Targeting Mitochondrial Network Architecture in Down Syndrome and Aging

 , , , and
, , , and
Abstract
:1. Introduction
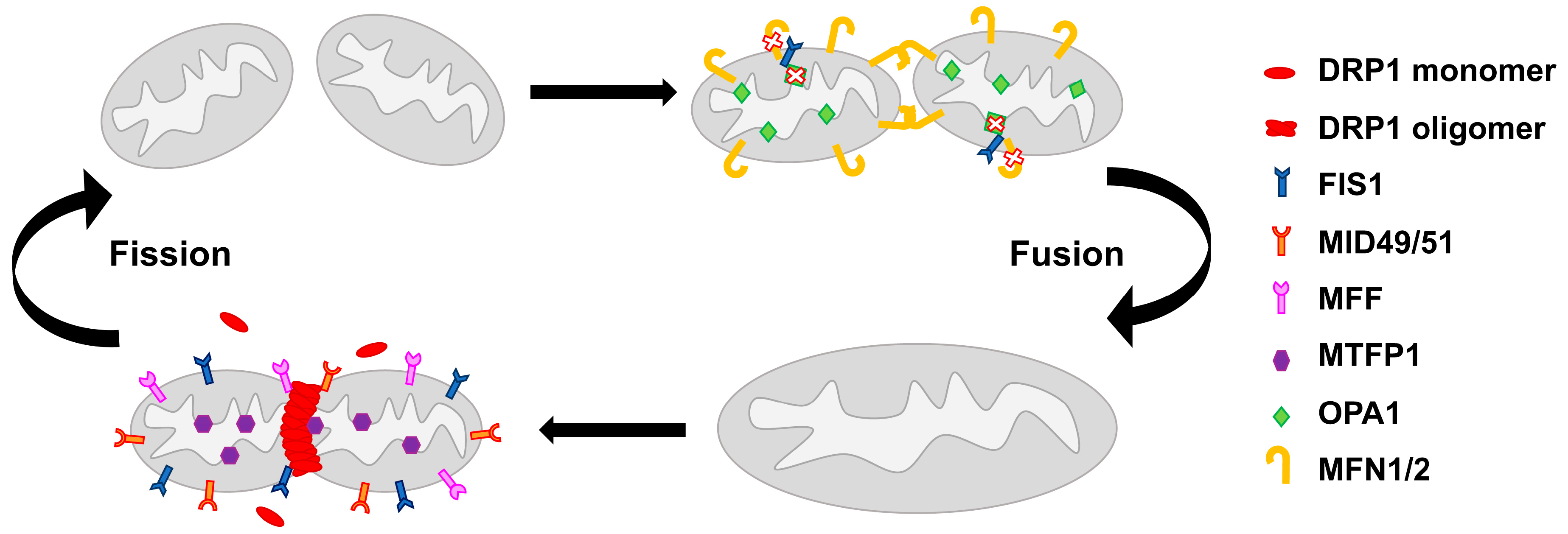
2. Mitochondrial Network Architecture
3. Mitochondrial Homeostasis
4. Accelerated Aging in Down Syndrome
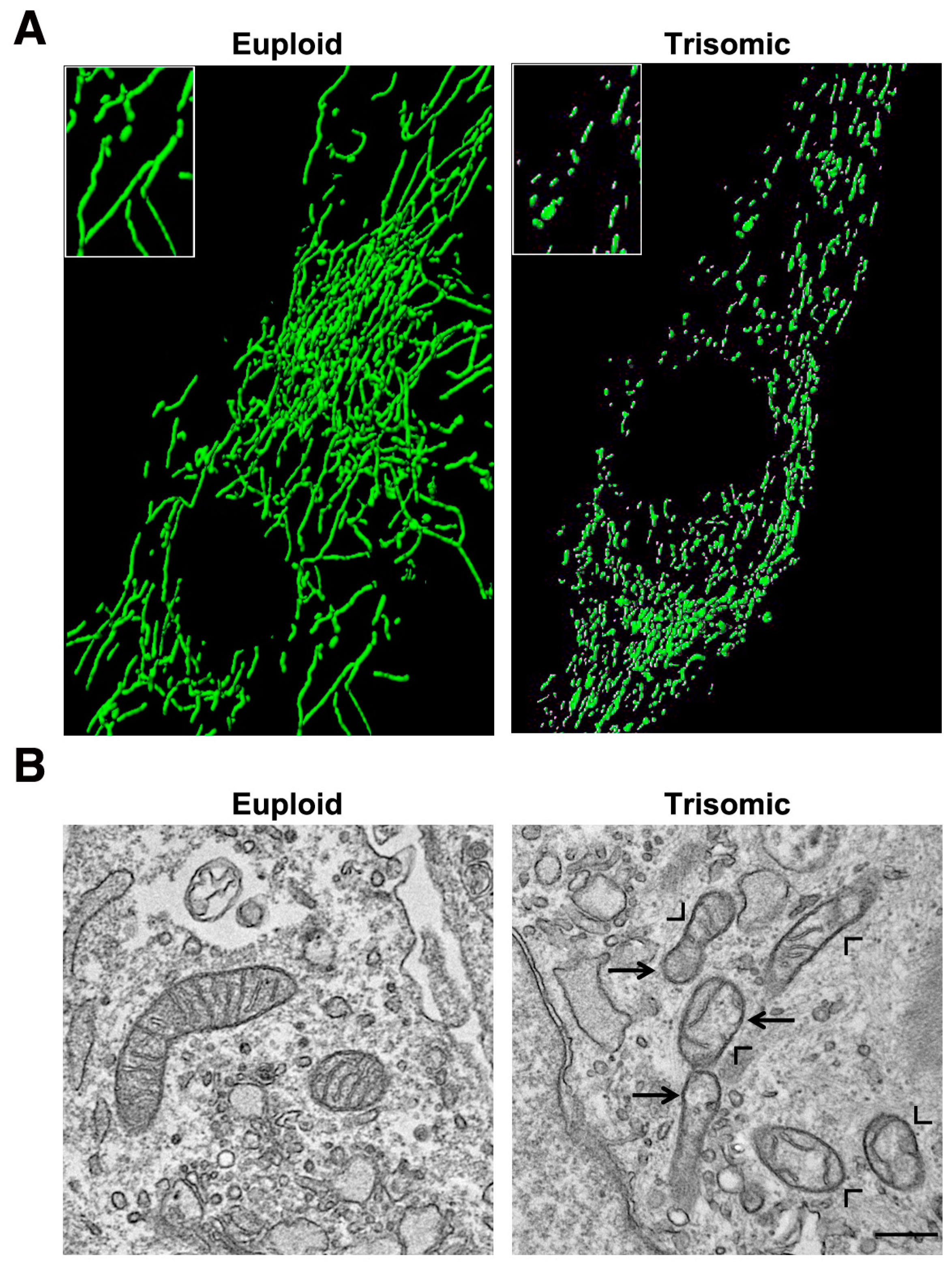
5. Mitochondrial Homeostasis Is Altered in DS and Aging
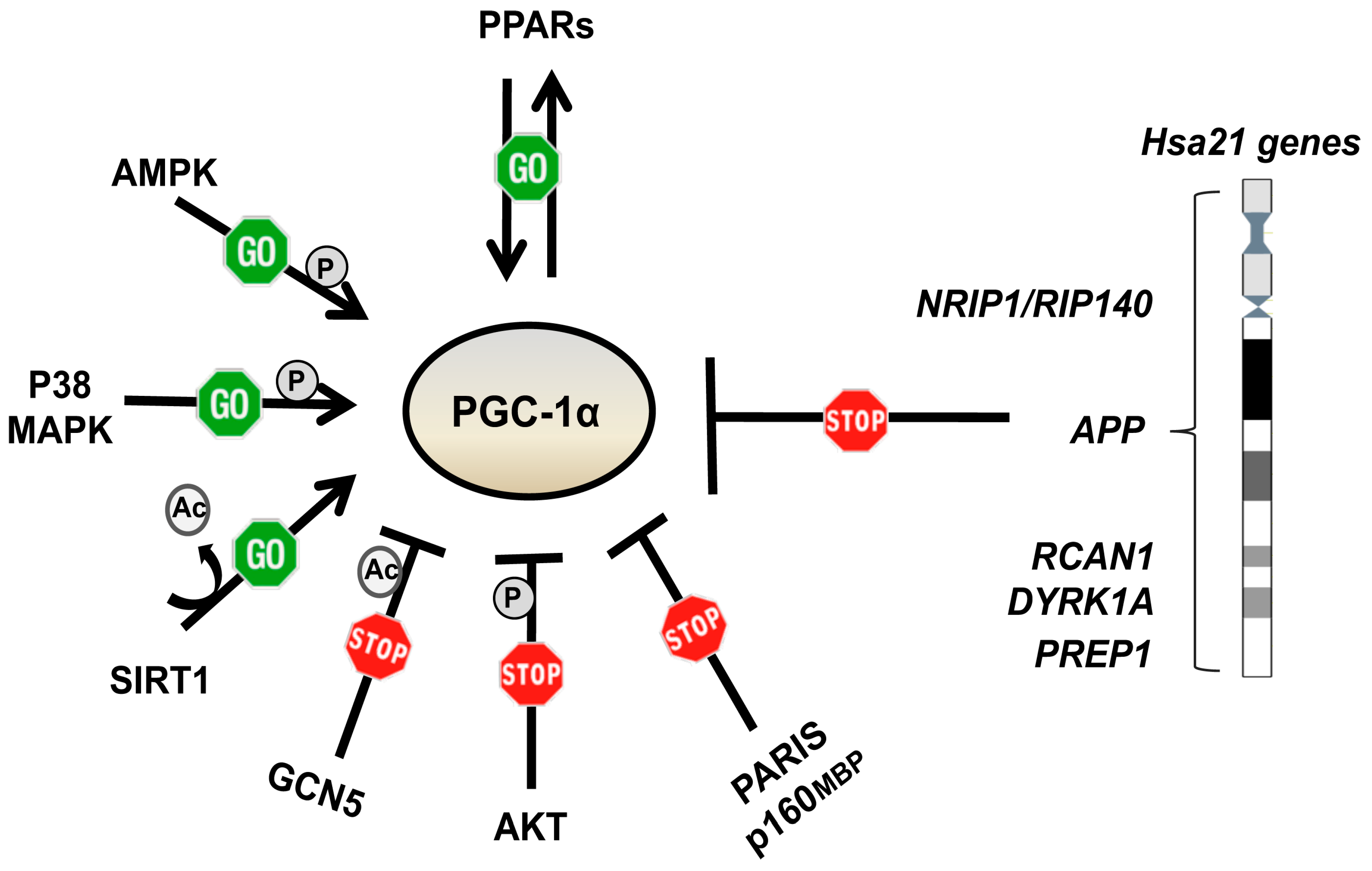
6. Pharmacological Strategies to Target Mitochondrial Network Architecture
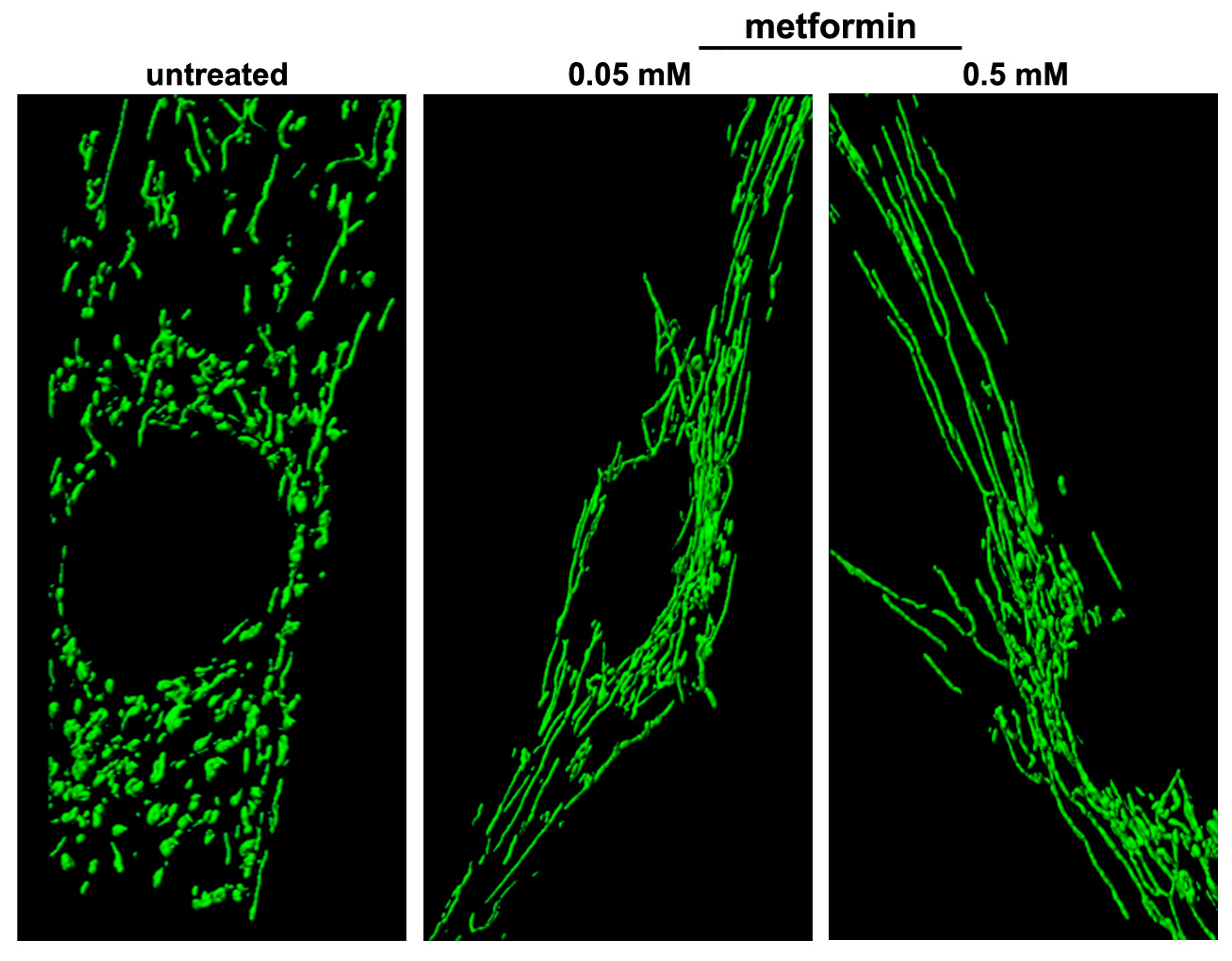
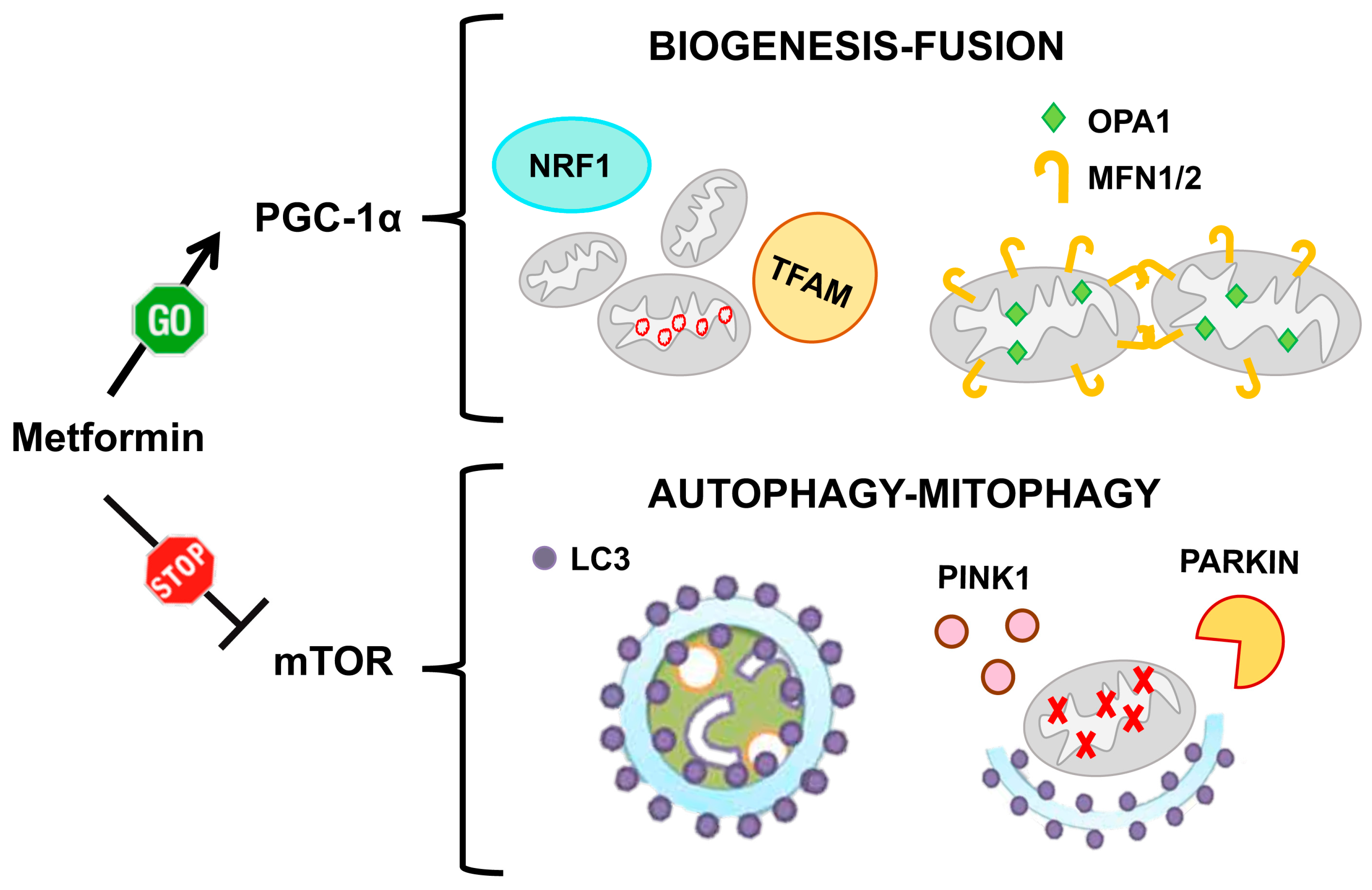
6.1. Metformin
6.2. Other PGC-1α Activating and/or mTOR Inhibiting Drugs
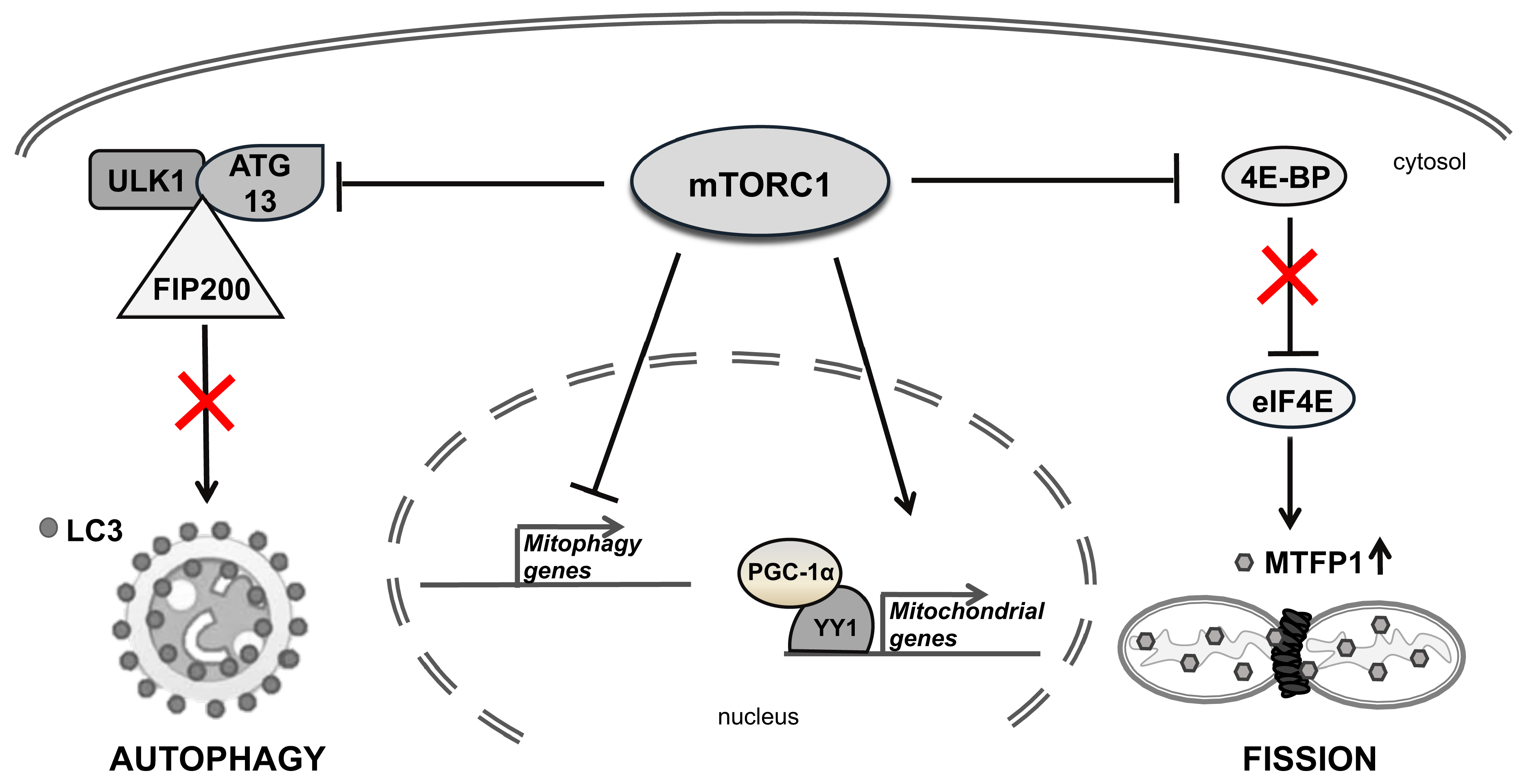
6.3. mTOR Direct Inhibitors
6.4. Drugs Affecting Mitochondrial Fission/Fusion Machinery
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Spinelli, J.B.; Haigis, M.C. The multifaceted contributions of mitochondria to cellular metabolism. Nat. Cell Biol. 2018, 20, 745–754. [Google Scholar] [CrossRef]
- Duchen, M.R. Mitochondria and calcium: From cell signalling to cell death. J. Physiol. 2000, 529, 57–68. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Youle, R.J. The role of mitochondria in apoptosis*. Ann. Rev. Genet. 2009, 43, 95–118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maeda, Y.; Chida, J. Control of cell differentiation by mitochondria, typically evidenced in dictyostelium development. Biomolecules 2013, 3, 943–966. [Google Scholar] [CrossRef] [PubMed]
- Buck, M.D.; O’Sullivan, D.; Klein Geltink, R.I.; Curtis, J.D.; Chang, C.H.; Sanin, D.E.; Qiu, J.; Kretz, O.; Braas, D.; van der Windt, G.J.; et al. Mitochondrial Dynamics Controls T Cell Fate through Metabolic Programming. Cell 2016, 166, 63–76. [Google Scholar] [CrossRef] [Green Version]
- Yu, S.B.; Pekkurnaz, G. Mechanisms Orchestrating Mitochondrial Dynamics for Energy Homeostasis. J. Mol. Biol. 2018, 430, 3922–3941. [Google Scholar] [CrossRef]
- Liesa, M.; Shirihai, O.S. Mitochondrial dynamics in the regulation of nutrient utilization and energy expenditure. Cell Metab. 2013, 17, 491–506. [Google Scholar] [CrossRef] [Green Version]
- Patten, D.A.; Wong, J.; Khacho, M.; Soubannier, V.; Mailloux, R.J.; Pilon-Larose, K.; MacLaurin, J.G.; Park, D.S.; McBride, H.M.; Trinkle-Mulcahy, L.; et al. OPA1-dependent cristae modulation is essential for cellular adaptation to metabolic demand. EMBO J. 2014, 33, 2676–2691. [Google Scholar] [CrossRef] [Green Version]
- Rambold, A.S.; Kostelecky, B.; Elia, N.; Lippincott-Schwartz, J. Tubular network formation protects mitochondria from autophagosomal degradation during nutrient starvation. Proc. Natl. Acad. Sci. USA 2011, 108, 10190–10195. [Google Scholar] [CrossRef] [Green Version]
- Gao, Z.; Li, Y.; Wang, F.; Huang, T.; Fan, K.; Zhang, Y.; Zhong, J.; Cao, Q.; Chao, T.; Jia, J.; et al. Mitochondrial dynamics controls anti-tumour innate immunity by regulating CHIP-IRF1 axis stability. Nat. Commun. 2017, 8, 1805. [Google Scholar] [CrossRef] [Green Version]
- Mitra, K.; Wunder, C.; Roysam, B.; Lin, G.; Lippincott-Schwartz, J. A hyperfused mitochondrial state achieved at G1-S regulates cyclin E buildup and entry into S phase. Proc. Natl. Acad. Sci. USA 2009, 106, 11960–11965. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gatta, A.T.; Levine, T.P. Piecing Together the Patchwork of Contact Sites. Trends Cell Biol. 2017, 27, 214–229. [Google Scholar] [CrossRef] [PubMed]
- Schorr, S.; van der Laan, M. Integrative functions of the mitochondrial contact site and cristae organizing system. Semin. Cell Dev. Biol. 2018, 76, 191–200. [Google Scholar] [CrossRef] [PubMed]
- Dolman, N.J.; Gerasimenko, J.V.; Gerasimenko, O.V.; Voronina, S.G.; Petersen, O.H.; Tepikin, A.V. Stable Golgi-mitochondria complexes and formation of Golgi Ca(2+) gradients in pancreatic acinar cells. J. Biol. Chem. 2005, 280, 15794–15799. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ribeiro, C.M.; Paradiso, A.M.; Livraghi, A.; Boucher, R.C. The mitochondrial barriers segregate agonist-induced calcium-dependent functions in human airway epithelia. J. Gen. Physiol. 2003, 122, 377–387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bruce, J.I.; Giovannucci, D.R.; Blinder, G.; Shuttleworth, T.J.; Yule, D.I. Modulation of [Ca2+]i signaling dynamics and metabolism by perinuclear mitochondria in mouse parotid acinar cells. J. Biol. Chem. 2004, 279, 12909–12917. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ureshino, R.P.; Erustes, A.G.; Bassani, T.B.; Wachilewski, P.; Guarache, G.C.; Nascimento, A.C.; Costa, A.J.; Smaili, S.S.; Pereira, G. The Interplay between Ca(2+) Signaling Pathways and Neurodegeneration. Int. J. Mol. Sci. 2019, 20, 4. [Google Scholar] [CrossRef] [Green Version]
- Cortes, L.; Malva, J.; Rego, A.C.; Pereira, C.F. Calcium Signaling in Aging and Neurodegenerative Diseases 2019. Int. J. Mol. Sci. 2020, 21, 1125. [Google Scholar] [CrossRef] [Green Version]
- Friedman, J.R.; Lackner, L.L.; West, M.; DiBenedetto, J.R.; Nunnari, J.; Voeltz, G.K. ER tubules mark sites of mitochondrial division. Science 2011, 334, 358–362. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Z.; Torres, M.; Sha, H.; Halbrook, C.J.; Van den Bergh, F.; Reinert, R.B.; Yamada, T.; Wang, S.; Luo, Y.; Hunter, A.H.; et al. Endoplasmic reticulum-associated degradation regulates mitochondrial dynamics in brown adipocytes. Science 2020, 368, 54–60. [Google Scholar] [CrossRef]
- Moltedo, O.; Remondelli, P.; Amodio, G. The Mitochondria-Endoplasmic Reticulum Contacts and Their Critical Role in Aging and Age-Associated Diseases. Front. Cell Dev. Biol. 2019, 7, 172. [Google Scholar] [CrossRef]
- Shai, N.; Yifrach, E.; van Roermund, C.W.T.; Cohen, N.; Bibi, C.; Ijlst, L.; Cavellini, L.; Meurisse, J.; Schuster, R.; Zada, L.; et al. Systematic mapping of contact sites reveals tethers and a function for the peroxisome-mitochondria contact. Nat. Commun. 2018, 9, 1761. [Google Scholar] [CrossRef] [PubMed]
- Piccoli, C.; Izzo, A.; Scrima, R.; Bonfiglio, F.; Manco, R.; Negri, R.; Quarato, G.; Cela, O.; Ripoli, M.; Prisco, M.; et al. Chronic pro-oxidative state and mitochondrial dysfunctions are more pronounced in fibroblasts from Down syndrome foeti with congenital heart defects. Hum. Mol. Genet. 2013, 22, 1218–1232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Izzo, A.; Nitti, M.; Mollo, N.; Paladino, S.; Procaccini, C.; Faicchia, D.; Cali, G.; Genesio, R.; Bonfiglio, F.; Cicatiello, R.; et al. Metformin restores the mitochondrial network and reverses mitochondrial dysfunction in Down syndrome cells. Hum. Mol. Genet. 2017, 26, 1056–1069. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Izzo, A.; Mollo, N.; Nitti, M.; Paladino, S.; Cali, G.; Genesio, R.; Bonfiglio, F.; Cicatiello, R.; Barbato, M.; Sarnataro, V.; et al. Mitochondrial dysfunction in down syndrome: Molecular mechanisms and therapeutic targets. Mol. Med. 2018, 24, 2. [Google Scholar] [CrossRef] [Green Version]
- Son, J.M.; Lee, C. Mitochondria: Multifaceted regulators of aging. BMB Rep. 2019, 52, 13–23. [Google Scholar] [CrossRef] [Green Version]
- Bordi, M.; Darji, S.; Sato, Y.; Mellen, M.; Berg, M.J.; Kumar, A.; Jiang, Y.; Nixon, R.A. mTOR hyperactivation in Down Syndrome underlies deficits in autophagy induction, autophagosome formation, and mitophagy. Cell Death Dis. 2019, 10, 563. [Google Scholar] [CrossRef] [Green Version]
- Kowald, A.; Kirkwood, T.B. The evolution and role of mitochondrial fusion and fission in aging and disease. Commun. Integr. Biol. 2011, 4, 627–629. [Google Scholar] [CrossRef]
- Cogliati, S.; Enriquez, J.A.; Scorrano, L. Mitochondrial Cristae: Where Beauty Meets Functionality. Trends Biochem. Sci. 2016, 41, 261–273. [Google Scholar] [CrossRef] [Green Version]
- Scott, I.; Youle, R.J. Mitochondrial fission and fusion. Essays Biochem. 2010, 47, 85–98. [Google Scholar] [CrossRef] [Green Version]
- Tilokani, L.; Nagashima, S.; Paupe, V.; Prudent, J. Mitochondrial dynamics: Overview of molecular mechanisms. Essays Biochem. 2018, 62, 341–360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smirnova, E.; Griparic, L.; Shurland, D.L.; van der Bliek, A.M. Dynamin-related protein Drp1 is required for mitochondrial division in mammalian cells. Mol. Biol. Cell 2001, 12, 2245–2256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loson, O.C.; Song, Z.; Chen, H.; Chan, D.C. Fis1, Mff, MiD49, and MiD51 mediate Drp1 recruitment in mitochondrial fission. Mol. Biol. Cell 2013, 24, 659–667. [Google Scholar] [CrossRef] [PubMed]
- Otsuga, D.; Keegan, B.R.; Brisch, E.; Thatcher, J.W.; Hermann, G.J.; Bleazard, W.; Shaw, J.M. The dynamin-related GTPase, Dnm1p, controls mitochondrial morphology in yeast. J. Cell Biol. 1998, 143, 333–349. [Google Scholar] [CrossRef] [Green Version]
