Immunological Mechanisms in Inflammation-Associated Colon Carcinogenesis

 , , and
, , and 
{kind=link}
{kind=link}
{kind=link}
Abstract
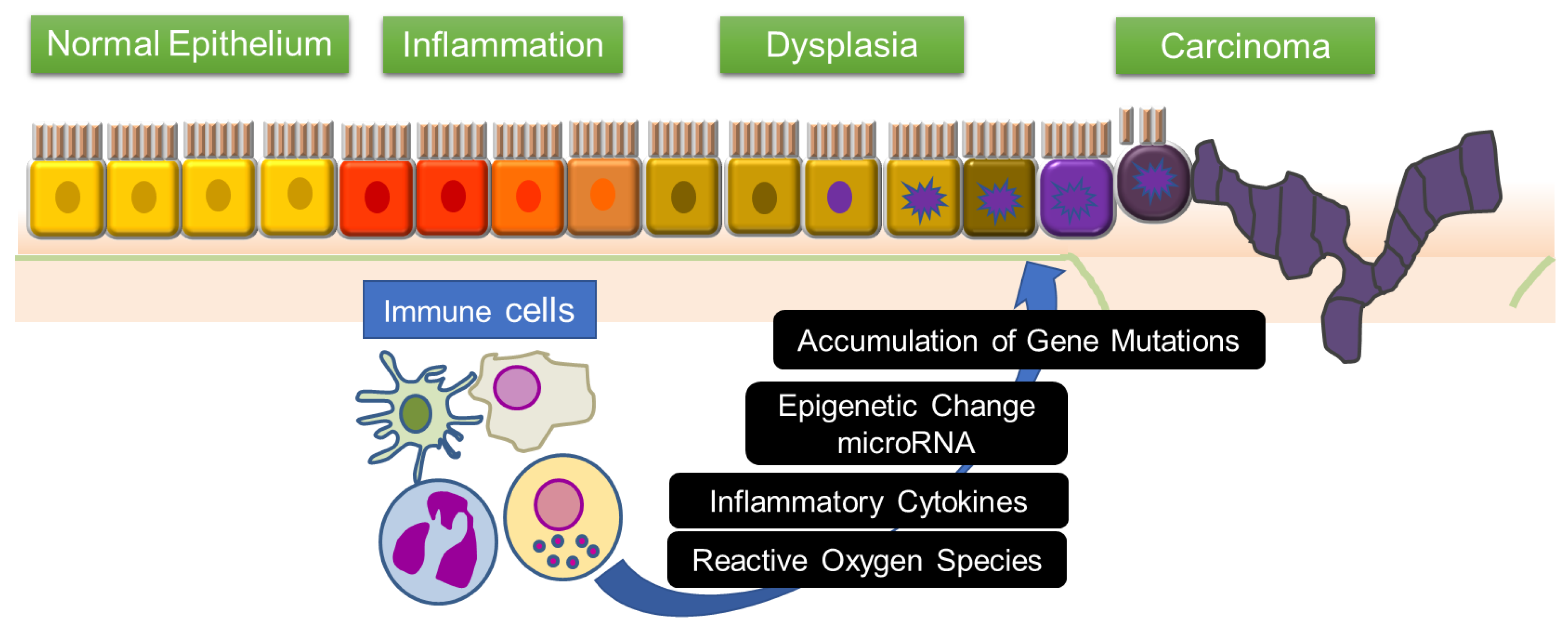
:1. Introduction
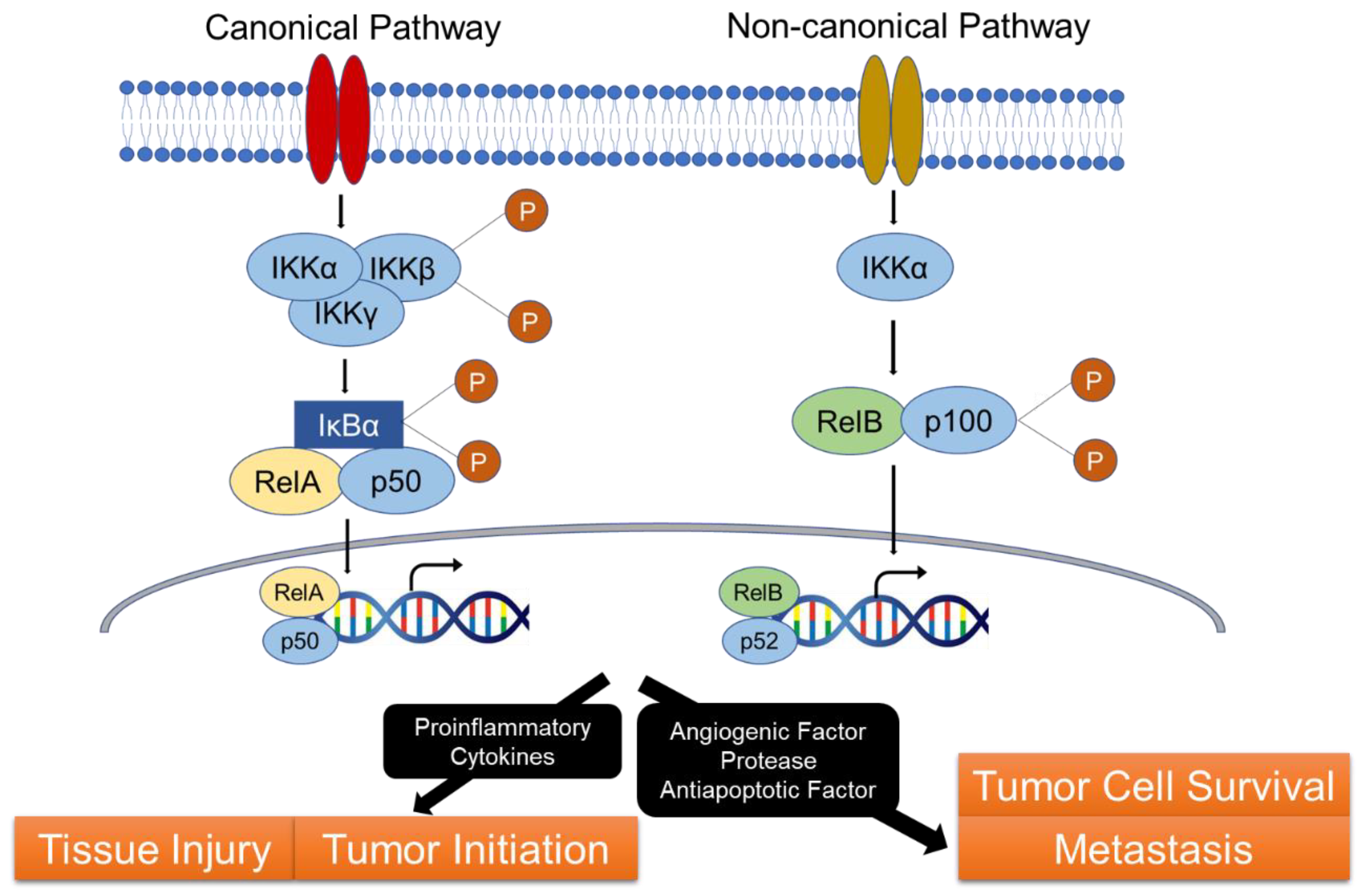
2. NF-κB Pathway
3. PGE2/COX-2 Pathway
4. IL-6/STAT3 Pathway
5. IL-23/Th17 Pathway
6. Purinergic Signaling
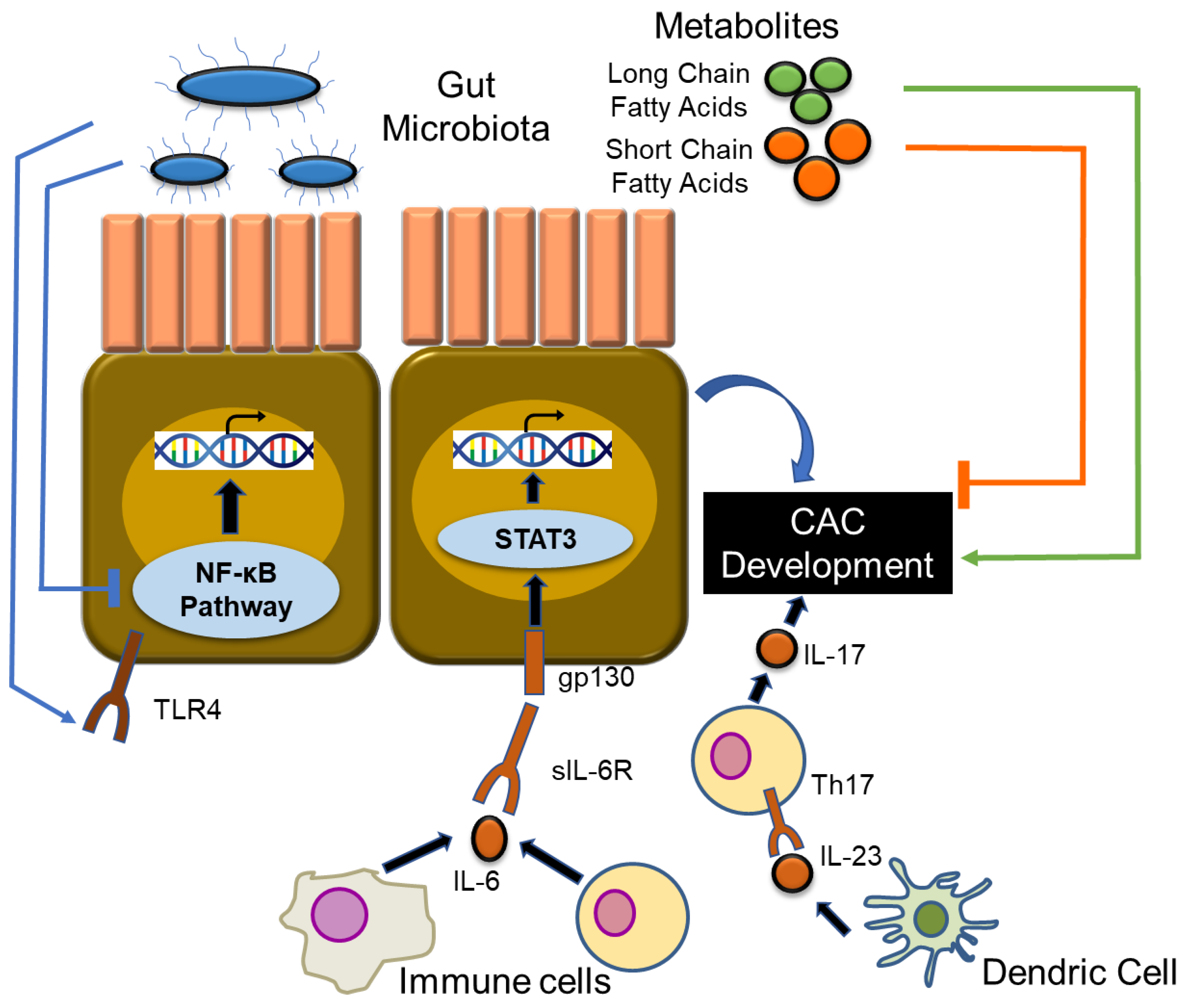
7. CAC and Gut Microbiota
8. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| IBD | Inflammatory Bowel Disease |
| UC | Ulcerative Colitis |
| CD | Crohn’s Disease |
| CRC | Colorectal Cancer |
| CAC | Colitis-Associated Cancer |
| AOM | Azoxymethane |
| DSS | Dextran Sulfate Sodium |
| WT | Wild-Type |
| NLR | Nod-Like Receptor |
| TNFR1 | TNF receptor type 1 |
| TNFR2 | TNF receptor type 2 |
| KO | Knockout |
| FAP | Familial Adenomatous Polyposis |
| IEC | Intestinal Epithelial Cell |
| SC | Sporadic Cancer |
| Treg | Regulatory T cell |
| PPARδ | Peroxisome Proliferator-Activated Receptor δ |
| JAK | Janus kinase |
References
- Eaden, J.A.; Abrams, K.R.; Mayberry, J.F. The risk of colorectal cancer in ulcerative colitis: A meta-analysis. Gut 2001, 48, 526–535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zitvogel, L.; Pietrocola, F.; Kroemer, G. Nutrition, inflammation and cancer. Nat. Immunol. 2017, 18, 843–850. [Google Scholar] [CrossRef]
- Choi, C.H.; Rutter, M.D.; Askari, A.; Lee, G.H.; Warusavitarne, J.; Moorghen, M.; Thomas-Gibson, S.; Saunders, B.P.; Graham, T.A.; Hart, A.L. Forty-year analysis of colonoscopic surveillance program for neoplasia in ulcerative colitis: An updated overview. Am. J. Gastroenterol. 2015, 110, 1022–1034. [Google Scholar] [CrossRef] [Green Version]
- Sugita, A.; Greenstein, A.J.; Ribeiro, M.B.; Sachar, D.B.; Bodian, C.; Panday, A.K.; Szporn, A.; Pozner, J.; Heimann, T.; Palmer, M. Survival with colorectal cancer in ulcerative colitis. A study of 102 cases. Ann. Surg. 1993, 218, 189–195. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, T.; Konishi, T.; Kishimoto, J.; Kotake, K.; Muto, T.; Sugihara, K. Ulcerative colitis-associated colorectal cancer shows a poorer survival than sporadic colorectal cancer: A nationwide Japanese study. Inflamm. Bowel Dis. 2011, 17, 802–808. [Google Scholar] [CrossRef] [PubMed]
- Leowardi, C.; Schneider, M.L.; Hinz, U.; Harnoss, J.M.; Tarantino, I.; Lasitschka, F.; Ulrich, A.; Büchler, M.W.; Kadmon, M. Prognosis of Ulcerative Colitis-Associated Colorectal Carcinoma Compared to Sporadic Colorectal Carcinoma: A Matched Pair Analysis. Ann. Surg. Oncol. 2016, 23, 870–876. [Google Scholar] [CrossRef]
- Canavan, C.; Abrams, K.R.; Mayberry, J. Meta-analysis: Colorectal and small bowel cancer risk in patients with Crohn′s disease. Aliment. Pharmacol. Ther. 2006, 23, 1097–1104. [Google Scholar] [CrossRef]
- Olén, O.; Erichsen, R.; Sachs, M.C.; Pedersen, L.; Halfvarson, J.; Askling, J.; Ekbom, A.; Sørensen, H.T.; Ludvigsson, J.F. Colorectal cancer in Crohn′s disease: A Scandinavian population-based cohort study. Lancet Gastroenterol. Hepatol. 2020, in press. [Google Scholar] [CrossRef]
