A Novel Zinc Chelator, 1H10, Ameliorates Experimental Autoimmune Encephalomyelitis by Modulating Zinc Toxicity and AMPK Activation



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. 1H10 Has Zinc-Chelating Capacity
2.2. 1H10 Reduces EAE-Induced MS Symptoms, Demyelination, and Microglial/Macrophage Activation
2.3. Suppression of EAE-Induced Aberrant Zinc Patches, MMP-9 Activation, and BBB Breakdown in 1H10-Treated Mice
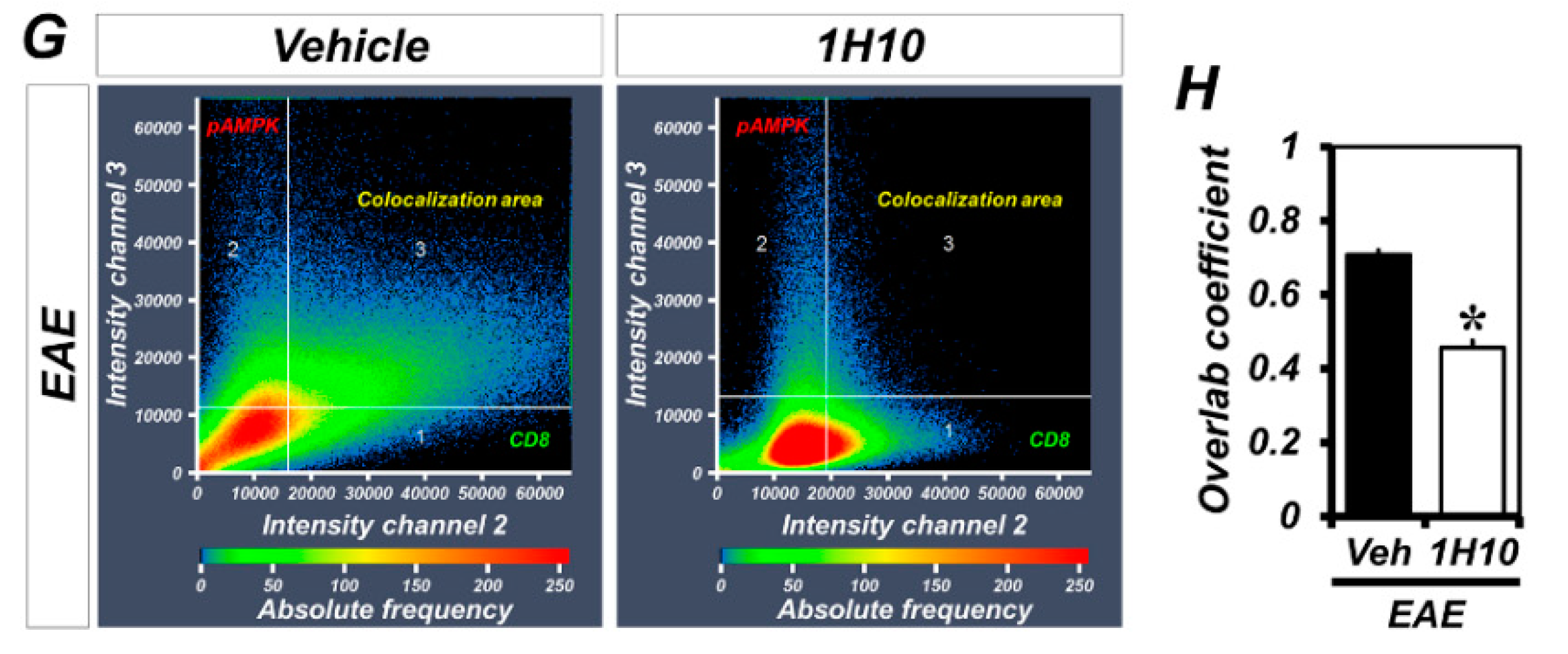
2.4. 1H10 Decreases Immune Cell Infiltration and the Phosphorylation of AMPK in Infiltrated CD8+ T Cells Following EAE Induction
2.5. Long-Term Protective Effects of 1H10 Following EAE Induction
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. EAE Induction and Clinical Evaluation
4.3. 1H10 Administration and Experimental Design
4.4. Tissue Preparation and Cresyl Violet Staining
4.5. Immunohistochemistry
4.6. Immunofluorescence Analysis
4.7. Zinc Staining (TSQ Method)
4.8. Cell Culture
4.9. Detection of Zinc-Chelating Capacity in Cell Cultures or in Test Tubes
4.10. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Noseworthy, J.H.; Lucchinetti, C.; Rodriguez, M.; Weinshenker, B.G. Multiple sclerosis. N. Engl. J. Med. 2000, 343, 938–952. [Google Scholar] [CrossRef] [PubMed]
- Trapp, B.D.; Nave, K.A. Multiple sclerosis: An immune or neurodegenerative disorder? Annu. Rev. Neurosci. 2008, 31, 247–269. [Google Scholar] [CrossRef] [PubMed]
- Constantinescu, C.S.; Farooqi, N.; O’Brien, K.; Gran, B. Experimental autoimmune encephalomyelitis (EAE) as a model for multiple sclerosis (MS). Br. J. Pharmacol. 2011, 164, 1079–1106. [Google Scholar] [CrossRef] [PubMed]
- Bennett, J.; Basivireddy, J.; Kollar, A.; Biron, K.E.; Reickmann, P.; Jefferies, W.A.; McQuaid, S. Blood-brain barrier disruption and enhanced vascular permeability in the multiple sclerosis model EAE. J. Neuroimmunol. 2010, 229, 180–191. [Google Scholar] [CrossRef]
- Zhang, S.; Su, Y.; Ying, Z.; Guo, D.; Pan, C.; Guo, J.; Zou, Z.; Wang, L.; Zhang, Z.; Jiang, Z.; et al. RIP1 kinase inhibitor halts the progression of an immune-induced demyelination disease at the stage of monocyte elevation. Proc. Natl. Acad. Sci. USA 2019, 116, 5675–5680. [Google Scholar] [CrossRef] [Green Version]
- Compston, A.; Coles, A. Multiple sclerosis. Lancet 2008, 372, 1502–1517. [Google Scholar] [CrossRef]
- Choi, B.Y.; Kim, J.H.; Kho, A.R.; Kim, I.Y.; Lee, S.H.; Lee, B.E.; Choi, E.; Sohn, M.; Stevenson, M.; Chung, T.N.; et al. Inhibition of NADPH oxidase activation reduces EAE-induced white matter damage in mice. J. NeuroInflamm. 2015, 12, 104. [Google Scholar] [CrossRef] [Green Version]
- Choi, B.Y.; Jang, B.G.; Kim, J.H.; Seo, J.N.; Wu, G.; Sohn, M.; Chung, T.N.; Suh, S.W. Copper/zinc chelation by clioquinol reduces spinal cord white matter damage and behavioral deficits in a murine MOG-induced multiple sclerosis model. Neurobiol. Dis. 2013, 54, 382–391. [Google Scholar] [CrossRef]