- Taguchi, N.; Ishihara, N.; Jofuku, A.; Oka, T.; Mihara, K. Mitotic phosphorylation of dynamin-related GTPase Drp1 participates in mitochondrial fission. J. Biol. Chem. 2007, 282, 11521–11529. [Google Scholar] [CrossRef] [Green Version]
- Palmer, C.S.; Osellame, L.D.; Laine, D.; Koutsopoulos, O.S.; Frazier, A.E.; Ryan, M.T. MiD49 and MiD51, new components of the mitochondrial fission machinery. EMBO Rep. 2011, 12, 565–573. [Google Scholar] [CrossRef]
- Otera, H.; Wang, C.; Cleland, M.M.; Setoguchi, K.; Yokota, S.; Youle, R.J.; Mihara, K. Mff is an essential factor for mitochondrial recruitment of Drp1 during mitochondrial fission in mammalian cells. J. Cell Biol. 2010, 191, 1141–1158. [Google Scholar] [CrossRef] [Green Version]
- Simula, L.; Pacella, I.; Colamatteo, A.; Procaccini, C.; Cancila, V.; Bordi, M.; Tregnago, C.; Corrado, M.; Pigazzi, M.; Barnaba, V.; et al. Drp1 Controls Effective T Cell Immune-Surveillance by Regulating T Cell Migration, Proliferation, and cMyc-Dependent Metabolic Reprogramming. Cell Rep. 2018, 25, 3059–3073.e10. [Google Scholar] [CrossRef] [Green Version]
- Tondera, D.; Czauderna, F.; Paulick, K.; Schwarzer, R.; Kaufmann, J.; Santel, A. The mitochondrial protein MTP18 contributes to mitochondrial fission in mammalian cells. J. Cell Sci. 2005, 118, 3049–3059. [Google Scholar] [CrossRef] [Green Version]
- Tondera, D.; Santel, A.; Schwarzer, R.; Dames, S.; Giese, K.; Klippel, A.; Kaufmann, J. Knockdown of MTP18, a novel phosphatidylinositol 3-kinase-dependent protein, affects mitochondrial morphology and induces apoptosis. J. Biol. Chem. 2004, 279, 31544–31555. [Google Scholar] [CrossRef] [Green Version]
- Ehses, S.; Raschke, I.; Mancuso, G.; Bernacchia, A.; Geimer, S.; Tondera, D.; Martinou, J.C.; Westermann, B.; Rugarli, E.I.; Langer, T. Regulation of OPA1 processing and mitochondrial fusion by m-AAA protease isoenzymes and OMA1. J. Cell Biol. 2009, 187, 1023–1036. [Google Scholar] [CrossRef] [PubMed]
- Song, Z.; Chen, H.; Fiket, M.; Alexander, C.; Chan, D.C. OPA1 processing controls mitochondrial fusion and is regulated by mRNA splicing, membrane potential, and Yme1L. J. Cell Biol. 2007, 178, 749–755. [Google Scholar] [CrossRef] [PubMed]
- Cipolat, S.; Rudka, T.; Hartmann, D.; Costa, V.; Serneels, L.; Craessaerts, K.; Metzger, K.; Frezza, C.; Annaert, W.; D’Adamio, L.; et al. Mitochondrial rhomboid PARL regulates cytochrome c release during apoptosis via OPA1-dependent cristae remodeling. Cell 2006, 126, 163–175. [Google Scholar] [CrossRef] [PubMed]
- Frezza, C.; Cipolat, S.; Martins de Brito, O.; Micaroni, M.; Beznoussenko, G.V.; Rudka, T.; Bartoli, D.; Polishuck, R.S.; Danial, N.N.; De Strooper, B.; et al. OPA1 controls apoptotic cristae remodeling independently from mitochondrial fusion. Cell 2006, 126, 177–189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duvezin-Caubet, S.; Jagasia, R.; Wagener, J.; Hofmann, S.; Trifunovic, A.; Hansson, A.; Chomyn, A.; Bauer, M.F.; Attardi, G.; Larsson, N.G.; et al. Proteolytic processing of OPA1 links mitochondrial dysfunction to alterations in mitochondrial morphology. J. Biol. Chem. 2006, 281, 37972–37979. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pellegrini, L.; Scorrano, L. A cut short to death: Parl and Opa1 in the regulation of mitochondrial morphology and apoptosis. Cell Death Differ. 2007, 14, 1275–1284. [Google Scholar] [CrossRef] [Green Version]
- Ryu, S.W.; Choi, K.; Park, J.H.; Park, Y.M.; Kim, S.; Choi, C. Mitofusin 1 inhibits an apoptosis-associated amino-terminal conformational change in Bax, but not its mitochondrial translocation, in a GTPase-dependent manner. Cancer Lett. 2012, 323, 62–68. [Google Scholar] [CrossRef]
- Escobar-Henriques, M. Mitofusins: Ubiquitylation promotes fusion. Cell Res. 2014, 24, 387–388. [Google Scholar] [CrossRef] [Green Version]
- Leboucher, G.P.; Tsai, Y.C.; Yang, M.; Shaw, K.C.; Zhou, M.; Veenstra, T.D.; Glickman, M.H.; Weissman, A.M. Stress-induced phosphorylation and proteasomal degradation of mitofusin 2 facilitates mitochondrial fragmentation and apoptosis. Mol. Cell 2012, 47, 547–557. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Yang, L.; Gao, Y.F.; Fan, Z.M.; Cai, X.Y.; Liu, M.Y.; Guo, X.R.; Gao, C.L.; Xia, Z.K. MicroRNA-106b induces mitochondrial dysfunction and insulin resistance in C2C12 myotubes by targeting mitofusin-2. Mol. Cell. Endocrinol. 2013, 381, 230–240. [Google Scholar] [CrossRef]
- Chen, Y.; Dorn, G.W. PINK1-phosphorylated mitofusin 2 is a Parkin receptor for culling damaged mitochondria. Science 2013, 340, 471–475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eura, Y.; Ishihara, N.; Oka, T.; Mihara, K. Identification of a novel protein that regulates mitochondrial fusion by modulating mitofusin (Mfn) protein function. J. Cell Sci. 2006, 119, 4913–4925. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.; Detmer, S.A.; Ewald, A.J.; Griffin, E.E.; Fraser, S.E.; Chan, D.C. Mitofusins Mfn1 and Mfn2 coordinately regulate mitochondrial fusion and are essential for embryonic development. J. Cell Biol. 2003, 160, 189–200. [Google Scholar] [CrossRef] [PubMed]
- Davies, V.J.; Hollins, A.J.; Piechota, M.J.; Yip, W.; Davies, J.R.; White, K.E.; Nicols, P.P.; Boulton, M.E.; Votruba, M. Opa1 deficiency in a mouse model of autosomal dominant optic atrophy impairs mitochondrial morphology, optic nerve structure and visual function. Hum. Mol. Genet. 2007, 16, 1307–1318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, R.; Jin, S.B.; Lendahl, U.; Nister, M.; Zhao, J. Human Fis1 regulates mitochondrial dynamics through inhibition of the fusion machinery. EMBO J. 2019, 38. [Google Scholar] [CrossRef] [PubMed]
- Twig, G.; Elorza, A.; Molina, A.J.; Mohamed, H.; Wikstrom, J.D.; Walzer, G.; Stiles, L.; Haigh, S.E.; Katz, S.; Las, G.; et al. Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J. 2008, 27, 433–446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klionsky, D.J.; Abdelmohsen, K.; Abe, A.; Abedin, M.J.; Abeliovich, H.; Acevedo Arozena, A.; Adachi, H.; Adams, C.M.; Adams, P.D.; Adeli, K.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy 2016, 12, 1–222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, Y.; Lee, H.Y.; Hanna, R.A.; Gustafsson, A.B. Mitochondrial autophagy by Bnip3 involves Drp1-mediated mitochondrial fission and recruitment of Parkin in cardiac myocytes. Am. J. Physiol. Heart Circ. Physiol. 2011, 301, H1924–H1931. [Google Scholar] [CrossRef]
- Palikaras, K.; Tavernarakis, N. Mitochondrial homeostasis: The interplay between mitophagy and mitochondrial biogenesis. Exp. Gerontol. 2014, 56, 182–188. [Google Scholar] [CrossRef]
- Jornayvaz, F.R.; Shulman, G.I. Regulation of mitochondrial biogenesis. Essays Biochem. 2010, 47, 69–84. [Google Scholar] [CrossRef] [Green Version]
- Fernandez-Marcos, P.J.; Auwerx, J. Regulation of PGC-1alpha, a nodal regulator of mitochondrial biogenesis. Am. J. Clin. Nutr. 2011, 93, 884S–890S. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scarpulla, R.C.; Vega, R.B.; Kelly, D.P. Transcriptional integration of mitochondrial biogenesis. Trends Endocrinol. Metab. TEM 2012, 23, 459–466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ekstrand, M.I.; Falkenberg, M.; Rantanen, A.; Park, C.B.; Gaspari, M.; Hultenby, K.; Rustin, P.; Gustafsson, C.M.; Larsson, N.G. Mitochondrial transcription factor A regulates mtDNA copy number in mammals. Hum. Mol. Genet. 2004, 13, 935–944. [Google Scholar] [CrossRef] [PubMed]
- Canugovi, C.; Maynard, S.; Bayne, A.C.; Sykora, P.; Tian, J.; de Souza-Pinto, N.C.; Croteau, D.L.; Bohr, V.A. The mitochondrial transcription factor A functions in mitochondrial base excision repair. DNA Repair 2010, 9, 1080–1089. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dabrowska, A.; Venero, J.L.; Iwasawa, R.; Hankir, M.K.; Rahman, S.; Boobis, A.; Hajji, N. PGC-1alpha controls mitochondrial biogenesis and dynamics in lead-induced neurotoxicity. Aging 2015, 7, 629–647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valenti, D.; de Bari, L.; de Rasmo, D.; Signorile, A.; Henrion-Caude, A.; Contestabile, A.; Vacca, R.A. The polyphenols resveratrol and epigallocatechin-3-gallate restore the severe impairment of mitochondria in hippocampal progenitor cells from a Down syndrome mouse model. Biochim. Biophys. Acta 2016, 1862, 1093–1104. [Google Scholar] [CrossRef]
- Mollo, N.; Nitti, M.; Zerillo, L.; Faicchia, D.; Micillo, T.; Accarino, R.; Secondo, A.; Petrozziello, T.; Cali, G.; Cicatiello, R.; et al. Pioglitazone Improves Mitochondrial Organization and Bioenergetics in Down Syndrome Cells. Front. Genet. 2019, 10, 606. [Google Scholar] [CrossRef] [Green Version]
- Rai, M.; Katti, P.; Nongthomba, U. Drosophila Erect wing (Ewg) controls mitochondrial fusion during muscle growth and maintenance by regulation of the Opa1-like gene. J. Cell Sci. 2014, 127, 191–203. [Google Scholar] [CrossRef] [Green Version]