- Hayden, M.S.; Ghosh, S. Shared principles in NF-kappaB signaling. Cell 2008, 132, 344–362. [Google Scholar] [CrossRef] [Green Version]
- Sun, S.C. The non-canonical NF-κB pathway in immunity and inflammation. Nat. Rev. Immunol. 2017, 17, 545–558. [Google Scholar] [CrossRef]
- Dejardin, E. The alternative NF-kappaB pathway from biochemistry to biology: Pitfalls and promises for future drug development. Biochem. Pharmacol. 2006, 72, 1161–1179. [Google Scholar] [CrossRef] [PubMed]
- Dejardin, E.; Droin, N.M.; Delhase, M.; Haas, E.; Cao, Y.; Makris, C.; Li, Z.W.; Karin, M.; Ware, C.F.; Green, D.R. The lymphotoxin-beta receptor induces different patterns of gene expression via two NF-kappaB pathways. Immunity 2002, 17, 525–535. [Google Scholar] [CrossRef] [Green Version]
- Coope, H.J.; Atkinson, P.G.; Huhse, B.; Belich, M.; Janzen, J.; Holman, M.J.; Klaus, G.G.; Johnston, L.H.; Ley, S.C. CD40 regulates the processing of NF-kappaB2 p100 to p52. EMBO J. 2002, 21, 5375–5385. [Google Scholar] [CrossRef]
- Claudio, E.; Brown, K.; Park, S.; Wang, H.; Siebenlist, U. BAFF-induced NEMO-independent processing of NF-kappa B2 in maturing B cells. Nat. Immunol. 2002, 3, 958–965. [Google Scholar] [CrossRef] [PubMed]
- Kayagaki, N.; Yan, M.; Seshasayee, D.; Wang, H.; Lee, W.; French, D.M.; Grewal, I.S.; Cochran, A.G.; Gordon, N.C.; Yin, J.; et al. BAFF/BLyS receptor 3 binds the B cell survival factor BAFF ligand through a discrete surface loop and promotes processing of NF-kappaB2. Immunity 2002, 17, 515–524. [Google Scholar] [CrossRef] [Green Version]
- Atreya, I.; Atreya, R.; Neurath, M.F. NF-kappaB in inflammatory bowel disease. J. Intern. Med. 2008, 263, 591–596. [Google Scholar] [CrossRef]
- O′Connor, P.M.; Lapointe, T.K.; Beck, P.L.; Buret, A.G. Mechanisms by which inflammation may increase intestinal cancer risk in inflammatory bowel disease. Inflamm. Bowel Dis. 2010, 16, 1411–1420. [Google Scholar] [CrossRef] [PubMed]
- Karin, M. Nuclear factor-kappaB in cancer development and progression. Nature 2006, 441, 431–436. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Liu, Z.; Wang, L.; Zhang, X. NF-kappaB signaling pathway, inflammation and colorectal cancer. Cell. Mol. Immunol. 2009, 6, 327–334. [Google Scholar] [CrossRef] [Green Version]
- Karin, M.; Greten, F.R. NF-kappaB: Linking inflammation and immunity to cancer development and progression. Nat. Rev. Immunol. 2005, 5, 749–759. [Google Scholar] [CrossRef]
- Greten, F.R.; Eckmann, L.; Greten, T.F.; Park, J.M.; Li, Z.W.; Egan, L.J.; Kagnoff, M.F.; Karin, M. IKKbeta links inflammation and tumorigenesis in a mouse model of colitis-associated cancer. Cell 2004, 118, 285–296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allen, I.C.; Wilson, J.E.; Schneider, M.; Lich, J.D.; Roberts, R.A.; Arthur, J.C.; Woodford, R.M.; Davis, B.K.; Uronis, J.M.; Herfarth, H.H.; et al. NLRP12 suppresses colon inflammation and tumorigenesis through the negative regulation of noncanonical NF-κB signaling. Immunity 2012, 36, 742–754. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cooks, T.; Pateras, I.S.; Tarcic, O.; Solomon, H.; Schetter, A.J.; Wilder, S.; Lozano, G.; Pikarsky, E.; Forshew, T.; Rosenfeld, N.; et al. Mutant p53 prolongs NF-κB activation and promotes chronic inflammation and inflammation-associated colorectal cancer. Cancer Cell 2013, 23, 634–646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwitalla, S.; Ziegler, P.K.; Horst, D.; Becker, V.; Kerle, I.; Begus-Nahrmann, Y.; Lechel, A.; Rudolph, K.L.; Langer, R.; Slotta-Huspenina, J.; et al. Loss of p53 in enterocytes generates an inflammatory microenvironment enabling invasion and lymph node metastasis of carcinogen-induced colorectal tumors. Cancer Cell 2013, 23, 93–106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bradley, J.R. TNF-mediated inflammatory disease. J. Pathol. 2008, 214, 149–160. [Google Scholar] [CrossRef]
- Faustman, D.; Davis, M. TNF receptor 2 pathway: Drug target for autoimmune diseases. Nat. Rev. Drug Discov. 2010, 9, 482–493. [Google Scholar] [CrossRef]
- Popivanova, B.K.; Kitamura, K.; Wu, Y.; Kondo, T.; Kagaya, T.; Kaneko, S.; Oshima, M.; Fujii, C.; Mukaida, N. Blocking TNF-alpha in mice reduces colorectal carcinogenesis associated with chronic colitis. J. Clin. Investig. 2008, 118, 560–570. [Google Scholar] [CrossRef]
- Onizawa, M.; Nagaishi, T.; Kanai, T.; Nagano, K.; Oshima, S.; Nemoto, Y.; Yoshioka, A.; Totsuka, T.; Okamoto, R.; Nakamura, T.; et al. Signaling pathway via TNF-alpha/NF-kappaB in intestinal epithelial cells may be directly involved in colitis-associated carcinogenesis. Am. J. Physiol. Gastrointest. Liver Physiol. 2009, 296, G850–G859. [Google Scholar] [CrossRef]
- Friis, S.; Riis, A.H.; Erichsen, R.; Baron, J.A.; Sørensen, H.T. Low-Dose Aspirin or Nonsteroidal Anti-inflammatory Drug Use and Colorectal Cancer Risk: A Population-Based, Case-Control Study. Ann. internal Med. 2015, 163, 347–355. [Google Scholar] [CrossRef]
- Oshima, M.; Dinchuk, J.E.; Kargman, S.L.; Oshima, H.; Hancock, B.; Kwong, E.; Trzaskos, J.M.; Evans, J.F.; Taketo, M.M. Suppression of intestinal polyposis in Apc delta716 knockout mice by inhibition of cyclooxygenase 2 (COX-2). Cell 1996, 87, 803–809. [Google Scholar] [CrossRef] [Green Version]
- Chulada, P.C.; Thompson, M.B.; Mahler, J.F.; Doyle, C.M.; Gaul, B.W.; Lee, C.; Tiano, H.F.; Morham, S.G.; Smithies, O.; Langenbach, R. Genetic disruption of Ptgs-1, as well as Ptgs-2, reduces intestinal tumorigenesis in Min mice. Cancer Res. 2000, 60, 4705–4708. [Google Scholar] [PubMed]
- Wang, D.; DuBois, R.N. PPARδ and PGE2 signaling pathways communicate and connect inflammation to colorectal cancer. Inflamm. Cell Signal. 2014, 1. [Google Scholar] [CrossRef] [Green Version]
- Sheng, H.; Shao, J.; Morrow, J.D.; Beauchamp, R.D.; DuBois, R.N. Modulation of apoptosis and Bcl-2 expression by prostaglandin E2 in human colon cancer cells. Cancer Res. 1998, 58, 362–366. [Google Scholar] [PubMed]