- Choi, B.Y.; Kim, I.Y.; Kim, J.H.; Kho, A.R.; Lee, S.H.; Lee, B.E.; Sohn, M.; Koh, J.Y.; Suh, S.W. Zinc transporter 3 (ZnT3) gene deletion reduces spinal cord white matter damage and motor deficits in a murine MOG-induced multiple sclerosis model. Neurobiol. Dis. 2016, 94, 205–212. [Google Scholar] [CrossRef]
- Ronnett, G.V.; Ramamurthy, S.; Kleman, A.M.; Landree, L.E.; Aja, S. AMPK in the brain: Its roles in energy balance and neuroprotection. J. Neurochem. 2009, 109 (Suppl. 1), 17–23. [Google Scholar] [CrossRef] [Green Version]
- Hardie, D.G. AMP-activated protein kinase: An energy sensor that regulates all aspects of cell function. Genes Dev. 2011, 25, 1895–1908. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Zeng, Z.; Viollet, B.; Ronnett, G.V.; McCullough, L.D. Neuroprotective effects of adenosine monophosphate-activated protein kinase inhibition and gene deletion in stroke. Stroke 2007, 38, 2992–2999. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ju, T.C.; Chen, H.M.; Lin, J.T.; Chang, C.P.; Chang, W.C.; Kang, J.J.; Sun, C.P.; Tao, M.H.; Tu, P.H.; Chang, C.; et al. Nuclear translocation of AMPK-alpha1 potentiates striatal neurodegeneration in Huntington’s disease. J. Cell Biol. 2011, 194, 209–227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salminen, A.; Kaarniranta, K. AMP-activated protein kinase (AMPK) controls the aging process via an integrated signaling network. Ageing Res. Rev. 2012, 11, 230–241. [Google Scholar] [CrossRef] [PubMed]
- Eom, J.W.; Kim, T.Y.; Seo, B.R.; Park, H.; Koh, J.Y.; Kim, Y.H. Identifying New AMP-Activated Protein Kinase Inhibitors That Protect against Ischemic Brain Injury. ACS Chem. Neurosci. 2019, 10, 2345–2354. [Google Scholar] [CrossRef] [PubMed]
- Di Vaira, M.; Bazzicalupi, C.; Orioli, P.; Messori, L.; Bruni, B.; Zatta, P. Clioquinol, a drug for Alzheimer’s disease specifically interfering with brain metal metabolism: Structural characterization of its zinc(II) and copper(II) complexes. Inorg. Chem. 2004, 43, 3795–3797. [Google Scholar] [CrossRef]
- Bolos, M.; Perea, J.R.; Avila, J. Alzheimer’s disease as an inflammatory disease. Biomol. Concepts 2017, 8, 37–43. [Google Scholar] [CrossRef]
- Zotova, E.; Bharambe, V.; Cheaveau, M.; Morgan, W.; Holmes, C.; Harris, S.; Neal, J.W.; Love, S.; Nicoll, J.A.; Boche, D. Inflammatory components in human Alzheimer’s disease and after active amyloid-beta42 immunization. Brain 2013, 136, 2677–2696. [Google Scholar] [CrossRef] [Green Version]
- Gudi, V.; Gai, L.; Herder, V.; Tejedor, L.S.; Kipp, M.; Amor, S.; Suhs, K.W.; Hansmann, F.; Beineke, A.; Baumgartner, W.; et al. Synaptophysin Is a Reliable Marker for Axonal Damage. J. Neuropathol. Exp. Neurol. 2017, 76, 109–125. [Google Scholar] [CrossRef]
- Tyszka-Czochara, M.; Grzywacz, A.; Gdula-Argasinska, J.; Librowski, T.; Wilinski, B.; Opoka, W. The role of zinc in the pathogenesis and treatment of central nervous system (CNS) diseases. Implications of zinc homeostasis for proper CNS function. Acta Pol. Pharm. 2014, 71, 369–377. [Google Scholar]