- Cartoni, R.; Leger, B.; Hock, M.B.; Praz, M.; Crettenand, A.; Pich, S.; Ziltener, J.L.; Luthi, F.; Deriaz, O.; Zorzano, A.; et al. Mitofusins 1/2 and ERRalpha expression are increased in human skeletal muscle after physical exercise. J. Physiol. 2005, 567, 349–358. [Google Scholar] [CrossRef]
- Soriano, F.X.; Liesa, M.; Bach, D.; Chan, D.C.; Palacin, M.; Zorzano, A. Evidence for a mitochondrial regulatory pathway defined by peroxisome proliferator-activated receptor-gamma coactivator-1 alpha, estrogen-related receptor-alpha, and mitofusin 2. Diabetes 2006, 55, 1783–1791. [Google Scholar] [CrossRef] [Green Version]
- Hondares, E.; Mora, O.; Yubero, P.; Rodriguez de la Concepcion, M.; Iglesias, R.; Giralt, M.; Villarroya, F. Thiazolidinediones and rexinoids induce peroxisome proliferator-activated receptor-coactivator (PGC)-1alpha gene transcription: An autoregulatory loop controls PGC-1alpha expression in adipocytes via peroxisome proliferator-activated receptor-gamma coactivation. Endocrinology 2006, 147, 2829–2838. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hondares, E.; Rosell, M.; Diaz-Delfin, J.; Olmos, Y.; Monsalve, M.; Iglesias, R.; Villarroya, F.; Giralt, M. Peroxisome proliferator-activated receptor alpha (PPARalpha) induces PPARgamma coactivator 1alpha (PGC-1alpha) gene expression and contributes to thermogenic activation of brown fat: Involvement of PRDM16. J. Biol. Chem. 2011, 286, 43112–43122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Puigserver, P.; Wu, Z.; Park, C.W.; Graves, R.; Wright, M.; Spiegelman, B.M. A cold-inducible coactivator of nuclear receptors linked to adaptive thermogenesis. Cell 1998, 92, 829–839. [Google Scholar] [CrossRef] [Green Version]
- Izzo, A.; Manco, R.; de Cristofaro, T.; Bonfiglio, F.; Cicatiello, R.; Mollo, N.; De Martino, M.; Genesio, R.; Zannini, M.; Conti, A.; et al. Overexpression of Chromosome 21 miRNAs May Affect Mitochondrial Function in the Hearts of Down Syndrome Fetuses. Int. J. Genom. 2017, 2017, 8737649. [Google Scholar] [CrossRef] [PubMed]
- Fan, M.; Rhee, J.; St-Pierre, J.; Handschin, C.; Puigserver, P.; Lin, J.; Jaeger, S.; Erdjument-Bromage, H.; Tempst, P.; Spiegelman, B.M. Suppression of mitochondrial respiration through recruitment of p160 myb binding protein to PGC-1alpha: Modulation by p38 MAPK. Genes Dev. 2004, 18, 278–289. [Google Scholar] [CrossRef] [Green Version]
- Shin, J.H.; Ko, H.S.; Kang, H.; Lee, Y.; Lee, Y.I.; Pletinkova, O.; Troconso, J.C.; Dawson, V.L.; Dawson, T.M. PARIS (ZNF746) repression of PGC-1alpha contributes to neurodegeneration in Parkinson’s disease. Cell 2011, 144, 689–702. [Google Scholar] [CrossRef] [Green Version]
- Jager, S.; Handschin, C.; St-Pierre, J.; Spiegelman, B.M. AMP-activated protein kinase (AMPK) action in skeletal muscle via direct phosphorylation of PGC-1alpha. Proc. Natl. Acad. Sci. USA 2007, 104, 12017–12022. [Google Scholar] [CrossRef] [Green Version]
- Puigserver, P.; Rhee, J.; Lin, J.; Wu, Z.; Yoon, J.C.; Zhang, C.Y.; Krauss, S.; Mootha, V.K.; Lowell, B.B.; Spiegelman, B.M. Cytokine stimulation of energy expenditure through p38 MAP kinase activation of PPARgamma coactivator-1. Mol. Cell 2001, 8, 971–982. [Google Scholar] [CrossRef]
- Li, X.; Monks, B.; Ge, Q.; Birnbaum, M.J. Akt/PKB regulates hepatic metabolism by directly inhibiting PGC-1alpha transcription coactivator. Nature 2007, 447, 1012–1016. [Google Scholar] [CrossRef]
- Anderson, R.M.; Barger, J.L.; Edwards, M.G.; Braun, K.H.; O’Connor, C.E.; Prolla, T.A.; Weindruch, R. Dynamic regulation of PGC-1alpha localization and turnover implicates mitochondrial adaptation in calorie restriction and the stress response. Aging Cell 2008, 7, 101–111. [Google Scholar] [CrossRef] [Green Version]
- Cunningham, J.T.; Rodgers, J.T.; Arlow, D.H.; Vazquez, F.; Mootha, V.K.; Puigserver, P. mTOR controls mitochondrial oxidative function through a YY1-PGC-1alpha transcriptional complex. Nature 2007, 450, 736–740. [Google Scholar] [CrossRef] [PubMed]
- Wenz, T. Mitochondria and PGC-1alpha in Aging and Age-Associated Diseases. J. Aging Res. 2011, 2011, 810619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laplante, M.; Sabatini, D.M. Regulation of mTORC1 and its impact on gene expression at a glance. J. Cell Sci. 2013, 126, 1713–1719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morita, M.; Gravel, S.P.; Chenard, V.; Sikstrom, K.; Zheng, L.; Alain, T.; Gandin, V.; Avizonis, D.; Arguello, M.; Zakaria, C.; et al. mTORC1 controls mitochondrial activity and biogenesis through 4E-BP-dependent translational regulation. Cell Metab. 2013, 18, 698–711. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.; Zhang, Y.; Ni, M.; Cao, H.; Signer, R.A.J.; Li, D.; Li, M.; Gu, Z.; Hu, Z.; Dickerson, K.E.; et al. Regulation of mitochondrial biogenesis in erythropoiesis by mTORC1-mediated protein translation. Nat. Cell Biol. 2017, 19, 626–638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gandin, V.; Masvidal, L.; Hulea, L.; Gravel, S.P.; Cargnello, M.; McLaughlan, S.; Cai, Y.; Balanathan, P.; Morita, M.; Rajakumar, A.; et al. nanoCAGE reveals 5’ UTR features that define specific modes of translation of functionally related MTOR-sensitive mRNAs. Genome Res. 2016, 26, 636–648. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morita, M.; Prudent, J.; Basu, K.; Goyon, V.; Katsumura, S.; Hulea, L.; Pearl, D.; Siddiqui, N.; Strack, S.; McGuirk, S.; et al. mTOR Controls Mitochondrial Dynamics and Cell Survival via MTFP1. Mol. Cell 2017, 67, 922–935. [Google Scholar] [CrossRef] [Green Version]
- Pickles, S.; Vigie, P.; Youle, R.J. Mitophagy and Quality Control Mechanisms in Mitochondrial Maintenance. Curr. Biol. CB 2018, 28, R170–R185. [Google Scholar] [CrossRef]
- Youle, R.J.; Narendra, D.P. Mechanisms of mitophagy. Nat. Rev. Mol. Cell Biol. 2011, 12, 9–14. [Google Scholar] [CrossRef]
- Rakovic, A.; Shurkewitsch, K.; Seibler, P.; Grunewald, A.; Zanon, A.; Hagenah, J.; Krainc, D.; Klein, C. Phosphatase and tensin homolog (PTEN)-induced putative kinase 1 (PINK1)-dependent ubiquitination of endogenous Parkin attenuates mitophagy: Study in human primary fibroblasts and induced pluripotent stem cell-derived neurons. J. Biol. Chem. 2013, 288, 2223–2237. [Google Scholar] [CrossRef] [Green Version]
- Gegg, M.E.; Cooper, J.M.; Chau, K.Y.; Rojo, M.; Schapira, A.H.; Taanman, J.W. Mitofusin 1 and mitofusin 2 are ubiquitinated in a PINK1/parkin-dependent manner upon induction of mitophagy. Hum. Mol. Genet. 2010, 19, 4861–4870. [Google Scholar] [CrossRef] [PubMed]
- Poole, A.C.; Thomas, R.E.; Yu, S.; Vincow, E.S.; Pallanck, L. The mitochondrial fusion-promoting factor mitofusin is a substrate of the PINK1/parkin pathway. PLoS ONE 2010, 5, e10054. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takikita, S.; Schreiner, C.; Baum, R.; Xie, T.; Ralston, E.; Plotz, P.H.; Raben, N. Fiber type conversion by PGC-1alpha activates lysosomal and autophagosomal biogenesis in both unaffected and Pompe skeletal muscle. PLoS ONE 2010, 5, e15239. [Google Scholar] [CrossRef] [PubMed]
- Settembre, C.; Di Malta, C.; Polito, V.A.; Garcia Arencibia, M.; Vetrini, F.; Erdin, S.; Erdin, S.U.; Huynh, T.; Medina, D.; Colella, P.; et al. TFEB links autophagy to lysosomal biogenesis. Science 2011, 332, 1429–1433. [Google Scholar] [CrossRef] [Green Version]
- Zheng, L.; Bernard-Marissal, N.; Moullan, N.; D’Amico, D.; Auwerx, J.; Moore, D.J.; Knott, G.; Aebischer, P.; Schneider, B.L. Parkin functionally interacts with PGC-1alpha to preserve mitochondria and protect dopaminergic neurons. Hum. Mol. Genet. 2017, 26, 582–598. [Google Scholar] [CrossRef] [Green Version]
- Jiang, H.; Kang, S.U.; Zhang, S.; Karuppagounder, S.; Xu, J.; Lee, Y.K.; Kang, B.G.; Lee, Y.; Zhang, J.; Pletnikova, O.; et al. Adult Conditional Knockout of PGC-1alpha Leads to Loss of Dopamine Neurons. eNeuro 2016, 3. [Google Scholar] [CrossRef] [Green Version]
- Chan, E.Y. mTORC1 phosphorylates the ULK1-mAtg13-FIP200 autophagy regulatory complex. Sci. Signal. 2009, 2, pe51. [Google Scholar] [CrossRef]
- Bartolome, A.; Garcia-Aguilar, A.; Asahara, S.I.; Kido, Y.; Guillen, C.; Pajvani, U.B.; Benito, M. MTORC1 Regulates both General Autophagy and Mitophagy Induction after Oxidative Phosphorylation Uncoupling. Mol. Cell. Biol. 2017, 37, e00441-17. [Google Scholar] [CrossRef] [Green Version]
- Englund, A.; Jonsson, B.; Zander, C.S.; Gustafsson, J.; Anneren, G. Changes in mortality and causes of death in the Swedish Down syndrome population. Am. J. Med. Genet. Part A 2013, 161, 642–649. [Google Scholar] [CrossRef]
- Bayen, E.; Possin, K.L.; Chen, Y.; Cleret de Langavant, L.; Yaffe, K. Prevalence of Aging, Dementia, and Multimorbidity in Older Adults with Down Syndrome. JAMA Neurol. 2018, 75, 1399–1406. [Google Scholar] [CrossRef]
