, E.P.; Weeks, J.C. Clinicopathologic Features, Patterns of Recurrence, and Survival Among Women With Triple-Negative Breast Cancer in the National Comprehensive Cancer Network. Cancer 2012, 118, 5463–5472. [Google Scholar] [CrossRef] [Green Version]
- Collignon, J.; Lousberg, L.; Schroeder, H.; Jerusalem, G. Triple-negative breast cancer: Treatment challenges and solutions. Breast Cancer-Targets Ther. 2016, 8, 93–107. [Google Scholar] [CrossRef] [Green Version]
- Lee, D.H.; Kim, S.; Nam, K.S. Protective effects of deep sea water against doxorubicin-induced cardiotoxicity in H9c2 cardiac muscle cells. Int. J. Oncol. 2014, 45, 2569–2575. [Google Scholar] [CrossRef] [Green Version]
- Minotti, G.; Menna, P.; Salvatorelli, E.; Cairo, G.; Gianni, L. Anthracyclines: Molecular advances and pharmacologic developments in antitumor activity and cardiotoxicity. Pharmacol. Rev. 2004, 56, 185–229. [Google Scholar] [CrossRef] [Green Version]
- Carvalho, C.; Santos, R.X.; Cardoso, S.; Correia, S.; Oliveira, P.J.; Santos, M.S.; Moreira, P.I. Doxorubicin: The good, the bad and the ugly effect. Curr. Med. Chem. 2009, 16, 3267–3285. [Google Scholar] [CrossRef]
- Hayashi, K.; Narutaki, K.; Nagaoka, Y.; Hayashi, T.; Uesato, S. Therapeutic Effect of Arctiin and Arctigenin in Immunocompetent and Immunocompromised Mice Infected with Influenza A Virus. Biol. Pharm. Bull. 2010, 33, 1199–1205. [Google Scholar] [CrossRef] [Green Version]
- Swarup, V.; Ghosh, J.; Mishra, M.K.; Basu, A. Novel strategy for treatment of Japanese encephalitis using arctigenin, a plant lignan. J. Antimicrob. Chemother. 2008, 61, 679–688. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.; Hollenbaugh, J.A.; Kim, D.H.; Kim, B. Novel PI3K/Akt Inhibitors Screened by the Cytoprotective Function of Human Immunodeficiency Virus Type 1 Tat. PLoS ONE 2011, 6, e21781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, H.S.; Lee, J.Y.; Kim, C.J. Anti-inflammatory activity of arctigenin from Forsythiae Fructus. J. Ethnopharmacol. 2008, 116, 305–312. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.Y.; Kim, C.J. Arctigenin, a Phenylpropanoid Dibenzylbutyrolactone Lignan, Inhibits Type I-IV Allergic Inflammation and Pro-inflammatory Enzymes. Arch. Pharmacal Res. 2010, 33, 947–957. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.C.; Liang, Y.; Tian, Y.; Hu, G.R. Arctigenin induces apoptosis in colon cancer cells through ROS/p38MAPK pathway. J. BUON 2016, 21, 87–94. [Google Scholar]
- Hsieh, C.J.; Kuo, P.L.; Hsu, Y.C.; Huang, Y.F.; Tsai, E.M.; Hsu, Y.L. Arctigenin, a dietary phytoestrogen, induces apoptosis of estrogen receptor-negative breast cancer cells through the ROS/p38 MAPK pathway and epigenetic regulation. Free Radic. Biol. Med. 2014, 67, 159–170. [Google Scholar] [CrossRef]
- Yang, S.C.; Ma, J.; Xiao, J.B.; Lv, X.H.; Li, X.L.; Yang, H.K.; Liu, Y.; Feng, S.J.; Zhang, Y.F. Arctigenin Anti-Tumor Activity in Bladder Cancer T24 Cell Line Through Induction of Cell-Cycle Arrest and Apoptosis. Anat. Rec. Adv. Integr. Anat. Evol. Biol. 2012, 295, 1260–1266. [Google Scholar] [CrossRef]
- Jiang, X.X.; Zeng, L.P.; Huang, J.F.; Zhou, H.; Liu, Y.B. Arctigenin, a Natural Lignan Compound, Induces Apoptotic Death of Hepatocellular Carcinoma Cells via Suppression of PI3-K/Akt Signaling. J. Biochem. Mol. Toxicol. 2015, 29, 458–464. [Google Scholar] [CrossRef]