- Takeda, A.; Hanajima, T.; Ijiro, H.; Ishige, A.; Iizuka, S.; Okada, S.; Oku, N. Release of zinc from the brain of El (epilepsy) mice during seizure induction. Brain Res. 1999, 828, 174–178. [Google Scholar] [CrossRef]
- Koh, J.Y.; Suh, S.W.; Gwag, B.J.; He, Y.Y.; Hsu, C.Y.; Choi, D.W. The role of zinc in selective neuronal death after transient global cerebral ischemia. Science 1996, 272, 1013–1016. [Google Scholar] [CrossRef] [PubMed]
- Suh, S.W.; Chen, J.W.; Motamedi, M.; Bell, B.; Listiak, K.; Pons, N.F.; Danscher, G.; Frederickson, C.J. Evidence that synaptically-released zinc contributes to neuronal injury after traumatic brain injury. Brain Res. 2000, 852, 268–273. [Google Scholar] [CrossRef]
- Choi, B.Y.; Lee, S.H.; Choi, H.C.; Lee, S.K.; Yoon, H.S.; Park, J.B.; Chung, W.S.; Suh, S.W. Alcohol dependence treating agent, acamprosate, prevents traumatic brain injury-induced neuron death through vesicular zinc depletion. Transl. Res. 2019, 207, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Suh, S.W.; Garnier, P.; Aoyama, K.; Chen, Y.; Swanson, R.A. Zinc release contributes to hypoglycemia-induced neuronal death. Neurobiol. Dis. 2004, 16, 538–545. [Google Scholar] [PubMed]
- Noh, K.M.; Kim, Y.H.; Koh, J.Y. Mediation by membrane protein kinase C of zinc-induced oxidative neuronal injury in mouse cortical cultures. J. Neurochem. 1999, 72, 1609–1616. [Google Scholar] [CrossRef]
- Murakami, K.; Whiteley, M.K.; Routtenberg, A. Regulation of protein kinase C activity by cooperative interaction of Zn2+ and Ca2+. J. Biol. Chem. 1987, 262, 13902–13906. [Google Scholar]
- Noh, K.M.; Koh, J.Y. Induction and activation by zinc of NADPH oxidase in cultured cortical neurons and astrocytes. J. Neurosci. 2000, 20, RC111. [Google Scholar] [CrossRef]
- Suh, S.W.; Shin, B.S.; Ma, H.; Van Hoecke, M.; Brennan, A.M.; Yenari, M.A.; Swanson, R.A. Glucose and NADPH oxidase drive neuronal superoxide formation in stroke. Ann. Neurol. 2008, 64, 654–663. [Google Scholar] [CrossRef]
- Choi, B.Y.; Jang, B.G.; Kim, J.H.; Lee, B.E.; Sohn, M.; Song, H.K.; Suh, S.W. Prevention of traumatic brain injury-induced neuronal death by inhibition of NADPH oxidase activation. Brain Res. 2012, 1481, 49–58. [Google Scholar] [CrossRef]
- Balabanov, R.; Dore-Duffy, P. Role of the CNS microvascular pericyte in the blood-brain barrier. J. Neurosci. Res. 1998, 53, 637–644. [Google Scholar] [CrossRef]
- Wolburg, H.; Wolburg-Buchholz, K.; Kraus, J.; Rascher-Eggstein, G.; Liebner, S.; Hamm, S.; Duffner, F.; Grote, E.H.; Risau, W.; Engelhardt, B. Localization of claudin-3 in tight junctions of the blood-brain barrier is selectively lost during experimental autoimmune encephalomyelitis and human glioblastoma multiforme. Acta Neuropathol. 2003, 105, 586–592. [Google Scholar] [CrossRef] [PubMed]