- Wang, D.; Wang, H.; Brown, J.; Daikoku, T.; Ning, W.; Shi, Q.; Richmond, A.; Strieter, R.; Dey, S.K.; DuBois, R.N. CXCL1 induced by prostaglandin E2 promotes angiogenesis in colorectal cancer. J. Exp. Med. 2006, 203, 941–951. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katoh, H.; Wang, D.; Daikoku, T.; Sun, H.; Dey, S.K.; Dubois, R.N. CXCR2-expressing myeloid-derived suppressor cells are essential to promote colitis-associated tumorigenesis. Cancer Cell 2013, 24, 631–644. [Google Scholar] [CrossRef] [Green Version]
- Buchert, M.; Burns, C.J.; Ernst, M. Targeting JAK kinase in solid tumors: Emerging opportunities and challenges. Oncogene 2016, 35, 939–951. [Google Scholar] [CrossRef]
- Waldner, M.J.; Neurath, M.F. Master regulator of intestinal disease: IL-6 in chronic inflammation and cancer development. Semin. Immunol. 2014, 26, 75–79. [Google Scholar] [CrossRef]
- Schaper, F.; Gendo, C.; Eck, M.; Schmitz, J.; Grimm, C.; Anhuf, D.; Kerr, I.M.; Heinrich, P.C. Activation of the protein tyrosine phosphatase SHP2 via the interleukin-6 signal transducing receptor protein gp130 requires tyrosine kinase Jak1 and limits acute-phase protein expression. Biochem. J. 1998, 335 Pt 3, 557–565. [Google Scholar] [CrossRef]
- Zegeye, M.M.; Lindkvist, M.; Fälker, K.; Kumawat, A.K.; Paramel, G.; Grenegård, M.; Sirsjö, A.; Ljungberg, L.U. Activation of the JAK/STAT3 and PI3K/AKT pathways are crucial for IL-6 trans-signaling-mediated pro-inflammatory response in human vascular endothelial cells. Cell Commun. Signal. CCS 2018, 16, 55. [Google Scholar] [CrossRef]
- Johnson, D.E.; O′Keefe, R.A.; Grandis, J.R. Targeting the IL-6/JAK/STAT3 signalling axis in cancer. Nat. Rev. Clin. Oncol. 2018, 15, 234–248. [Google Scholar] [CrossRef]
- Matsumoto, S.; Hara, T.; Mitsuyama, K.; Yamamoto, M.; Tsuruta, O.; Sata, M.; Scheller, J.; Rose-John, S.; Kado, S.; Takada, T. Essential roles of IL-6 trans-signaling in colonic epithelial cells, induced by the IL-6/soluble-IL-6 receptor derived from lamina propria macrophages, on the development of colitis-associated premalignant cancer in a murine model. J. Immunol. 2010, 184, 1543–1551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, J.; Nagahashi, M.; Kim, E.Y.; Harikumar, K.B.; Yamada, A.; Huang, W.C.; Hait, N.C.; Allegood, J.C.; Price, M.M.; Avni, D.; et al. Sphingosine-1-phosphate links persistent STAT3 activation, chronic intestinal inflammation, and development of colitis-associated cancer. Cancer Cell 2013, 23, 107–120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, H.; Deng, J.; Kujawski, M.; Yang, C.; Liu, Y.; Herrmann, A.; Kortylewski, M.; Horne, D.; Somlo, G.; Forman, S.; et al. STAT3-induced S1PR1 expression is crucial for persistent STAT3 activation in tumors. Nat. Med. 2010, 16, 1421–1428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knüpfer, H.; Preiss, R. Serum interleukin-6 levels in colorectal cancer patients—A summary of published results. Int. J. Colorectal Dis. 2010, 25, 135–140. [Google Scholar] [CrossRef]
- Han, J.; Theiss, A.L. Stat3: Friend or foe in colitis and colitis-associated cancer? Inflamm. Bowel Dis. 2014, 20, 2405–2411. [Google Scholar] [CrossRef] [Green Version]