- Carfi, A.; Liperoti, R.; Fusco, D.; Giovannini, S.; Brandi, V.; Vetrano, D.L.; Meloni, E.; Mascia, D.; Villani, E.R.; Manes Gravina, E.; et al. Bone mineral density in adults with Down syndrome. Osteoporos. Int. 2017, 28, 2929–2934. [Google Scholar] [CrossRef] [PubMed]
- Kinnear, D.; Morrison, J.; Allan, L.; Henderson, A.; Smiley, E.; Cooper, S.A. Prevalence of physical conditions and multimorbidity in a cohort of adults with intellectual disabilities with and without Down syndrome: Cross-sectional study. BMJ Open 2018, 8, e018292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prasher, V.P. Down syndrome and thyroid disorders: A review. Down Syndr. Res. Pract. 1999, 6, 25–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Esbensen, A.J. Health conditions associated with aging and end of life of adults with Down syndrome. Int. Rev. Res. Ment. Retard. 2010, 39, 107–126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kusters, M.A.; Verstegen, R.H.; de Vries, E. Down syndrome: Is it really characterized by precocious immunosenescence? Aging Dis. 2011, 2, 538–545. [Google Scholar] [PubMed]
- Capone, G.T.; Chicoine, B.; Bulova, P.; Stephens, M.; Hart, S.; Crissman, B.; Videlefsky, A.; Myers, K.; Roizen, N.; Esbensen, A.; et al. Co-occurring medical conditions in adults with Down syndrome: A systematic review toward the development of health care guidelines. Am. J. Med. Genet. Part A 2018, 176, 116–133. [Google Scholar] [CrossRef]
- Rachidi, M.; Lopes, C. Mental retardation in Down syndrome: From gene dosage imbalance to molecular and cellular mechanisms. Neurosci. Res. 2007, 59, 349–369. [Google Scholar] [CrossRef]
- Coppus, A.; Evenhuis, H.; Verberne, G.J.; Visser, F.; van Gool, P.; Eikelenboom, P.; van Duijin, C. Dementia and mortality in persons with Down’s syndrome. J. Intellect. Disabil. Res. JIDR 2006, 50, 768–777. [Google Scholar] [CrossRef]
- Zigman, W.B.; Devenny, D.A.; Krinsky-McHale, S.J.; Jenkins, E.C.; Urv, T.K.; Wegiel, J.; Schupf, N.; Silverman, W. Alzheimer’s Disease in Adults with Down Syndrome. Int. Rev. Res. Ment. Retard. 2008, 36, 103–145. [Google Scholar] [CrossRef] [Green Version]
- Cipriani, G.; Danti, S.; Carlesi, C.; Di Fiorino, M. Aging with Down Syndrome: The Dual Diagnosis: Alzheimer’s Disease and Down Syndrome. Am. J. Alzheimer’s Dis. Other Dement. 2018, 33, 253–262. [Google Scholar] [CrossRef]
- Lott, I.T.; Doran, E.; Nguyen, V.Q.; Tournay, A.; Movsesyan, N.; Gillen, D.L. Down syndrome and dementia: Seizures and cognitive decline. J. Alzheimer’s Dis. JAD 2012, 29, 177–185. [Google Scholar] [CrossRef] [PubMed]
- Borelli, V.; Vanhooren, V.; Lonardi, E.; Reiding, K.R.; Capri, M.; Libert, C.; Garagnani, P.; Salvioli, S.; Franceschi, C.; Wuhrer, M. Plasma N-Glycome Signature of Down Syndrome. J. Proteome Res. 2015, 14, 4232–4245. [Google Scholar] [CrossRef]
- Sanders, J.L.; Newman, A.B. Telomere length in epidemiology: A biomarker of aging, age-related disease, both, or neither? Epidemiol. Rev. 2013, 35, 112–131. [Google Scholar] [CrossRef] [Green Version]
- Vaziri, H.; Schachter, F.; Uchida, I.; Wei, L.; Zhu, X.; Effros, R.; Cohen, D.; Harley, C.B. Loss of telomeric DNA during aging of normal and trisomy 21 human lymphocytes. Am. J. Hum. Genet. 1993, 52, 661–667. [Google Scholar] [PubMed]
- Jenkins, E.C.; Velinov, M.T.; Ye, L.; Gu, H.; Li, S.; Jenkins, E.C., Jr.; Brooks, S.S.; Pang, D.; Devenny, D.A.; Zigman, W.B.; et al. Telomere shortening in T lymphocytes of older individuals with Down syndrome and dementia. Neurobiol. Aging 2006, 27, 941–945. [Google Scholar] [CrossRef] [PubMed]
- Gimeno, A.; Garcia-Gimenez, J.L.; Audi, L.; Toran, N.; Andaluz, P.; Dasi, F.; Vina, J.; Pallardo, F.V. Decreased cell proliferation and higher oxidative stress in fibroblasts from Down Syndrome fetuses. Preliminary study. Biochim. Biophys. Acta 2014, 1842, 116–125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horvath, S.; Garagnani, P.; Bacalini, M.G.; Pirazzini, C.; Salvioli, S.; Gentilini, D.; Di Blasio, A.M.; Giuliani, C.; Tung, S.; Vinters, H.V.; et al. Accelerated epigenetic aging in Down syndrome. Aging Cell 2015, 14, 491–495. [Google Scholar] [CrossRef]
- Jin, S.; Lee, Y.K.; Lim, Y.C.; Zheng, Z.; Lin, X.M.; Ng, D.P.; Holbrook, J.D.; Law, H.Y.; Kwek, K.Y.; Yeo, G.S.; et al. Global DNA hypermethylation in down syndrome placenta. PLoS Genet. 2013, 9, e1003515. [Google Scholar] [CrossRef] [Green Version]
- Mendioroz, M.; Do, C.; Jiang, X.; Liu, C.; Darbary, H.K.; Lang, C.F.; Lin, J.; Thomas, A.; Abu-Amero, S.; Stanier, P.; et al. Trans effects of chromosome aneuploidies on DNA methylation patterns in human Down syndrome and mouse models. Genome Biol. 2015, 16, 263. [Google Scholar] [CrossRef] [Green Version]
- Bacalini, M.G.; Gentilini, D.; Boattini, A.; Giampieri, E.; Pirazzini, C.; Giuliani, C.; Fontanesi, E.; Scurti, M.; Remondini, D.; Capri, M.; et al. Identification of a DNA methylation signature in blood cells from persons with Down Syndrome. Aging 2015, 7, 82–96. [Google Scholar] [CrossRef] [Green Version]
- Ciccarone, F.; Valentini, E.; Malavolta, M.; Zampieri, M.; Bacalini, M.G.; Calabrese, R.; Guastafierro, T.; Reale, A.; Franceschi, C.; Capri, M.; et al. DNA Hydroxymethylation Levels Are Altered in Blood Cells from Down Syndrome Persons Enrolled in the MARK-AGE Project. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2018, 73, 737–744. [Google Scholar] [CrossRef] [PubMed]
- Cole, J.H.; Annus, T.; Wilson, L.R.; Remtulla, R.; Hong, Y.T.; Fryer, T.D.; Acosta-Cabronero, J.; Cardenas-Blanco, A.; Smith, R.; Menon, D.K.; et al. Brain-predicted age in Down syndrome is associated with beta amyloid deposition and cognitive decline. Neurobiol. Aging 2017, 56, 41–49. [Google Scholar] [CrossRef] [PubMed]
- Bambrick, L.L.; Fiskum, G. Mitochondrial dysfunction in mouse trisomy 16 brain. Brain Res. 2008, 1188, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Aburawi, E.H.; Souid, A.K. Lymphocyte respiration in children with Trisomy 21. BMC Pediatr. 2012, 12, 193. [Google Scholar] [CrossRef] [Green Version]
- Helguera, P.; Seiglie, J.; Rodriguez, J.; Hanna, M.; Helguera, G.; Busciglio, J. Adaptive downregulation of mitochondrial function in down syndrome. Cell Metab. 2013, 17, 132–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caracausi, M.; Ghini, V.; Locatelli, C.; Mericio, M.; Piovesan, A.; Antonaros, F.; Pelleri, M.C.; Vitale, L.; Vacca, R.A.; Bedetti, F.; et al. Plasma and urinary metabolomic profiles of Down syndrome correlate with alteration of mitochondrial metabolism. Sci. Rep. 2018, 8, 2977. [Google Scholar] [CrossRef] [Green Version]
- Valenti, D.; Tullo, A.; Caratozzolo, M.F.; Merafina, R.S.; Scartezzini, P.; Marra, E.; Vacca, R.A. Impairment of F1F0-ATPase, adenine nucleotide translocator and adenylate kinase causes mitochondrial energy deficit in human skin fibroblasts with chromosome 21 trisomy. Biochem. J. 2010, 431, 299–310. [Google Scholar] [CrossRef]
- Valenti, D.; Manente, G.A.; Moro, L.; Marra, E.; Vacca, R.A. Deficit of complex I activity in human skin fibroblasts with chromosome 21 trisomy and overproduction of reactive oxygen species by mitochondria: Involvement of the cAMP/PKA signalling pathway. Biochem. J. 2011, 435, 679–688. [Google Scholar] [CrossRef] [Green Version]
- Izzo, A.; Manco, R.; Bonfiglio, F.; Cali, G.; De Cristofaro, T.; Patergnani, S.; Cicatiello, R.; Scrima, R.; Zannini, M.; Pinton, P.; et al. NRIP1/RIP140 siRNA-mediated attenuation counteracts mitochondrial dysfunction in Down syndrome. Hum. Mol. Genet. 2014, 23, 4406–4419. [Google Scholar] [CrossRef]
- Valenti, D.; De Rasmo, D.; Signorile, A.; Rossi, L.; de Bari, L.; Scala, I.; Granese, B.; Papa, S.; Vacca, R.A. Epigallocatechin-3-gallate prevents oxidative phosphorylation deficit and promotes mitochondrial biogenesis in human cells from subjects with Down’s syndrome. Biochim. Biophys. Acta 2013, 1832, 542–552. [Google Scholar] [CrossRef] [Green Version]
- Valenti, D.; de Bari, L.; De Filippis, B.; Henrion-Caude, A.; Vacca, R.A. Mitochondrial dysfunction as a central actor in intellectual disability-related diseases: An overview of Down syndrome, autism, Fragile X and Rett syndrome. Neurosci. Biobehav. Rev. 2014, 46, 202–217. [Google Scholar] [CrossRef] [PubMed]
- Conti, A.; Fabbrini, F.; D’Agostino, P.; Negri, R.; Greco, D.; Genesio, R.; D’Armiento, M.; Olla, C.; Paladini, D.; Zannini, M.; et al. Altered expression of mitochondrial and extracellular matrix genes in the heart of human fetuses with chromosome 21 trisomy. BMC Genom. 2007, 8, 268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zamponi, N.; Zamponi, E.; Cannas, S.A.; Billoni, O.V.; Helguera, P.R.; Chialvo, D.R. Mitochondrial network complexity emerges from fission/fusion dynamics. Sci. Rep. 2018, 8, 363. [Google Scholar] [CrossRef] [Green Version]
