Phenolic Compounds Cannabidiol, Curcumin and Quercetin Cause Mitochondrial Dysfunction and Suppress Acute Lymphoblastic Leukemia Cells




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
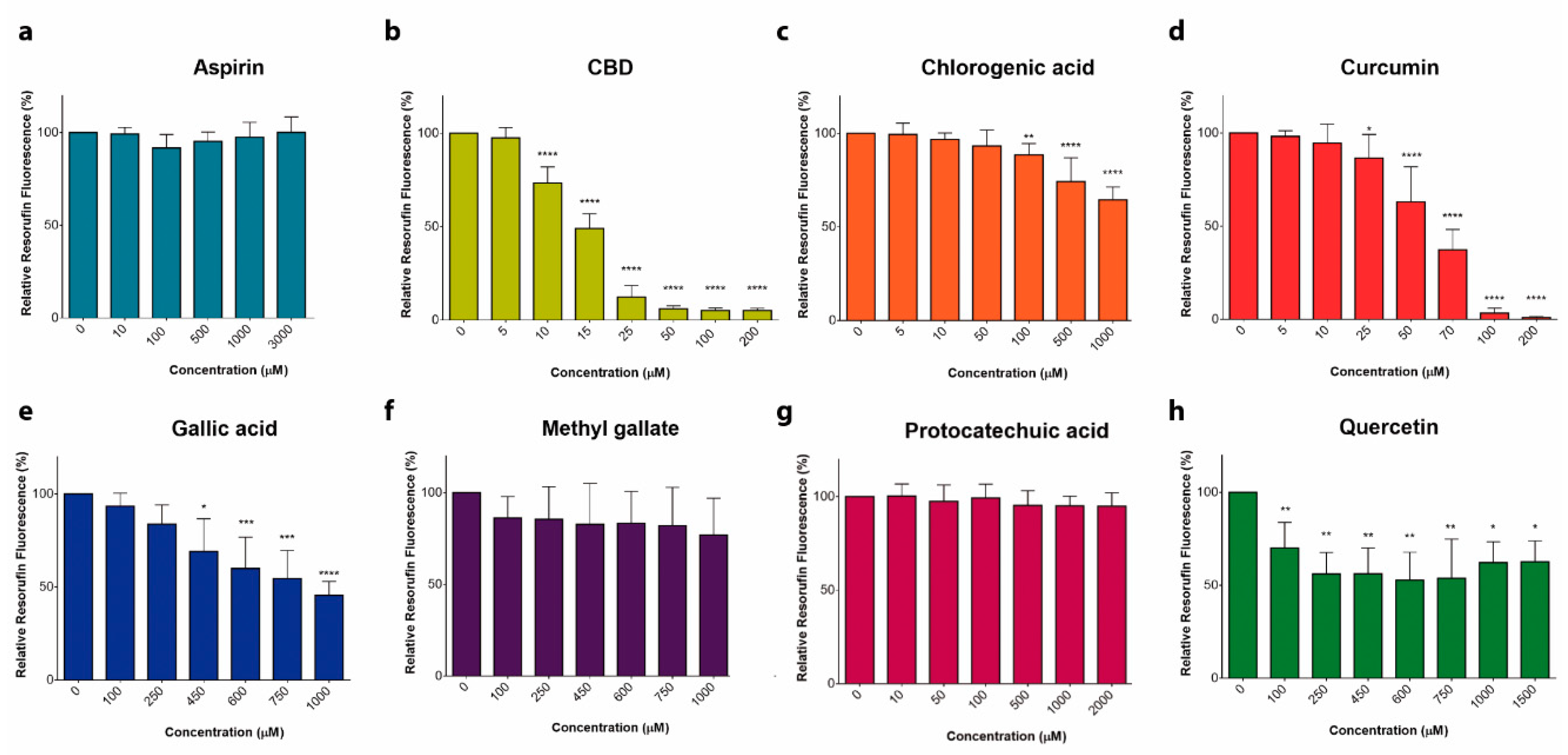
2.1. Comparison of Antileukemic Properties of Different Phenolic Compounds in T-ALL Model