- Leppert, D.; Ford, J.; Stabler, G.; Grygar, C.; Lienert, C.; Huber, S.; Miller, K.M.; Hauser, S.L.; Kappos, L. Matrix metalloproteinase-9 (gelatinase B) is selectively elevated in CSF during relapses and stable phases of multiple sclerosis. Brain 1998, 121, 2327–2334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kirk, J.; Plumb, J.; Mirakhur, M.; McQuaid, S. Tight junctional abnormality in multiple sclerosis white matter affects all calibres of vessel and is associated with blood-brain barrier leakage and active demyelination. J. Pathol. 2003, 201, 319–327. [Google Scholar] [CrossRef] [PubMed]
- Chang, A.; Tourtellotte, W.W.; Rudick, R.; Trapp, B.D. Premyelinating oligodendrocytes in chronic lesions of multiple sclerosis. N. Engl. J. Med. 2002, 346, 165–173. [Google Scholar] [CrossRef]
- Keirstead, H.S. Stem cells for the treatment of myelin loss. Trends Neurosci. 2005, 28, 677–683. [Google Scholar] [CrossRef]
- Kauppinen, T.M.; Higashi, Y.; Suh, S.W.; Escartin, C.; Nagasawa, K.; Swanson, R.A. Zinc triggers microglial activation. J. Neurosci. 2008, 28, 5827–5835. [Google Scholar] [CrossRef] [Green Version]
- Prasad, A.S. Clinical, immunological, anti-inflammatory and antioxidant roles of zinc. Exp. Gerontol. 2008, 43, 370–377. [Google Scholar] [CrossRef]
- Overbeck, S.; Rink, L.; Haase, H. Modulating the immune response by oral zinc supplementation: A single approach for multiple diseases. Arch. Immunol. Ther. Exp. (Warsz) 2008, 56, 15–30. [Google Scholar] [CrossRef]
- Steinman, L. Multiple sclerosis: A coordinated immunological attack against myelin in the central nervous system. Cell 1996, 85, 299–302. [Google Scholar] [CrossRef] [Green Version]
- Huseby, E.S.; Liggitt, D.; Brabb, T.; Schnabel, B.; Ohlen, C.; Goverman, J. A pathogenic role for myelin-specific CD8(+) T cells in a model for multiple sclerosis. J. Exp. Med. 2001, 194, 669–676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, R. HLA class I: Friend and foe of multiple sclerosis. Nat. Med. 2008, 14, 1150–1151. [Google Scholar] [CrossRef] [PubMed]
- Sabelko, K.A.; Kelly, K.A.; Nahm, M.H.; Cross, A.H.; Russell, J.H. Fas and Fas ligand enhance the pathogenesis of experimental allergic encephalomyelitis, but are not essential for immune privilege in the central nervous system. J. Immunol. 1997, 159, 3096–3099. [Google Scholar] [PubMed]
- Waldner, H.; Sobel, R.A.; Howard, E.; Kuchroo, V.K. Fas- and FasL-deficient mice are resistant to induction of autoimmune encephalomyelitis. J. Immunol. 1997, 159, 3100–3103. [Google Scholar]
- Sabelko-Downes, K.A.; Gimenez, M.T.; Suvannavejh, G.C.; Miller, S.D.; Russell, J.H. Genetic control of pathogenic mechanisms in autoimmune demyelinating disease. J. Neuroimmunol. 2000, 110, 168–176. [Google Scholar] [CrossRef]
- Yang, K.; Chi, H. AMPK helps T cells survive nutrient starvation. Immunity 2015, 42, 4–6. [Google Scholar] [CrossRef] [Green Version]
- Rolf, J.; Zarrouk, M.; Finlay, D.K.; Foretz, M.; Viollet, B.; Cantrell, D.A. AMPKalpha1: A glucose sensor that controls CD8 T-cell memory. Eur. J. Immunol. 2013, 43, 889–896. [Google Scholar] [CrossRef] [Green Version]