- Valenti, D.; Rossi, L.; Marzulli, D.; Bellomo, F.; De Rasmo, D.; Signorile, A.; Vacca, R.A. Inhibition of Drp1-mediated mitochondrial fission improves mitochondrial dynamics and bioenergetics stimulating neurogenesis in hippocampal progenitor cells from a Down syndrome mouse model. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 3117–3127. [Google Scholar] [CrossRef] [PubMed]
- Zick, M.; Rabl, R.; Reichert, A.S. Cristae formation-linking ultrastructure and function of mitochondria. Biochim. Biophys. Acta 2009, 1793, 5–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iyer, A.M.; van Scheppingen, J.; Milenkovic, I.; Anink, J.J.; Adle-Biassette, H.; Kovacs, G.G.; Aronica, E. mTOR Hyperactivation in down syndrome hippocampus appears early during development. J. Neuropathol. Exp. Neurol. 2014, 73, 671–683. [Google Scholar] [CrossRef] [Green Version]
- Perluigi, M.; Di Domenico, F.; Butterfield, D.A. mTOR signaling in aging and neurodegeneration: At the crossroad between metabolism dysfunction and impairment of autophagy. Neurobiol. Dis. 2015, 84, 39–49. [Google Scholar] [CrossRef] [PubMed]
- Tramutola, A.; Triplett, J.C.; Di Domenico, F.; Niedowicz, D.M.; Murphy, M.P.; Coccia, R.; Perluigi, M.; Butterfield, D.A. Alteration of mTOR signaling occurs early in the progression of Alzheimer disease (AD): Analysis of brain from subjects with pre-clinical AD, amnestic mild cognitive impairment and late-stage AD. J. Neurochem. 2015, 133, 739–749. [Google Scholar] [CrossRef]
- Aivazidis, S.; Jain, A.; Rauniyar, A.K.; Anderson, C.C.; Marentette, J.O.; Orlicky, D.J.; Fritz, K.S.; Harris, P.S.; Siegel, D.; Maclean, K.N.; et al. SNARE proteins rescue impaired autophagic flux in Down syndrome. PLoS ONE 2019, 14, e0223254. [Google Scholar] [CrossRef] [Green Version]
- Perluigi, M.; Butterfield, D.A. Oxidative Stress and Down Syndrome: A Route toward Alzheimer-Like Dementia. Curr. Gerontol. Geriatr. Res. 2012, 2012, 724904. [Google Scholar] [CrossRef] [Green Version]
- Kauppila, T.E.S.; Kauppila, J.H.K.; Larsson, N.G. Mammalian Mitochondria and Aging: An Update. Cell Metab. 2017, 25, 57–71. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.J.; McIntyre, R.L.; Janssens, G.E.; Houtkooper, R.H. Mitochondrial fission and fusion: A dynamic role in aging and potential target for age-related disease. Mech. Ageing Dev. 2020, 186, 111212. [Google Scholar] [CrossRef] [PubMed]
- Regmi, S.G.; Rolland, S.G.; Conradt, B. Age-dependent changes in mitochondrial morphology and volume are not predictors of lifespan. Aging 2014, 6, 118–130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, H.C.; Hsu, J.M.; Yen, C.P.; Chao, C.C.; Chen, R.H.; Pan, C.L. Neural activity and CaMKII protect mitochondria from fragmentation in aging Caenorhabditis elegans neurons. Proc. Natl. Acad. Sci. USA 2015, 112, 8768–8773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jendrach, M.; Pohl, S.; Voth, M.; Kowald, A.; Hammerstein, P.; Bereiter-Hahn, J. Morpho-dynamic changes of mitochondria during ageing of human endothelial cells. Mech. Ageing Dev. 2005, 126, 813–821. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ron-Harel, N.; Notarangelo, G.; Ghergurovich, J.M.; Paulo, J.A.; Sage, P.T.; Santos, D.; Satterstrom, F.K.; Gygi, S.P.; Rabinowitz, J.D.; Sharpe, A.H.; et al. Defective respiration and one-carbon metabolism contribute to impaired naive T cell activation in aged mice. Proc. Natl. Acad. Sci. USA 2018, 115, 13347–13352. [Google Scholar] [CrossRef] [Green Version]
- Short, K.R.; Bigelow, M.L.; Kahl, J.; Singh, R.; Coenen-Schimke, J.; Raghavakaimal, S.; Nair, K.S. Decline in skeletal muscle mitochondrial function with aging in humans. Proc. Natl. Acad. Sci. USA 2005, 102, 5618–5623. [Google Scholar] [CrossRef] [Green Version]
- Kang, C.; Ji, L.L. Muscle immobilization and remobilization downregulates PGC-1alpha signaling and the mitochondrial biogenesis pathway. J. Appl. Physiol. 2013, 115, 1618–1625. [Google Scholar] [CrossRef]
- Sahin, E.; Colla, S.; Liesa, M.; Moslehi, J.; Muller, F.L.; Guo, M.; Cooper, M.; Kotton, D.; Fabian, A.J.; Walkey, C.; et al. Telomere dysfunction induces metabolic and mitochondrial compromise. Nature 2011, 470, 359–365. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Chen, X.; Osland, J.; Gerber, S.J.; Luan, C.; Delfino, K.; Goodwin, L.; Yuan, R. Deletion of Nrip1 Extends Female Mice Longevity, Increases Autophagy, and Delays Cell Senescence. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2018, 73, 882–892. [Google Scholar] [CrossRef]
- Leidal, A.M.; Levine, B.; Debnath, J. Autophagy and the cell biology of age-related disease. Nat. Cell Biol. 2018, 20, 1338–1348. [Google Scholar] [CrossRef] [PubMed]
- Roque, W.; Cuevas-Mora, K.; Romero, F. Mitochondrial Quality Control in Age-Related Pulmonary Fibrosis. Int. J. Mol. Sci. 2020, 21, 643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rana, A.; Rera, M.; Walker, D.W. Parkin overexpression during aging reduces proteotoxicity, alters mitochondrial dynamics, and extends lifespan. Proc. Natl. Acad. Sci. USA 2013, 110, 8638–8643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saxton, R.A.; Sabatini, D.M. mTOR Signaling in Growth, Metabolism, and Disease. Cell 2017, 169, 361–371. [Google Scholar] [CrossRef]
- Hsieh, C.C.; Papaconstantinou, J. Akt/PKB and p38 MAPK signaling, translational initiation and longevity in Snell dwarf mouse livers. Mech. Ageing Dev. 2004, 125, 785–798. [Google Scholar] [CrossRef]
- Papadopoli, D.; Boulay, K.; Kazak, L.; Pollak, M.; Mallette, F.; Topisirovic, I.; Hulea, L. mTOR as a central regulator of lifespan and aging. F1000Research 2019, 8. [Google Scholar] [CrossRef]
- Johnson, S.C.; Rabinovitch, P.S.; Kaeberlein, M. mTOR is a key modulator of ageing and age-related disease. Nature 2013, 493, 338–345. [Google Scholar] [CrossRef] [Green Version]
- Zhou, G.; Myers, R.; Li, Y.; Chen, Y.; Shen, X.; Fenyk-Melody, J.; Wu, M.; Ventre, J.; Doebber, T.; Fujii, N.; et al. Role of AMP-activated protein kinase in mechanism of metformin action. J. Clin. Investig. 2001, 108, 1167–1174. [Google Scholar] [CrossRef]
- Owen, M.R.; Doran, E.; Halestrap, A.P. Evidence that metformin exerts its anti-diabetic effects through inhibition of complex 1 of the mitochondrial respiratory chain. Biochem. J. 2000, 348, 607–614. [Google Scholar] [CrossRef]
- Hawley, S.A.; Gadalla, A.E.; Olsen, G.S.; Hardie, D.G. The antidiabetic drug metformin activates the AMP-activated protein kinase cascade via an adenine nucleotide-independent mechanism. Diabetes 2002, 51, 2420–2425. [Google Scholar] [CrossRef] [Green Version]
- Howell, J.J.; Hellberg, K.; Turner, M.; Talbott, G.; Kolar, M.J.; Ross, D.S.; Hoxhaj, G.; Saghatelian, A.; Shaw, R.J.; Manning, B.D. Metformin Inhibits Hepatic mTORC1 Signaling via Dose-Dependent Mechanisms Involving AMPK and the TSC Complex. Cell Metab. 2017, 25, 463–471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ben Sahra, I.; Regazzetti, C.; Robert, G.; Laurent, K.; Le Marchand-Brustel, Y.; Auberger, P.; Tanti, J.F.; Giorgetti-Peraldi, S.; Bost, F. Metformin, independent of AMPK, induces mTOR inhibition and cell-cycle arrest through REDD1. Cancer Res. 2011, 71, 4366–4372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.; Kundu, M.; Viollet, B.; Guan, K.L. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol. 2011, 13, 132–141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, W.; Tian, W.; Hu, Z.; Chen, G.; Huang, L.; Li, W.; Zhang, X.; Xue, P.; Zhou, C.; Liu, L.; et al. ULK1 translocates to mitochondria and phosphorylates FUNDC1 to regulate mitophagy. EMBO Rep. 2014, 15, 566–575. [Google Scholar] [CrossRef] [Green Version]
- Bhansali, S.; Bhansali, A.; Dhawan, V. Metformin promotes mitophagy in mononuclear cells: A potential in vitro model for unraveling metformin’s mechanism of action. Ann. N. Y. Acad. Sci. 2019. [Google Scholar] [CrossRef]
- Bhansali, S.; Bhansali, A.; Dutta, P.; Walia, R.; Dhawan, V. Metformin upregulates mitophagy in patients with T2DM: A randomized placebo-controlled study. J. Cell. Mol. Med. 2020. [Google Scholar] [CrossRef]
- Martin-Montalvo, A.; Mercken, E.M.; Mitchell, S.J.; Palacios, H.H.; Mote, P.L.; Scheibye-Knudsen, M.; Gomes, A.P.; Ward, T.M.; Minor, R.K.; Blouin, M.J.; et al. Metformin improves healthspan and lifespan in mice. Nat. Commun. 2013, 4, 2192. [Google Scholar] [CrossRef]
- Cabreiro, F.; Au, C.; Leung, K.Y.; Vergara-Irigaray, N.; Cocheme, H.M.; Noori, T.; Weinkove, D.; Schuster, E.; Greene, N.D.; Gems, D. Metformin retards aging in C. elegans by altering microbial folate and methionine metabolism. Cell 2013, 153, 228–239. [Google Scholar] [CrossRef] [Green Version]
- Soukas, A.A.; Hao, H.; Wu, L. Metformin as Anti-Aging Therapy: Is It for Everyone? Trends Endocrinol. Metab. TEM 2019, 30, 745–755. [Google Scholar] [CrossRef]
- Barzilai, N.; Crandall, J.P.; Kritchevsky, S.B.; Espeland, M.A. Metformin as a Tool to Target Aging. Cell Metab. 2016, 23, 1060–1065. [Google Scholar] [CrossRef] [Green Version]