- Jeong, J.B.; Hong, S.C.; Jeong, H.J.; Koo, J.S. Arctigenin induces cell cycle arrest by blocking the phosphorylation of Rb via the modulation of cell cycle regulatory proteins in human gastric cancer cells. Int. Immunopharmacol. 2011, 11, 1573–1577. [Google Scholar] [CrossRef]
- Maxwell, T.; Lee, K.S.; Kim, S.; Nam, K.S. Arctigenin inhibits the activation of the mTOR pathway, resulting in autophagic cell death and decreased ER expression in ER-positive human breast cancer cells. Int. J. Oncol. 2018, 52, 1339–1349. [Google Scholar] [CrossRef] [Green Version]
- Maxwell, T.; Chun, S.Y.; Lee, K.S.; Kim, S.; Nam, K.S. The anti-metastatic effects of the phytoestrogen arctigenin on human breast cancer cell lines regardless of the status of ER expression. Int. J. Oncol. 2017, 50, 727–735. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.Q.; Jin, J.J.; Wang, J. Arctigenin Enhances Chemosensitivity to Cisplatin in Human Nonsmall Lung Cancer H460 Cells through Downregulation of Survivin Expression. J. Biochem. Mol. Toxicol. 2014, 28, 39–45. [Google Scholar] [CrossRef]
- Mayer, L.D.; Janoff, A.S. Optimizing combination chemotherapy by controlling drug ratios. Mol. Interv. 2007, 7, 216–223. [Google Scholar] [CrossRef]
- Zoli, W.; Ricotti, L.; Tesei, A.; Barzanti, F.; Amadori, D. In vitro preclinical models for a rational design of chemotherapy combinations in human tumors. Crit. Rev. Oncol. Hematol. 2001, 37, 69–82. [Google Scholar] [CrossRef]
- Leslie, E.M.; Deeley, R.G.; Cole, S.P.C. Multidrug resistance proteins: Role of P-glycoprotein, MRP1, MRP2, and BCRP (ABCG2) in tissue defense. Toxicol. Appl. Pharmacol. 2005, 204, 216–237. [Google Scholar] [CrossRef]
- Yamamoto, H.; Ngan, C.Y.; Monden, M. Cancer cells survive with survivin. Cancer Sci. 2008, 99, 1709–1714. [Google Scholar] [CrossRef]
- Jaiswal, P.K.; Goel, A.; Mittal, R.D. Survivin: A molecular biomarker in cancer. Indian J. Med Res. 2015, 141, 389–397. [Google Scholar] [CrossRef] [PubMed]
- Sherr, C.J.; Roberts, J.M. CDK inhibitors: Positive and negative regulators of G1-phase progression. Genes Dev. 1999, 13, 1501–1512. [Google Scholar] [CrossRef] [Green Version]
- Masgras, I.; Carrera, S.; de Verdier, P.J.; Brennan, P.; Majid, A.; Makhtar, W.; Tulchinsky, E.; Jones, G.D.; Roninson, I.B.; Macip, S. Reactive oxygen species and mitochondrial sensitivity to oxidative stress determine induction of cancer cell death by p21. J. Biol. Chem. 2012, 287, 9845–9854. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Li, X.; Wang, J.; Ye, Z.; Li, J.-C. Oridonin up-regulates expression of P21 and induces autophagy and apoptosis in human prostate cancer cells. Int. J. Biol. Sci. 2012, 8, 901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, J.Y.; Park, S.I.; Oh, J.H.; Kim, S.M.; Jeong, C.H.; Jun, J.A.; Lee, K.S.; Oh, W.; Lee, J.K.; Jeun, S.S. Brain-derived neurotrophic factor stimulates the neural differentiation of human umbilical cord blood-derived mesenchymal stem cells and survival of differentiated cells through MAPK/ERK and PI3K/Akt-dependent signaling pathways. J. Neurosci. Res. 2008, 86, 2168–2178. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.J.; Byun, J.Y.; Yun, C.H.; Park, I.C.; Lee, K.H.; Lee, S.J. c-Src-p38 Mitogen-Activated Protein Kinase Signaling Is Required for Akt Activation in Response to Ionizing Radiation. Mol. Cancer Res. 2008, 6, 1872–1880. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ji, J.P.; Zhang, Y.P.; Redon, C.E.; Reinhold, W.C.; Chen, A.P.; Fogli, L.K.; Holbeck, S.L.; Parchment, R.E.; Hollingshead, M.; Tomaszewski, J.E.; et al. Phosphorylated fraction of H2AX as a measurement for DNA damage in cancer cells and potential applications of a novel assay. PLoS ONE 2017, 12, e0171582. [Google Scholar] [CrossRef] [PubMed]