- Leppkes, M.; Becker, C.; Ivanov, I.I.; Hirth, S.; Wirtz, S.; Neufert, C.; Pouly, S.; Murphy, A.J.; Valenzuela, D.M.; Yancopoulos, G.D.; et al. RORγ-expressing Th17 cells induce murine chronic intestinal inflammation via redundant effects of IL-17A and IL-17F. Gastroenterology 2009, 136, 257–267. [Google Scholar] [CrossRef]
- Sugimoto, K.; Ogawa, A.; Mizoguchi, E.; Shimomura, Y.; Andoh, A.; Bhan, A.K.; Blumberg, R.S.; Xavier, R.J.; Mizoguchi, A. IL-22 ameliorates intestinal inflammation in a mouse model of ulcerative colitis. J. Clin. Investig. 2008, 118, 534–544. [Google Scholar] [CrossRef] [Green Version]
- Eyerich, S.; Eyerich, K.; Pennino, D.; Carbone, T.; Nasorri, F.; Pallotta, S.; Cianfarani, F.; Odorisio, T.; Traidl-Hoffmann, C.; Behrendt, H.; et al. Th22 cells represent a distinct human T cell subset involved in epidermal immunity and remodeling. J. Clin. Investig. 2009, 119, 3573–3585. [Google Scholar] [CrossRef] [Green Version]
- Sugimoto, K. Role of STAT3 in inflammatory bowel disease. World J. Gastroenterol. 2008, 14, 5110–5114. [Google Scholar] [CrossRef] [Green Version]
- Oppmann, B.; Lesley, R.; Blom, B.; Timans, J.C.; Xu, Y.; Hunte, B.; Vega, F.; Yu, N.; Wang, J.; Singh, K.; et al. Novel p19 protein engages IL-12p40 to form a cytokine, IL-23, with biological activities similar as well as distinct from IL-12. Immunity 2012, 13, 715–725. [Google Scholar] [CrossRef] [Green Version]
- Parham, C.; Chirica, M.; Timans, J.; Vaisberg, E.; Travis, M.; Cheung, J.; Pflanz, S.; Zhang, R.; Singh, K.P.; Vega, F.; et al. A receptor for the heterodimeric cytokine IL-23 is composed of IL-12Rbeta1 and a novel cytokine receptor subunit, IL-23R. J. Immunol. 2002, 168, 5699–5708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Samuels, J.S.; Holland, L.; López, M.; Meyers, K.; Cumbie, W.G.; McClain, A.; Ignatowicz, A.; Nelson, D.; Shashidharamurthy, R. Prostaglandin E2 and IL-23 interconnects STAT3 and RoRγ pathways to initiate Th17 CD4+ T-cell development during rheumatoid arthritis. Inflamm. Res. Off. J. Eur. Histamine Res. Soc. 2018, 67, 589–596. [Google Scholar] [CrossRef] [PubMed]
- Iwakura, Y.; Ishigame, H. The IL-23/IL-17 axis in inflammation. J. Clin. Investig. 2006, 116, 1218–1222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujino, S.; Andoh, A.; Bamba, S.; Ogawa, A.; Hata, K.; Araki, Y.; Bamba, T.; Fujiyama, Y. Increased expression of interleukin 17 in inflammatory bowel disease. Gut 2003, 52, 65–70. [Google Scholar] [CrossRef]
- Moschen, A.R.; Tilg, H.; Raine, T. IL-12, IL-23 and IL-17 in IBD: Immunobiology and therapeutic targeting. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 185–196. [Google Scholar] [CrossRef] [PubMed]
- Grivennikov, S.I.; Wang, K.; Mucida, D.; Stewart, C.A.; Schnabl, B.; Jauch, D.; Taniguchi, K.; Yu, G.Y.; Osterreicher, C.H.; Hung, K.E.; et al. Adenoma-linked barrier defects and microbial products drive IL-23/IL-17-mediated tumour growth. Nature 2012, 491, 254–258. [Google Scholar] [CrossRef] [Green Version]