- de Kreutzenberg, S.V.; Ceolotto, G.; Cattelan, A.; Pagnin, E.; Mazzucato, M.; Garagnani, P.; Borelli, V.; Bacalini, M.G.; Franceschi, C.; Fadini, G.P.; et al. Metformin improves putative longevity effectors in peripheral mononuclear cells from subjects with prediabetes. A randomized controlled trial. Nutr. Metab. Cardiovasc. Dis. 2015, 25, 686–693. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, A.S.; Brutsaert, E.F.; Anghel, V.; Zhang, K.; Bloomgarden, N.; Pollak, M.; Mar, J.C.; Hawkins, M.; Crandall, J.P.; Barzilai, N. Metformin regulates metabolic and nonmetabolic pathways in skeletal muscle and subcutaneous adipose tissues of older adults. Aging Cell 2018, 17. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, S.; Xavier, D.; George, B.; Umesh, S.; Fathima, S.; Bantwal, G. Effect of intensive lifestyle modification & metformin on cardiovascular risk in prediabetes: A pilot randomized control trial. Indian J. Med. Res. 2018, 148, 705–712. [Google Scholar] [CrossRef]
- Walton, R.G.; Dungan, C.M.; Long, D.E.; Tuggle, S.C.; Kosmac, K.; Peck, B.D.; Bush, H.M.; Villasante Tezanos, A.G.; McGwin, G.; Windham, S.T.; et al. Metformin blunts muscle hypertrophy in response to progressive resistance exercise training in older adults: A randomized, double-blind, placebo-controlled, multicenter trial: The MASTERS trial. Aging Cell 2019, 18, e13039. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Espinoza, S.E.; Jiwani, R.; Wang, J.; Wang, C.P. Review of Interventions for the Frailty Syndrome and the Role of Metformin as a Potential Pharmacologic Agent for Frailty Prevention. Clin. Ther. 2019, 41, 376–386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shaw, R.J. LKB1 and AMP-activated protein kinase control of mTOR signalling and growth. Acta Physiol. 2009, 196, 65–80. [Google Scholar] [CrossRef]
- Kang, S.W.; Haydar, G.; Taniane, C.; Farrell, G.; Arias, I.M.; Lippincott-Schwartz, J.; Fu, D. AMPK Activation Prevents and Reverses Drug-Induced Mitochondrial and Hepatocyte Injury by Promoting Mitochondrial Fusion and Function. PLoS ONE 2016, 11, e0165638. [Google Scholar] [CrossRef]
- Khorraminejad-Shirazi, M.; Sani, M.; Talaei-Khozani, T.; Dorvash, M.; Mirzaei, M.; Faghihi, M.A.; Monabati, A.; Attar, A. AICAR and nicotinamide treatment synergistically augment the proliferation and attenuate senescence-associated changes in mesenchymal stromal cells. Stem Cell Res. Ther. 2020, 11, 45. [Google Scholar] [CrossRef]
- Cieslik, K.A.; Trial, J.; Entman, M.L. Aicar treatment reduces interstitial fibrosis in aging mice: Suppression of the inflammatory fibroblast. J. Mol. Cell. Cardiol. 2017, 111, 81–85. [Google Scholar] [CrossRef]
- Kobilo, T.; Guerrieri, D.; Zhang, Y.; Collica, S.C.; Becker, K.G.; van Praag, H. AMPK agonist AICAR improves cognition and motor coordination in young and aged mice. Learn. Mem. 2014, 21, 119–126. [Google Scholar] [CrossRef] [Green Version]
- Sauerbeck, A.; Gao, J.; Readnower, R.; Liu, M.; Pauly, J.R.; Bing, G.; Sullivan, P.G. Pioglitazone attenuates mitochondrial dysfunction, cognitive impairment, cortical tissue loss, and inflammation following traumatic brain injury. Exp. Neurol. 2011, 227, 128–135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jafari, M.; Khodayari, B.; Felgner, J.; Bussel, I.I.; Rose, M.R.; Mueller, L.D. Pioglitazone: An anti-diabetic compound with anti-aging properties. Biogerontology 2007, 8, 639–651. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, D.; Li, H.; Zhou, R.; Liu, M.J.; Yu, H.; Wu, D.F. Pioglitazone attenuates aging-related disorders in aged apolipoprotein E deficient mice. Exp. Gerontol. 2018, 102, 101–108. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Li, B.; Cai, G.; Huang, M.; Jiang, L.; Pu, J.; Li, L.; Wu, Q.; Zuo, L.; Wang, Q.; et al. Activation of PPAR-gamma by pioglitazone attenuates oxidative stress in aging rat cerebral arteries through upregulating UCP2. J. Cardiovasc. Pharmacol. 2014, 64, 497–506. [Google Scholar] [CrossRef]
- Blalock, E.M.; Phelps, J.T.; Pancani, T.; Searcy, J.L.; Anderson, K.L.; Gant, J.C.; Popovic, J.; Avdiushko, M.G.; Cohen, D.A.; Chen, K.C.; et al. Effects of long-term pioglitazone treatment on peripheral and central markers of aging. PLoS ONE 2010, 5, e10405. [Google Scholar] [CrossRef]
- Seok, H.; Lee, M.; Shin, E.; Yun, M.R.; Lee, Y.H.; Moon, J.H.; Kim, E.; Lee, P.H.; Lee, B.W.; Kang, E.S.; et al. Low-dose pioglitazone can ameliorate learning and memory impairment in a mouse model of dementia by increasing LRP1 expression in the hippocampus. Sci. Rep. 2019, 9, 4414. [Google Scholar] [CrossRef]
- Kulkarni, S.S.; Canto, C. The molecular targets of resveratrol. Biochim. Biophys. Acta 2015, 1852, 1114–1123. [Google Scholar] [CrossRef] [Green Version]
- Giovannelli, L.; Pitozzi, V.; Jacomelli, M.; Mulinacci, N.; Laurenzana, A.; Dolara, P.; Mocali, A. Protective effects of resveratrol against senescence-associated changes in cultured human fibroblasts. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2011, 66, 9–18. [Google Scholar] [CrossRef] [Green Version]
- Farhadnejad, H.; Emamat, H.; Zand, H. The Effect of Resveratrol on Cellular Senescence in Normal and Cancer Cells: Focusing on Cancer and Age-Related Diseases. Nutr. Cancer 2019, 71, 1175–1180. [Google Scholar] [CrossRef]
- Howitz, K.T.; Bitterman, K.J.; Cohen, H.Y.; Lamming, D.W.; Lavu, S.; Wood, J.G.; Zipkin, R.E.; Chung, P.; Kisielewski, A.; Zhang, L.L.; et al. Small molecule activators of sirtuins extend Saccharomyces cerevisiae lifespan. Nature 2003, 425, 191–196. [Google Scholar] [CrossRef]
- Wood, J.G.; Rogina, B.; Lavu, S.; Howitz, K.; Helfand, S.L.; Tatar, M.; Sinclair, D. Sirtuin activators mimic caloric restriction and delay ageing in metazoans. Nature 2004, 430, 686–689. [Google Scholar] [CrossRef]
- Baur, J.A.; Pearson, K.J.; Price, N.L.; Jamieson, H.A.; Lerin, C.; Kalra, A.; Prabhu, V.V.; Allard, J.S.; Lopez-Lluch, G.; Lewis, K.; et al. Resveratrol improves health and survival of mice on a high-calorie diet. Nature 2006, 444, 337–342. [Google Scholar] [CrossRef]
- Wang, C.; Wheeler, C.T.; Alberico, T.; Sun, X.; Seeberger, J.; Laslo, M.; Spangler, E.; Kern, B.; de Cabo, R.; Zou, S. The effect of resveratrol on lifespan depends on both gender and dietary nutrient composition in Drosophila melanogaster. Age 2013, 35, 69–81. [Google Scholar] [CrossRef] [PubMed]
- Brasnyo, P.; Molnar, G.A.; Mohas, M.; Marko, L.; Laczy, B.; Cseh, J.; Mikolas, E.; Szijarto, I.A.; Merei, A.; Halmai, R.; et al. Resveratrol improves insulin sensitivity, reduces oxidative stress and activates the Akt pathway in type 2 diabetic patients. Br. J. Nutr. 2011, 106, 383–389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crandall, J.P.; Oram, V.; Trandafirescu, G.; Reid, M.; Kishore, P.; Hawkins, M.; Cohen, H.W.; Barzilai, N. Pilot study of resveratrol in older adults with impaired glucose tolerance. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2012, 67, 1307–1312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, K.; Tao, Y.; Zhang, J.; Wang, J.; Ye, F.; Dan, G.; Zhao, Y.; Cai, Y.; Zhao, J.; Wu, Q.; et al. Resveratrol Regulates Mitochondrial Biogenesis and Fission/Fusion to Attenuate Rotenone-Induced Neurotoxicity. Oxid. Med. Cell. Longev. 2016, 2016, 6705621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Das, S.; Mitrovsky, G.; Vasanthi, H.R.; Das, D.K. Antiaging properties of a grape-derived antioxidant are regulated by mitochondrial balance of fusion and fission leading to mitophagy triggered by a signaling network of Sirt1-Sirt3-Foxo3-PINK1-PARKIN. Oxid. Med. Cell. Longev. 2014, 2014, 345105. [Google Scholar] [CrossRef]
- Ren, X.; Chen, L.; Xie, J.; Zhang, Z.; Dong, G.; Liang, J.; Liu, L.; Zhou, H.; Luo, P. Resveratrol Ameliorates Mitochondrial Elongation via Drp1/Parkin/PINK1 Signaling in Senescent-Like Cardiomyocytes. Oxid. Med. Cell. Longev. 2017, 2017, 4175353. [Google Scholar] [CrossRef]
- Park, D.; Jeong, H.; Lee, M.N.; Koh, A.; Kwon, O.; Yang, Y.R.; Noh, J.; Suh, P.G.; Park, H.; Ryu, S.H. Resveratrol induces autophagy by directly inhibiting mTOR through ATP competition. Sci. Rep. 2016, 6, 21772. [Google Scholar] [CrossRef] [Green Version]
- Guo, Y.J.; Dong, S.Y.; Cui, X.X.; Feng, Y.; Liu, T.; Yin, M.; Kuo, S.H.; Tan, E.K.; Zhao, W.J.; Wu, Y.C. Resveratrol alleviates MPTP-induced motor impairments and pathological changes by autophagic degradation of alpha-synuclein via SIRT1-deacetylated LC3. Mol. Nutr. Food Res. 2016, 60, 2161–2175. [Google Scholar] [CrossRef]
- Lees, J.G.; Kong, A.M.; Chen, Y.C.; Sivakumaran, P.; Hernandez, D.; Pebay, A.; Harvey, A.J.; Gardner, D.K.; Lim, S.Y. Mitochondrial Fusion by M1 Promotes Embryoid Body Cardiac Differentiation of Human Pluripotent Stem Cells. Stem Cells Int. 2019, 2019, 6380135. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Chen, J.; Sun, X.; Shi, X.; Wang, L.; Huang, L.; Zhou, W. Evaluation of the neuroprotective effect of EGCG: A potential mechanism of mitochondrial dysfunction and mitochondrial dynamics after subarachnoid hemorrhage. Food Funct. 2018, 9, 6349–6359. [Google Scholar] [CrossRef] [PubMed]