- Fukuchi, K.; Tomoyasu, S.; Nakamaki, T.; Tsuruoka, N.; Gomi, K. DNA damage induces p21 protein expression by inhibiting ubiquitination in ML-1 cells. Biochim. Biophys. Acta Mol. Cell Res. 1998, 1404, 405–411. [Google Scholar] [CrossRef] [Green Version]
- Yang, Q.; Pan, Q.; Li, C.; Xu, Y.; Wen, C.; Sun, F. NRAGE is involved in homologous recombination repair to resist the DNA-damaging chemotherapy and composes a ternary complex with RNF8-BARD1 to promote cell survival in squamous esophageal tumorigenesis. Cell Death Differ. 2016, 23, 1406–1416. [Google Scholar] [CrossRef] [Green Version]
- Lee, M.G.; Lee, K.S.; Nam, K.S. The association of changes in RAD51 and survivin expression levels with the proton beam sensitivity of Capan-1 and Panc-1 human pancreatic cancer cells. Int. J. Oncol. 2019, 54, 744–752. [Google Scholar] [CrossRef] [Green Version]
- Vequaud, E.; Desplanques, G.; Jezequel, P.; Juin, P.; Barille-Nion, S. Survivin contributes to DNA repair by homologous recombination in breast cancer cells. Breast Cancer Res. Treat. 2016, 155, 53–63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gildemeister, O.S.; Sage, J.M.; Knight, K.L. Cellular Redistribution of Rad51 in Response to DNA Damage NOVEL ROLE FOR Rad51C. J. Biol. Chem. 2009, 284, 31945–31952. [Google Scholar] [CrossRef] [Green Version]
- Banerjee, K.; Resat, H. Constitutive activation of STAT3 in breast cancer cells: A review. Int. J. Cancer 2016, 138, 2570–2578. [Google Scholar] [CrossRef]
- Kunigal, S.; Lakka, S.S.; Sodadasu, P.K.; Estes, N.; Rao, J.S. Stat3-siRNA induces Fas-mediated apoptosis in vitro and in vivo in breast cancer. Int. J. Oncol. 2009, 34, 1209–1220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.F.; Zhang, Y.; Zhang, X.W.; Tian, W.X.; Feng, W.L.; Chen, T.M. The curcumin analogue hydrazinocurcumin exhibits potent suppressive activity on carcinogenicity of breast cancer cells via STAT3 inhibition. Int. J. Oncol. 2012, 40, 1189–1195. [Google Scholar] [CrossRef]
- Oseguera, C.A.V.; Spencer, J.V. cmvIL-10 Stimulates the Invasive Potential of MDA-MB-231 Breast Cancer Cells. PLoS ONE 2014, 9, e88708. [Google Scholar] [CrossRef] [PubMed]
- Sehara, Y.; Sawicka, K.; Hwang, J.Y.; Latuszek-Barrantes, A.; Etgen, A.M.; Zukin, R.S. Survivin Is a Transcriptional Target of STAT3 Critical to Estradiol Neuroprotection in Global Ischemia. J. Neurosci. 2013, 33, 12364–12374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinou, J.C.; Youle, R.J. Mitochondria in Apoptosis: Bcl-2 Family Members and Mitochondrial Dynamics. Dev. Cell 2011, 21, 92–101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Donovan, M.; Cotter, T.G. Control of mitochondrial integrity by Bcl-2 family members and caspase-independent cell death. Biochim. Biophys. Acta Mol. Cell Res. 2004, 1644, 133–147. [Google Scholar] [CrossRef]
- Daugas, E.; Susin, S.A.; Zamzami, N.; Ferri, K.F.; Irinopoulou, T.; Larochette, N.; Prevost, M.C.; Leber, B.; Andrews, D.; Penninger, J.; et al. Mitochondrio-nuclear translocation of AIF in apoptosis and necrosis. FASEB J. 2000, 14, 729–739. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.; Fraser, M.; Abedini, M.R.; Bai, T.; Tsang, B.K. Regulation of apoptosis-inducing factor-mediated, cisplatin-induced apoptosis by Akt. Br. J. Cancer 2008, 98, 803–808. [Google Scholar] [CrossRef] [PubMed]