- Punkenburg, E.; Vogler, T.; Büttner, M.; Amann, K.; Waldner, M.; Atreya, R.; Abendroth, B.; Mudter, J.; Merkel, S.; Gallmeier, E.; et al. Batf-dependent Th17 cells critically regulate IL-23 driven colitis-associated colon cancer. Gut 2016, 65, 1139–1150. [Google Scholar] [CrossRef]
- Richter, C.; Herrero San Juan, M.; Weigmann, B.; Bergis, D.; Dauber, K.; Muders, M.H.; Baretton, G.B.; Pfeilschifter, J.M.; Bonig, H.; Brenner, S.; et al. Defective IL-23/IL-17 Axis Protects p47phox-/- Mice from Colon Cancer. Front. Immunol. 2017, 8, 44. [Google Scholar] [CrossRef] [Green Version]
- Qi, H.; Yang, H.; Xu, G.; Ren, J.; Hua, W.; Shi, Y.; Torsvik, M.; Florholmen, J.; Cui, G. Therapeutic efficacy of IL-17A antibody injection in preventing the development of colitis associated carcinogenesis in mice. Immunobiology 2012, 220, 54–59. [Google Scholar] [CrossRef]
- Hyun, Y.S.; Han, D.S.; Lee, A.R.; Eun, C.S.; Youn, J.; Kim, H.Y. Role of IL-17A in the development of colitis-associated cancer. Carcinogenesis 2012, 33, 931–936. [Google Scholar] [CrossRef]
- Burnstock, G.; Williams, M. P2 purinergic receptors: Modulation of cell function and therapeutic potential. J. Pharmacol. Exp. Ther. 2000, 295, 862–869. [Google Scholar] [PubMed]
- Neves, A.R.; Castelo-Branco, M.T.; Figliuolo, V.R.; Bernardazzi, C.; Buongusto, F.; Yoshimoto, A.; Nanini, H.F.; Coutinho, C.M.; Carneiro, A.J.; Coutinho-Silva, R.; et al. Overexpression of ATP-activated P2X7 receptors in the intestinal mucosa is implicated in the pathogenesis of Crohn′s disease. Inflamm. Bowel Dis. 2014, 20, 444–457. [Google Scholar] [CrossRef] [PubMed]
- Figliuolo, V.R.; Savio, L.; Safya, H.; Nanini, H.; Bernardazzi, C.; Abalo, A.; de Souza, H.; Kanellopoulos, J.; Bobé, P.; Coutinho, C.; et al. P2X7 receptor promotes intestinal inflammation in chemically induced colitis and triggers death of mucosal regulatory T cells. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 1183–1194. [Google Scholar] [CrossRef]
- Hofman, P.; Cherfils-Vicini, J.; Bazin, M.; Ilie, M.; Juhel, T.; Hébuterne, X.; Gilson, E.; Schmid-Alliana, A.; Boyer, O.; Adriouch, S.; et al. Genetic and pharmacological inactivation of the purinergic P2RX7 receptor dampens inflammation but increases tumor incidence in a mouse model of colitis-associated cancer. Cancer Res. 2015, 75, 835–845. [Google Scholar] [CrossRef] [Green Version]
- Kado, S.; Uchida, K.; Funabashi, H.; Iwata, S.; Nagata, Y.; Ando, M.; Onoue, M.; Matsuoka, Y.; Ohwaki, M.; Morotomi, M. Intestinal microflora are necessary for development of spontaneous adenocarcinoma of the large intestine in T-cell receptor beta chain and p53 double-knockout mice. Cancer Res. 2001, 61, 2395–2398. [Google Scholar]
- Uronis, J.M.; Mühlbauer, M.; Herfarth, H.H.; Rubinas, T.C.; Jones, G.S.; Jobin, C. Modulation of the intestinal microbiota alters colitis-associated colorectal cancer susceptibility. PLoS ONE 2009, 4, e6026. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Appleyard, C.B.; Cruz, M.L.; Isidro, A.A.; Arthur, J.C.; Jobin, C.; De Simone, C. Pretreatment with the probiotic VSL#3 delays transition from inflammation to dysplasia in a rat model of colitis-associated cancer. Am. J. Physiol. Gastrointest. Liver Physiol. 2011, 301, G1004–G1013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Talero, E.; Bolivar, S.; Ávila-Román, J.; Alcaide, A.; Fiorucci, S.; Motilva, V. Inhibition of chronic ulcerative colitis-associated adenocarcinoma development in mice by VSL#3. Inflamm. Bowel Dis. 2015, 21, 1027–1037. [Google Scholar] [CrossRef] [Green Version]