- de la Torre, R.; de Sola, S.; Hernandez, G.; Farre, M.; Pujol, J.; Rodriguez, J.; Espadaler, J.M.; Langohr, K.; Cuenca-Royo, A.; Principe, A.; et al. Safety and efficacy of cognitive training plus epigallocatechin-3-gallate in young adults with Down’s syndrome (TESDAD): A double-blind, randomised, placebo-controlled, phase 2 trial. Lancet Neurol. 2016, 15, 801–810. [Google Scholar] [CrossRef]
- Qiao, Z.; Yue, G.; Fan, L.; Ying, L.; Yucun, N. Epigallocatechin gallate prevents senescence by alleviating oxidative stress and inflammation in WI38 human embryonic fibroblasts. R. Soc. Chem. Adv. 2019, 9, 26787–26798. [Google Scholar] [CrossRef] [Green Version]
- Xiong, L.G.; Chen, Y.J.; Tong, J.W.; Gong, Y.S.; Huang, J.A.; Liu, Z.H. Epigallocatechin-3-gallate promotes healthy lifespan through mitohormesis during early-to-mid adulthood in Caenorhabditis elegans. Redox Biol. 2018, 14, 305–315. [Google Scholar] [CrossRef]
- Ryu, D.; Mouchiroud, L.; Andreux, P.A.; Katsyuba, E.; Moullan, N.; Nicolet-Dit-Felix, A.A.; Williams, E.G.; Jha, P.; Lo Sasso, G.; Huzard, D.; et al. Urolithin A induces mitophagy and prolongs lifespan in C. elegans and increases muscle function in rodents. Nat. Med. 2016, 22, 879–888. [Google Scholar] [CrossRef]
- Lin, J.; Zhuge, J.; Zheng, X.; Wu, Y.; Zhang, Z.; Xu, T.; Meftah, Z.; Xu, H.; Wu, Y.; Tian, N.; et al. Urolithin A-induced mitophagy suppresses apoptosis and attenuates intervertebral disc degeneration via the AMPK signaling pathway. Free Radic. Biol. Med. 2020, 150, 109–119. [Google Scholar] [CrossRef]
- Fang, E.F.; Hou, Y.; Palikaras, K.; Adriaanse, B.A.; Kerr, J.S.; Yang, B.; Lautrup, S.; Hasan-Olive, M.M.; Caponio, D.; Dan, X.; et al. Mitophagy inhibits amyloid-beta and tau pathology and reverses cognitive deficits in models of Alzheimer’s disease. Nat. Neurosc. 2019, 22, 401–412. [Google Scholar] [CrossRef]
- Andreux, P.A.; Ryu, D.; Burdet, F.; Ibberson, M.; Aebischer, P.; Auwerx, J.; Singh, A.; Rinsch, C. The mitophagy activator urolithin A is safe and induces a molecular signature of improved mitochondrial and cellular health in humans. Nat. Metab. 2019, 1, 595–603. [Google Scholar] [CrossRef]
- Li, Y.; Wu, P.; Dai, J.; Zhang, T.; Bihl, J.; Wang, C.; Liu, Y.; Shi, H. Inhibition of mTOR Alleviates Early Brain Injury After Subarachnoid Hemorrhage Via Relieving Excessive Mitochondrial Fission. Cell. Mol. Neurobiol. 2019, 1–14. [Google Scholar] [CrossRef]
- Lamming, D.W. Inhibition of the Mechanistic Target of Rapamycin (mTOR)-Rapamycin and Beyond. Cold Spring Harb. Perspect. Med. 2016, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lamming, D.W.; Ye, L.; Sabatini, D.M.; Baur, J.A. Rapalogs and mTOR inhibitors as anti-aging therapeutics. J. Clin. Investig. 2013, 123, 980–989. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harrison, D.E.; Strong, R.; Sharp, Z.D.; Nelson, J.F.; Astle, C.M.; Flurkey, K.; Nadon, N.L.; Wilkinson, J.E.; Frenkel, K.; Carter, C.S.; et al. Rapamycin fed late in life extends lifespan in genetically heterogeneous mice. Nature 2009, 460, 392–395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, R.A.; Harrison, D.E.; Astle, C.M.; Baur, J.A.; Boyd, A.R.; de Cabo, R.; Fernandez, E.; Flurkey, K.; Javors, M.A.; Nelson, J.F.; et al. Rapamycin, but not resveratrol or simvastatin, extends life span of genetically heterogeneous mice. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2011, 66, 191–201. [Google Scholar] [CrossRef]
- Demidenko, Z.N.; Zubova, S.G.; Bukreeva, E.I.; Pospelov, V.A.; Pospelova, T.V.; Blagosklonny, M.V. Rapamycin decelerates cellular senescence. Cell Cycle 2009, 8, 1888–1895. [Google Scholar] [CrossRef]
- Li, Q.; Zhang, T.; Wang, J.; Zhang, Z.; Zhai, Y.; Yang, G.Y.; Sun, X. Rapamycin attenuates mitochondrial dysfunction via activation of mitophagy in experimental ischemic stroke. Biochem. Biophys. Res. Commun. 2014, 444, 182–188. [Google Scholar] [CrossRef]
- Blagosklonny, M.V. Rapamycin, proliferation and geroconversion to senescence. Cell Cycle 2018, 17, 2655–2665. [Google Scholar] [CrossRef] [Green Version]
- Horvath, S.; Lu, A.T.; Cohen, H.; Raj, K. Rapamycin retards epigenetic ageing of keratinocytes independently of its effects on replicative senescence, proliferation and differentiation. Aging 2019, 11, 3238–3249. [Google Scholar] [CrossRef]
- Di Domenico, F.; Tramutola, A.; Foppoli, C.; Head, E.; Perluigi, M.; Butterfield, D.A. mTOR in Down syndrome: Role in Ass and tau neuropathology and transition to Alzheimer disease-like dementia. Free Radic. Biol. Med. 2018, 114, 94–101. [Google Scholar] [CrossRef]
- Di Domenico, F.; Tramutola, A.; Barone, E.; Lanzillotta, C.; Defever, O.; Arena, A.; Zuliani, I.; Foppoli, C.; Iavarone, F.; Vincenzoni, F.; et al. Restoration of aberrant mTOR signaling by intranasal rapamycin reduces oxidative damage: Focus on HNE-modified proteins in a mouse model of down syndrome. Redox Biol. 2019, 23, 101162. [Google Scholar] [CrossRef]
- Duval, N.; Vacano, G.N.; Patterson, D. Rapamycin Treatment Ameliorates Age-Related Accumulation of Toxic Metabolic Intermediates in Brains of the Ts65Dn Mouse Model of Down Syndrome and Aging. Front. Aging Neurosci. 2018, 10, 263. [Google Scholar] [CrossRef] [PubMed]
- Chaudhari, S.N.; Kipreos, E.T. Increased mitochondrial fusion allows the survival of older animals in diverse C. elegans longevity pathways. Nat. Commun. 2017, 8, 182. [Google Scholar] [CrossRef] [PubMed]
- Reddy, P.H. Inhibitors of mitochondrial fission as a therapeutic strategy for diseases with oxidative stress and mitochondrial dysfunction. J. Alzheimer’s Dis. JAD 2014, 40, 245–256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cassidy-Stone, A.; Chipuk, J.E.; Ingerman, E.; Song, C.; Yoo, C.; Kuwana, T.; Kurth, M.J.; Shaw, J.T.; Hinshaw, J.E.; Green, D.R.; et al. Chemical inhibition of the mitochondrial division dynamin reveals its role in Bax/Bak-dependent mitochondrial outer membrane permeabilization. Dev. Cell 2008, 14, 193–204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manczak, M.; Kandimalla, R.; Yin, X.; Reddy, P.H. Mitochondrial division inhibitor 1 reduces dynamin-related protein 1 and mitochondrial fission activity. Hum. Mol. Genet. 2019, 28, 177–199. [Google Scholar] [CrossRef]
- Wang, W.; Yin, J.; Ma, X.; Zhao, F.; Siedlak, S.L.; Wang, Z.; Torres, S.; Fujioka, H.; Xu, Y.; Perry, G.; et al. Inhibition of mitochondrial fragmentation protects against Alzheimer’s disease in rodent model. Hum. Mol. Genet. 2017, 26, 4118–4131. [Google Scholar] [CrossRef] [Green Version]
- Reddy, P.H.; Manczak, M.; Yin, X. Mitochondria-Division Inhibitor 1 Protects Against Amyloid-beta induced Mitochondrial Fragmentation and Synaptic Damage in Alzheimer’s Disease. J. Alzheimer’s Dis. JAD 2017, 58, 147–162. [Google Scholar] [CrossRef] [Green Version]
- Qi, X.; Qvit, N.; Su, Y.C.; Mochly-Rosen, D. A novel Drp1 inhibitor diminishes aberrant mitochondrial fission and neurotoxicity. J. Cell Sci. 2013, 126, 789–802. [Google Scholar] [CrossRef] [Green Version]
- Joshi, A.U.; Saw, N.L.; Shamloo, M.; Mochly-Rosen, D. Drp1/Fis1 interaction mediates mitochondrial dysfunction, bioenergetic failure and cognitive decline in Alzheimer’s disease. Oncotarget 2018, 9, 6128–6143. [Google Scholar] [CrossRef] [Green Version]
- Macia, E.; Ehrlich, M.; Massol, R.; Boucrot, E.; Brunner, C.; Kirchhausen, T. Dynasore, a cell-permeable inhibitor of dynamin. Dev. Cell 2006, 10, 839–850. [Google Scholar] [CrossRef] [Green Version]
- Gao, D.; Zhang, L.; Dhillon, R.; Hong, T.T.; Shaw, R.M.; Zhu, J. Dynasore protects mitochondria and improves cardiac lusitropy in Langendorff perfused mouse heart. PLoS ONE 2013, 8, e60967. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fa, M.; Staniszewski, A.; Saeed, F.; Francis, Y.I.; Arancio, O. Dynamin 1 is required for memory formation. PLoS ONE 2014, 9, e91954. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.; Dasgupta, A.; Chen, K.H.; Neuber-Hess, M.; Patel, J.; Hurst, T.E.; Mewburn, J.D.; Lima, P.D.A.; Alizadeh, E.; Martin, A.; et al. Identification of novel dynamin-related protein 1 (Drp1) GTPase inhibitors: Therapeutic potential of Drpitor1 and Drpitor1a in cancer and cardiac ischemia-reperfusion injury. FASEB J. 2020, 34, 1447–1464. [Google Scholar] [CrossRef] [Green Version]
- Miret-Casals, L.; Sebastian, D.; Brea, J.; Rico-Leo, E.M.; Palacin, M.; Fernandez-Salguero, P.M.; Loza, M.I.; Albericio, F.; Zorzano, A. Identification of New Activators of Mitochondrial Fusion Reveals a Link between Mitochondrial Morphology and Pyrimidine Metabolism. Cell Chem. Biol. 2018, 25, 268–278. [Google Scholar] [CrossRef] [Green Version]