- Blagih, J.; Coulombe, F.; Vincent, E.E.; Dupuy, F.; Galicia-Vazquez, G.; Yurchenko, E.; Raissi, T.C.; van der Windt, G.J.; Viollet, B.; Pearce, E.L.; et al. The energy sensor AMPK regulates T cell metabolic adaptation and effector responses in vivo. Immunity 2015, 42, 41–54. [Google Scholar] [CrossRef] [Green Version]
- Rao, E.; Zhang, Y.; Zhu, G.; Hao, J.; Persson, X.M.; Egilmez, N.K.; Suttles, J.; Li, B. Deficiency of AMPK in CD8+ T cells suppresses their anti-tumor function by inducing protein phosphatase-mediated cell death. Oncotarget 2015, 6, 7944–7958. [Google Scholar] [CrossRef]
- Kilkenny, C.; Browne, W.J.; Cuthill, I.C.; Emerson, M.; Altman, D.G. Improving bioscience research reporting: The ARRIVE guidelines for reporting animal research. PLoS Biol. 2012, 8, e1000412. [Google Scholar] [CrossRef] [Green Version]
- Frederickson, C.J.; Kasarskis, E.J.; Ringo, D.; Frederickson, R.E. A quinoline fluorescence method for visualizing and assaying the histochemically reactive zinc (bouton zinc) in the brain. J. Neurosci. Methods 1987, 20, 91–103. [Google Scholar] [CrossRef]
- Suh, S.W.; Listiack, K.; Bell, B.; Chen, J.; Motamedi, M.; Silva, D.; Danscher, G.; Whetsell, W.; Thompson, R.; Frederickson, C. Detection of pathological zinc accumulation in neurons: Methods for autopsy, biopsy, and cultured tissue. J. Histochem. Cytochem. 1999, 47, 969–972. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, Y.H.; Kim, E.Y.; Gwag, B.J.; Sohn, S.; Koh, J.Y. Zinc-induced cortical neuronal death with features of apoptosis and necrosis: Mediation by free radicals. Neuroscience 1999, 89, 175–182. [Google Scholar] [CrossRef]








© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Choi, B.Y.; Jeong, J.H.; Eom, J.-W.; Koh, J.-Y.; Kim, Y.-H.; Suh, S.W. A Novel Zinc Chelator, 1H10, Ameliorates Experimental Autoimmune Encephalomyelitis by Modulating Zinc Toxicity and AMPK Activation. Int. J. Mol. Sci. 2020, 21, 3375. https://doi.org/10.3390/ijms21093375
Choi BY, Jeong JH, Eom J-W, Koh J-Y, Kim Y-H, Suh SW. A Novel Zinc Chelator, 1H10, Ameliorates Experimental Autoimmune Encephalomyelitis by Modulating Zinc Toxicity and AMPK Activation. International Journal of Molecular Sciences. 2020; 21(9):3375. https://doi.org/10.3390/ijms21093375
Chicago/Turabian StyleChoi, Bo Young, Jeong Hyun Jeong, Jae-Won Eom, Jae-Young Koh, Yang-Hee Kim, and Sang Won Suh. 2020. "A Novel Zinc Chelator, 1H10, Ameliorates Experimental Autoimmune Encephalomyelitis by Modulating Zinc Toxicity and AMPK Activation" International Journal of Molecular Sciences 21, no. 9: 3375. https://doi.org/10.3390/ijms21093375
APA StyleChoi, B. Y., Jeong, J. H., Eom, J. -W., Koh, J. -Y., Kim, Y. -H., & Suh, S. W. (2020). A Novel Zinc Chelator, 1H10, Ameliorates Experimental Autoimmune Encephalomyelitis by Modulating Zinc Toxicity and AMPK Activation. International Journal of Molecular Sciences, 21(9), 3375. https://doi.org/10.3390/ijms21093375