- Arthur, J.C.; Gharaibeh, R.Z.; Uronis, J.M.; Perez-Chanona, E.; Sha, W.; Tomkovich, S.; Mühlbauer, M.; Fodor, A.A.; Jobin, C. VSL#3 probiotic modifies mucosal microbial composition but does not reduce colitis-associated colorectal cancer. Sci. Rep. 2013, 3, 2868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kostic, A.D.; Xavier, R.J.; Gevers, D. The microbiome in inflammatory bowel disease: Current status and the future ahead. Gastroenterology 2014, 146, 1489–1499. [Google Scholar] [CrossRef] [Green Version]
- Yachida, S.; Mizutani, S.; Shiroma, H.; Shiba, S.; Nakajima, T.; Sakamoto, T.; Watanabe, H.; Masuda, K.; Nishimoto, Y.; Kubo, M.; et al. Metagenomic and metabolomic analyses reveal distinct stage-specific phenotypes of the gut microbiota in colorectal cancer. Nat. Med. 2019, 25, 968–976. [Google Scholar] [CrossRef] [PubMed]
- Abreu Maria, T. Toll-like receptor signalling in the intestinal epithelium: How bacterial recognition shapes intestinal function. Nat. Rev. Immunol. 2010, 10, 131–144. [Google Scholar] [CrossRef] [PubMed]
- Fukata, M.; Chen, A.; Vamadevan, A.S.; Cohen, J.; Breglio, K.; Krishnareddy, S.; Hsu, D.; Xu, R.; Harpaz, N.; Dannenberg, A.J.; et al. Toll-like receptor-4 promotes the development of colitis-associated colorectal tumors. Gastroenterology 2007, 133, 1869–1881. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rakoff-Nahoum, S.; Medzhitov, R. Regulation of spontaneous intestinal tumorigenesis through the adaptor protein MyD88. Science 2007, 317, 124–127. [Google Scholar] [CrossRef] [PubMed]
- Xiao, H.; Gulen, M.F.; Qin, J.; Yao, J.; Bulek, K.; Kish, D.; Altuntas, C.Z.; Wald, D.; Ma, C.; Zhou, H.; et al. The Toll-interleukin-1 receptor member SIGIRR regulates colonic epithelial homeostasis, inflammation, and tumorigenesis. Immunity 2007, 26, 461–475. [Google Scholar] [CrossRef] [Green Version]
- Kesselring, R.; Glaesner, J.; Hiergeist, A.; Naschberger, E.; Neumann, H.; Brunner, S.M.; Wege, A.K.; Seebauer, C.; Köhl, G.; Merkl, S.; et al. IRAK-M Expression in Tumor Cells Supports Colorectal Cancer Progression through Reduction of Antimicrobial Defense and Stabilization of STAT3. Cancer Cell 2016, 29, 684–696. [Google Scholar] [CrossRef] [Green Version]
- Richard, M.L.; Liguori, G.; Lamas, B.; Brandi, G.; da Costa, G.; Hoffmann, T.W.; Pierluigi Di Simone, M.; Calabrese, C.; Poggioli, G.; Langella, P.; et al. Mucosa-associated microbiota dysbiosis in colitis associated cancer. Gut Microbes 2018, 9, 131–142. [Google Scholar] [CrossRef] [Green Version]