- Li, E.K.; Tam, L.S.; Tomlinson, B. Leflunomide in the treatment of rheumatoid arthritis. Clin. Ther. 2004, 26, 447–459. [Google Scholar] [CrossRef]
- Wang, D.; Wang, J.; Bonamy, G.M.; Meeusen, S.; Brusch, R.G.; Turk, C.; Yang, P.; Schultz, P.G. A small molecule promotes mitochondrial fusion in mammalian cells. Angew. Chem. Int. Ed. 2012, 51, 9302–9305. [Google Scholar] [CrossRef]
- Ding, M.; Liu, C.; Shi, R.; Yu, M.; Zeng, K.; Kang, J.; Fu, F.; Mi, M. Mitochondrial fusion promoter restores mitochondrial dynamics balance and ameliorates diabetic cardiomyopathy in an optic atrophy 1-dependent way. Acta Physiol. 2019, 229, e13428. [Google Scholar] [CrossRef]
- Li, A.; Zhang, S.; Li, J.; Liu, K.; Huang, F.; Liu, B. Metformin and resveratrol inhibit Drp1-mediated mitochondrial fission and prevent ER stress-associated NLRP3 inflammasome activation in the adipose tissue of diabetic mice. Mol. Cell. Endocrinol. 2016, 434, 36–47. [Google Scholar] [CrossRef]
- Fang, J.; Yang, J.; Wu, X.; Zhang, G.; Li, T.; Wang, X.; Zhang, H.; Wang, C.C.; Liu, G.H.; Wang, L. Metformin alleviates human cellular aging by upregulating the endoplasmic reticulum glutathione peroxidase 7. Aging Cell 2018, 17, e12765. [Google Scholar] [CrossRef]
- Wang, R.; Yu, Z.; Sunchu, B.; Shoaf, J.; Dang, I.; Zhao, S.; Caples, K.; Bradley, L.; Beaver, L.M.; Ho, E.; et al. Rapamycin inhibits the secretory phenotype of senescent cells by a Nrf2-independent mechanism. Aging Cell 2017, 16, 564–574. [Google Scholar] [CrossRef]
- Lamming, D.W.; Ye, L.; Astle, C.M.; Baur, J.A.; Sabatini, D.M.; Harrison, D.E. Young and old genetically heterogeneous HET3 mice on a rapamycin diet are glucose intolerant but insulin sensitive. Aging Cell 2013, 12, 712–718. [Google Scholar] [CrossRef] [PubMed] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Title and Date | Objectives | Dose and Outcome Measures |
|---|---|---|
| Effects of metformin on longevity gene expression and inflammation in pre-diabetic individuals. June 2010–March 2013 | To study the role of the AMPK pathway on longevity genes and inflammation in pre-diabetic setting. Completed. | 500 mg tris in die Metformin induced changes in the expression of longevity genes SIRT1, p66Shc, mTOR, p53 in peripheral blood mononuclear cells [171]. |
| MILES Metformin in Longevity Study. October 2014–December 2017 | To determine if metformin restores the gene expression profile of old, glucose intolerant adults to that of young healthy subjects. Completed. | 1700 mg/day Metformin regulates metabolic and non-metabolic pathways in skeletal muscle and subcutaneous adipose tissues of older adults [172,173]. |
| MASTERS Metformin to augment resistance and training adaptations in older adults. January 2015–June 2018 | To determine whether metformin can enhance the benefits seen during resistance exercise, such as increased muscle mass and strength. Completed. | 1700 mg/day Results do not support the use of metformin to enhance the benefits of physical activity in healthy elderly people [174]. |
| Phase 1 Study of the effects of combining topical FDA-approved drugs on age-related pathways on the skin of healthy volunteers. March 2017–February 2019 | To examine the effects of FDA approved medications, including metformin, on skin aging when applied in topical form. Completed. | Topical metformin applied to the skin. Primary measure: profile of gene transcript changes. Secondary measure: wrinkle score. |
| MATE Metformin and Aging Trial in the Elderly: A pilot and feasibility study. May 2018–April 2020 | To test whether chronic metformin administration reduces aging-related biochemical parameters and improves physical performance. | 500 mg every 6 to 8 h Primary measure: frailty measured by the short physical performance battery, a group of measures that combines the results of the gait speed, chair stand and balance tests. Secondary measure: effect of metformin on senescent markers. |
| Anti-Aging, Pro-Autophagy effects of Metformin in Adults with Prediabetes. September 2017–July 2021 | To demonstrate that metformin therapy increases cellular senescence and autophagy. | 1500 mg/day Primary measure: change in Leucocyte LC3 Score. |
| Metformin for Preventing Frailty in High-risk Older Adults. April 2016–October 2022 | To demonstrate that metformin modulates diabetes/insulin resistance and inflammation will prevent and/or ameliorate the progression of frailty. | 1000 mg twice a day Primary measure: frailty, measured by validated standardized criteria [175]. |
| Metformin to prevent inactivity-induced loss of muscle health during aging. July 2017–July 2022 | To investigate metformin as a preventive strategy to maintain muscle and metabolic health in bed ridden older adults. | 2000 mg/day Primary measure: change in muscle size from baseline to 5 days of bed rest (determined by magnetic resonance imaging). |
| VA-IMPACT. Effects of Metformin on Atherosclerotic Cardiovascular Outcomes in Pre-Diabetes. February 2019–August 2024 | To demonstrate that metformin reduces the risk of death, heart attacks, and/or strokes in patients who have pre-diabetes and heart or blood vessel problems. | 1000 mg/day–2000 mg/day Primary measures: death; non-fatal myocardial infarction or stroke; unstable angina with acute myocardial ischemia; or coronary revascularization. Secondary measures: cumulative/recurrent incidence of the primary measures; time to new/recurrent diagnosis of a malignancy; time to new diagnosis of T2DM. |
| Does insulin sensitivity impact the potential of metformin to slow aging? March 2020–April 2024 | To demonstrate who may benefit from metformin treatment to slow aging. | 1500 mg/day Primary measures: change in insulin sensitivity (determined by a hyperinsulinemic-euglycemic clamp); evaluation of the mitochondrial function. Secondary measures: 5-day continuous glucose monitoring; change of aging biomarkers in blood. |
| Drug/ Compound | Activity on Mitochondrial Network | Tested in DS | Tested in Aging |
|---|---|---|---|
| Metformin (FDA approved) | Induces OPA1 and MFN2 [24] and inhibits DRP1 [238]. | In vitro: [24]. | In vitro: [239]. Animal models: [167,168]. Human: [169,171,172,173,174,175]. |
| AICAR | Induces OPA1 and MFN1 [177]. | Not tested | In vitro: [177,178]. Animal models: [179,180]. |
| Pioglitazone (FDA approved) | Induces OPA1 and MFN1 and inhibits DRP1 [67]. | In vitro: [67]. | Animal models: [182,183,184,185]. |
| Resveratrol (nutraceutic) | Inhibits DRP1 [198,238]. Regulates DRP1, FIS1, OPA1, and MFN2 [196]. | In vitro: [66]. | In vitro: [188,189]. Animal models: [190,191,192,193]. Human: [194,195]. |
| Epigallocatechin-3-gallate (EGCG) (nutraceutic) | Regulates Drp1, Fis1, Opa1, Mfn1, and Mfn2 [202]. | In vitro: [66,130]. Human: [203]. | In vitro: [204]. Animal models: [205]. |
| Urolithin A (UA) (nutraceutic) | Inhibits OPA1 and FZO1 [206]. | Not tested | Human: [209]. |
| Rapamycin (FDA approved) | Induces MTFP1 [87]. | In vitro: [218]. Animal models: [219,221]. | In vitro: [215,240]. Animal models: [213,214,216,241]. |
| Mdivi-1 | Inhibits DRP1 [134,224]. | In vitro: [134]. | Not tested |
| P110 | Inhibits DRP1 [228]. | Not tested | Not tested |
| Dynasore | Inhibits DRP1 [230]. | Not tested | Not tested |
| Drpitor1 | Inhibits DRP1 GTPase activity [233]. | Not tested | Not tested |
| Leflunomide (FDA approved) | Induces MFN1 and MFN2 and inhibits OPA1 short isoform [234]. | Not tested | Not tested |
| Hydrazone M1 | Induces MFN2 and inhibits DRP1 [236]. | Not tested | Not tested |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mollo, N.; Cicatiello, R.; Aurilia, M.; Scognamiglio, R.; Genesio, R.; Charalambous, M.; Paladino, S.; Conti, A.; Nitsch, L.; Izzo, A. Targeting Mitochondrial Network Architecture in Down Syndrome and Aging. Int. J. Mol. Sci. 2020, 21, 3134. https://doi.org/10.3390/ijms21093134
Mollo N, Cicatiello R, Aurilia M, Scognamiglio R, Genesio R, Charalambous M, Paladino S, Conti A, Nitsch L, Izzo A. Targeting Mitochondrial Network Architecture in Down Syndrome and Aging. International Journal of Molecular Sciences. 2020; 21(9):3134. https://doi.org/10.3390/ijms21093134
Chicago/Turabian StyleMollo, Nunzia, Rita Cicatiello, Miriam Aurilia, Roberta Scognamiglio, Rita Genesio, Maria Charalambous, Simona Paladino, Anna Conti, Lucio Nitsch, and Antonella Izzo. 2020. "Targeting Mitochondrial Network Architecture in Down Syndrome and Aging" International Journal of Molecular Sciences 21, no. 9: 3134. https://doi.org/10.3390/ijms21093134
APA StyleMollo, N., Cicatiello, R., Aurilia, M., Scognamiglio, R., Genesio, R., Charalambous, M., Paladino, S., Conti, A., Nitsch, L., & Izzo, A. (2020). Targeting Mitochondrial Network Architecture in Down Syndrome and Aging. International Journal of Molecular Sciences, 21(9), 3134. https://doi.org/10.3390/ijms21093134