- Murakami, Y.; Ikeda, Y.; Yonemitsu, Y.; Onimaru, M.; Nakagawa, K.; Kohno, R.; Miyazaki, M.; Hisatomi, T.; Nakamura, M.; Yabe, T.; et al. Inhibition of Nuclear Translocation of Apoptosis-Inducing Factor Is an Essential Mechanism of the Neuroprotective Activity of Pigment Epithelium-Derived Factor in a Rat Model of Retinal Degeneration. Am. J. Pathol. 2008, 173, 1326–1338. [Google Scholar] [CrossRef] [Green Version]
- Hong, S.J.; Dawson, T.M.; Dawson, V.L. Nuclear and mitochondrial conversations in cell death: PARP-1 and AIF signaling. Trends Pharmacol. Sci. 2004, 25, 259–264. [Google Scholar] [CrossRef]
- Salmond, R.J.; Brownlie, R.J.; Meyuhas, O.; Zamoyska, R. Mechanistic Target of Rapamycin Complex 1/S6 Kinase 1 Signals Influence T Cell Activation Independently of Ribosomal Protein S6 Phosphorylation. J. Immunol. 2015, 195, 4615–4622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khalaileh, A.; Dreazen, A.; Khatib, A.; Apel, R.; Swisa, A.; Kidess-Bassir, N.; Maitra, A.; Meyuhas, O.; Dor, Y.; Zamir, G. Phosphorylation of Ribosomal Protein S6 Attenuates DNA Damage and Tumor Suppression during Development of Pancreatic Cancer. Cancer Res. 2013, 73, 1811–1820. [Google Scholar] [CrossRef] [Green Version]
- Chen, B.J.; Tan, Z.; Gao, J.; Wu, W.; Liu, L.D.; Jin, W.; Cao, Y.D.; Zhao, S.; Zhang, W.; Qiu, Z.X.; et al. Hyperphosphorylation of ribosomal protein S6 predicts unfavorable clinical survival in non-small cell lung cancer. J. Exp. Clin. Cancer Res. 2015, 34, 16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El-Deiry, W.S.; Tokino, T.; Velculescu, V.E.; Levy, D.B.; Parsons, R.; Trent, J.M.; Lin, D.; Mercer, W.E.; Kinzler, K.W.; Vogelstein, B. WAF1, a potential mediator of p53 tumor suppression. Cell 1993, 75, 817–825. [Google Scholar] [CrossRef]
- Georgakilas, A.G.; Martin, O.A.; Bonner, W.M. p21: A two-faced genome guardian. Trends Mol. Med. 2017, 23, 310–319. [Google Scholar] [CrossRef]
- Richon, V.M.; Sandhoff, T.W.; Rifkind, R.A.; Marks, P.A. Histone deacetylase inhibitor selectively induces p21WAF1 expression and gene-associated histone acetylation. Proc. Natl. Acad. Sci. USA 2000, 97, 10014–10019. [Google Scholar] [CrossRef] [Green Version]
- Passos, J.F.; Nelson, G.; Wang, C.; Richter, T.; Simillion, C.; Proctor, C.J.; Miwa, S.; Olijslagers, S.; Hallinan, J.; Wipat, A. Feedback between p21 and reactive oxygen production is necessary for cell senescence. Mol. Syst. Biol. 2010, 6. [Google Scholar] [CrossRef]
- Kim, M.J.; Woo, J.S.; Kwon, C.H.; Kim, J.H.; Kim, Y.K.; Kim, K.H. Luteolin induces apoptotic cell death through AIF nuclear translocation mediated by activation of ERK and p38 in human breast cancer cell lines. Cell Biol. Int. 2012, 36, 339–344. [Google Scholar] [CrossRef] [PubMed]
- Ha, S.H.; Jin, F.; Kwak, C.H.; Abekura, F.; Park, J.Y.; Park, N.G.; Chang, Y.C.; Lee, Y.C.; Chung, T.W.; Ha, K.T.; et al. Jellyfish extract induces apoptotic cell death through the p38 pathway and cell cycle arrest in chronic myelogenous leukemia K562 cells. Peerj 2017, 5. [Google Scholar] [CrossRef] [Green Version]
- Tsuchiya, A.; Kaku, Y.; Nakano, T.; Nishizaki, T. Diarachidonoylphosphoethanolamine induces apoptosis of malignant pleural mesothelioma cells through a Trx/ASK1/p38 MAPK pathway. J. Pharmacol. Sci. 2015, 129, 160–168. [Google Scholar] [CrossRef] [Green Version]