- Vieira, E.L.; Leonel, A.J.; Sad, A.P.; Beltrão, N.R.; Costa, T.F.; Ferreira, T.M.; Gomes-Santos, A.C.; Faria, A.M.; Peluzio, M.C.; Cara, D.C.; et al. Oral administration of sodium butyrate attenuates inflammation and mucosal lesion in experimental acute ulcerative colitis. J. Nutr. Biochem. 2012, 23, 430–436. [Google Scholar] [CrossRef]
- Sivaprakasam, S.; Bhutia, Y.D.; Yang, S.; Ganapathy, V. Short-chain fatty acid transporters: Role in colonic homeostasis. Compr. Physiol. 2017, 8, 299–314. [Google Scholar] [CrossRef]
- Kim, I.W.; Myung, S.J.; Do, M.Y.; Ryu, Y.M.; Kim, M.J.; Do, E.J.; Park, S.; Yoon, S.M.; Ye, B.D.; Byeon, J.S.; et al. Western-style diets induce macrophage infiltration and contribute to colitis-associated carcinogenesis. J. Gastroenterol. Hepatol. 2010, 25, 1785–1794. [Google Scholar] [CrossRef]
- Maddocks, O.D.; Short, A.J.; Donnenberg, M.S.; Bader, S.; Harrison, D.J. Attaching and effacing Escherichia coli downregulate DNA mismatch repair protein in vitro and are associated with colorectal adenocarcinomas in humans. PLoS ONE 2009, 4, e5517. [Google Scholar] [CrossRef] [Green Version]
- Atarashi, K.; Tanoue, T.; Shima, T.; Imaoka, A.; Kuwahara, T.; Momose, Y.; Cheng, G.; Yamasaki, S.; Saito, T.; Ohba, Y.; et al. Induction of colonic regulatory T cells by indigenous Clostridium species. Science 2011, 331, 337–341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fukuda, N.N.; Murakami, S.; Miyauchi, E.; Hino, S.; Atarashi, K.; Onawa, S.; Fujimura, Y.; Lockett, T.; Clarke, J.M.; Ohno, H.; et al. Commensal microbe-derived butyrate induces the differentiation of colonic regulatory T cells. Nature 2013, 504, 446–450. [Google Scholar] [CrossRef]
- Ladoire, S.; Martin, F.; Ghiringhelli, F. Prognostic role of FOXP3+ regulatory T cells infiltrating human carcinomas: The paradox of colorectal cancer. Cancer Immunol. Immunother. CII 2011, 60, 909–918. [Google Scholar] [CrossRef] [PubMed]
- Nishikawa, H.; Sakaguchi, S. Regulatory T cells in tumor immunity. Int. J. Cancer 2010, 127, 759–767. [Google Scholar] [CrossRef] [PubMed]



© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hirano, T.; Hirayama, D.; Wagatsuma, K.; Yamakawa, T.; Yokoyama, Y.; Nakase, H. Immunological Mechanisms in Inflammation-Associated Colon Carcinogenesis. Int. J. Mol. Sci. 2020, 21, 3062. https://doi.org/10.3390/ijms21093062
Hirano T, Hirayama D, Wagatsuma K, Yamakawa T, Yokoyama Y, Nakase H. Immunological Mechanisms in Inflammation-Associated Colon Carcinogenesis. International Journal of Molecular Sciences. 2020; 21(9):3062. https://doi.org/10.3390/ijms21093062
Chicago/Turabian StyleHirano, Takehiro, Daisuke Hirayama, Kohei Wagatsuma, Tsukasa Yamakawa, Yoshihiro Yokoyama, and Hiroshi Nakase. 2020. "Immunological Mechanisms in Inflammation-Associated Colon Carcinogenesis" International Journal of Molecular Sciences 21, no. 9: 3062. https://doi.org/10.3390/ijms21093062
APA StyleHirano, T., Hirayama, D., Wagatsuma, K., Yamakawa, T., Yokoyama, Y., & Nakase, H. (2020). Immunological Mechanisms in Inflammation-Associated Colon Carcinogenesis. International Journal of Molecular Sciences, 21(9), 3062. https://doi.org/10.3390/ijms21093062