- Kwon, C.H.; Park, J.Y.; Kim, T.H.; Woo, J.S.; Kim, Y.K. Ciglitazone induces apoptosis via activation of p38 MAPK and AIF nuclear translocation mediated by reactive oxygen species and Ca2+ in opossum kidney cells. Toxicology 2009, 257, 1–9. [Google Scholar] [CrossRef]
- Guo, Y.X.; Lin, D.J.; Zhang, M.Z.; Zhang, X.W.; Li, Y.R.; Yang, R.; Lu, Y.; Jin, X.S.; Yang, M.L.; Wang, M.M.; et al. CLDN6-induced apoptosis via regulating ASK1-p38/JNK signaling in breast cancer MCF-7 cells. Int. J. Oncol. 2016, 48, 2435–2444. [Google Scholar] [CrossRef] [Green Version]
- Ho, T.C.; Yang, Y.C.; Cheng, H.C.; Wu, A.C.; Chen, S.L.; Chen, H.K.; Tsao, Y.P. Activation of mitogen-activated protein kinases is essential for hydrogen peroxide-induced apoptosis in retinal pigment epithelial cells. Apoptosis 2006, 11, 1899–1908. [Google Scholar] [CrossRef]
- Chen, W.X.; Liu, L.; Luo, Y.; Odaka, Y.; Awate, S.; Zhou, H.Y.; Shen, T.; Zheng, S.Z.; Lu, Y.; Huang, S.L. Cryptotanshinone Activates p38/JNK and Inhibits Erk1/2 Leading to Caspase-Independent Cell Death in Tumor Cells. Cancer Prev. Res. 2012, 5, 778–787. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Laethem, A.; Van Kelst, S.; Lippens, S.; Declercq, W.; Vandenabeele, P.; Janssens, S.; Vandenheede, J.R.; Garmyn, M.; Agostinis, P. Activation of p38 MAPK is required for Bax translocation to mitochondria, cytochrome c release and apoptosis induced by UVB irradiation in human keratinocytes. FASEB J. 2004, 18, 1946. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shou, Y.; Li, L.; Prabhakaran, K.; Borowitz, J.L.; Isom, G.E. p38 mitogen-activated protein kinase regulates Bax translocation in cyanide-induced apoptosis. Toxicol. Sci. 2003, 75, 99–107. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Gang, L.-Y.; Pan, L.-H.; Yao, F.-F.; Peng, T.; Tan, Y.-J.; Zhang, G.-M.; Liu, Z.; Yao, J.-C.; Ren, Y. Toxicity study of 28-day subcutaneous injection of Arctigenin in Beagle dogs. Front. Pharmacol. 2019, 10, 1218. [Google Scholar] [CrossRef] [PubMed]
- Huang, Q.; Qin, S.; Yuan, X.; Zhang, L.; Ji, J.; Liu, X.; Ma, W.; Zhang, Y.; Liu, P.; Sun, Z. Arctigenin inhibits triple-negative breast cancers by targeting CIP2A to reactivate protein phosphatase 2A. Oncol. Rep. 2017, 38, 598–606. [Google Scholar] [CrossRef]







© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, K.-S.; Lee, M.-G.; Kwon, Y.-S.; Nam, K.-S. Arctigenin Enhances the Cytotoxic Effect of Doxorubicin in MDA-MB-231 Breast Cancer Cells. Int. J. Mol. Sci. 2020, 21, 2997. https://doi.org/10.3390/ijms21082997
Lee K-S, Lee M-G, Kwon Y-S, Nam K-S. Arctigenin Enhances the Cytotoxic Effect of Doxorubicin in MDA-MB-231 Breast Cancer Cells. International Journal of Molecular Sciences. 2020; 21(8):2997. https://doi.org/10.3390/ijms21082997
Chicago/Turabian StyleLee, Kyu-Shik, Min-Gu Lee, Yun-Suk Kwon, and Kyung-Soo Nam. 2020. "Arctigenin Enhances the Cytotoxic Effect of Doxorubicin in MDA-MB-231 Breast Cancer Cells" International Journal of Molecular Sciences 21, no. 8: 2997. https://doi.org/10.3390/ijms21082997
APA StyleLee, K. -S., Lee, M. -G., Kwon, Y. -S., & Nam, K. -S. (2020). Arctigenin Enhances the Cytotoxic Effect of Doxorubicin in MDA-MB-231 Breast Cancer Cells. International Journal of Molecular Sciences, 21(8), 2997. https://doi.org/10.3390/ijms21082997