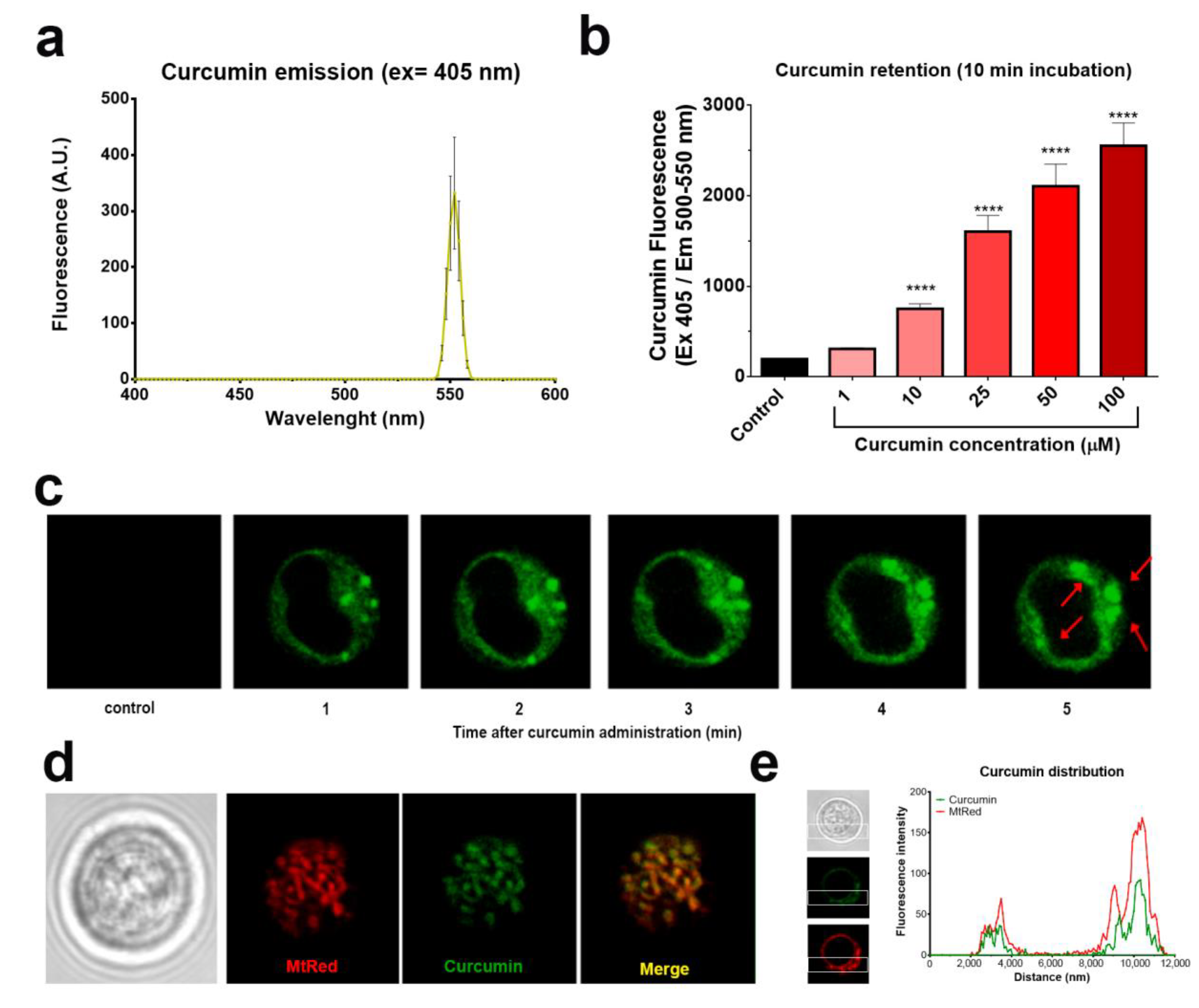
2.2. Curcumin is Rapidly Taken Up by Mitochondria
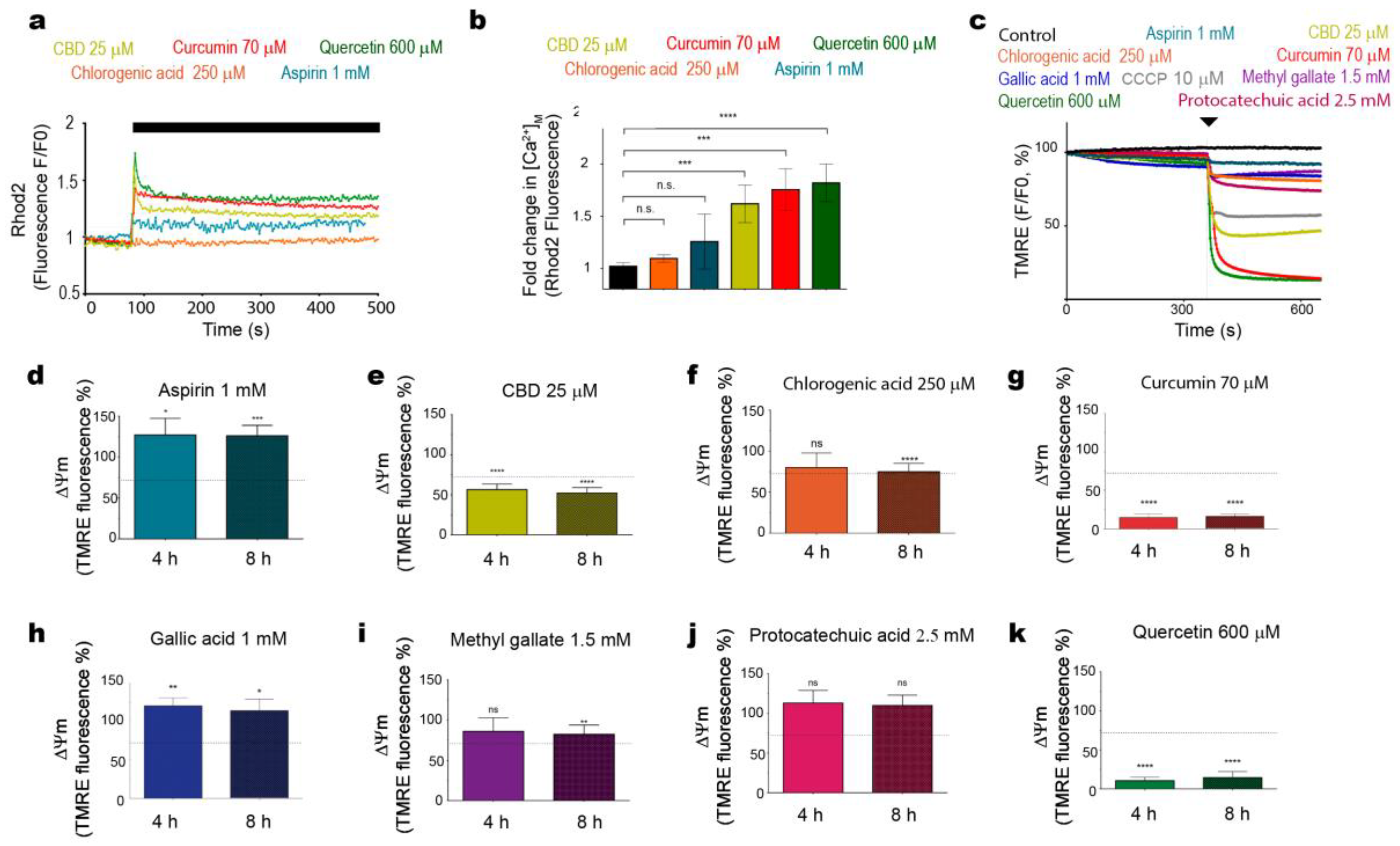
2.3. Cytotoxic Phenols Promote Mitochondrial [Ca2+] Overload and ∆Ψm Loss
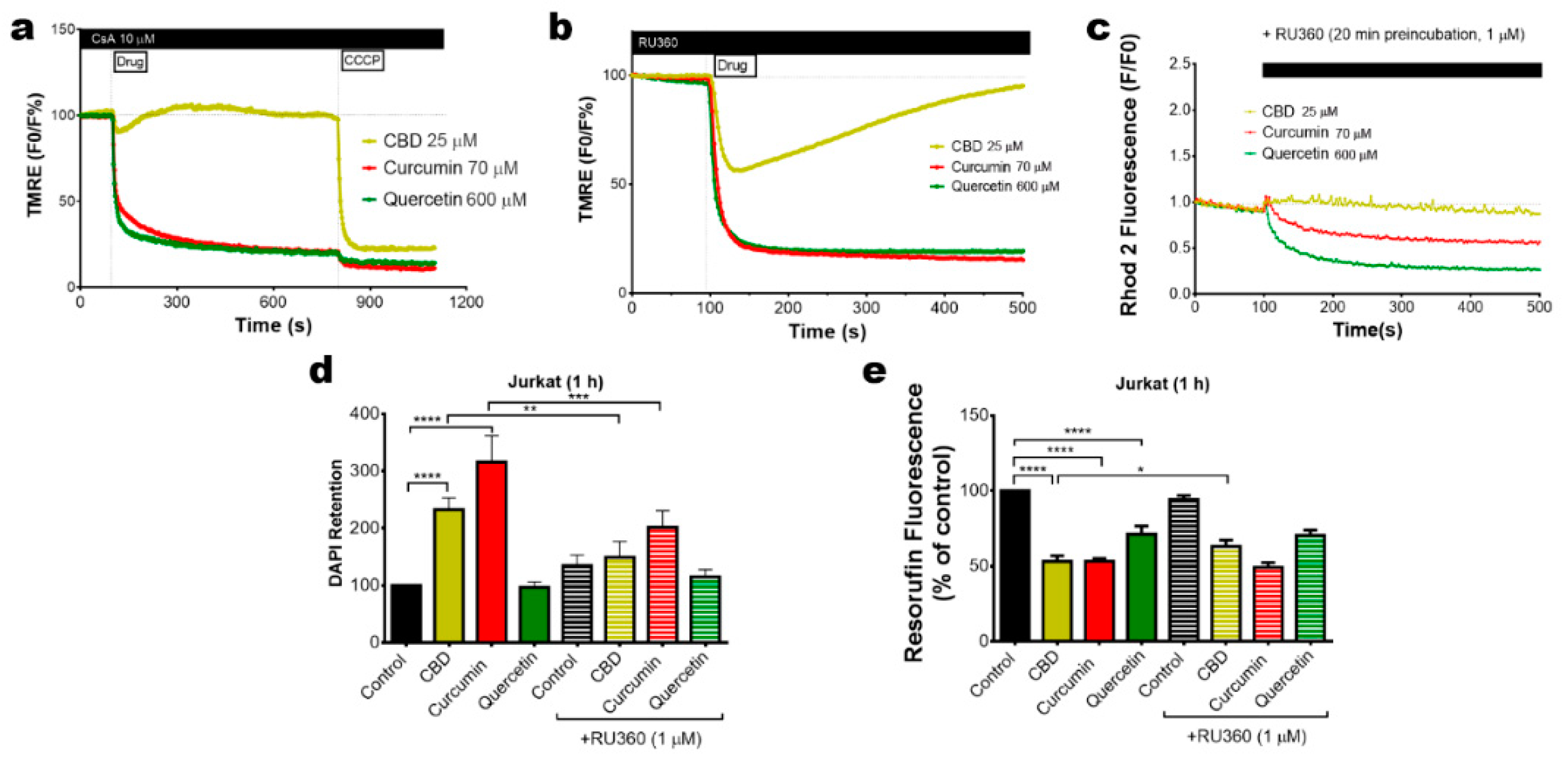
2.4. Mitochondrial Uncoupling and Ca2+ Overload Mediate the Cytotoxic Effect of Bioactive Phenols
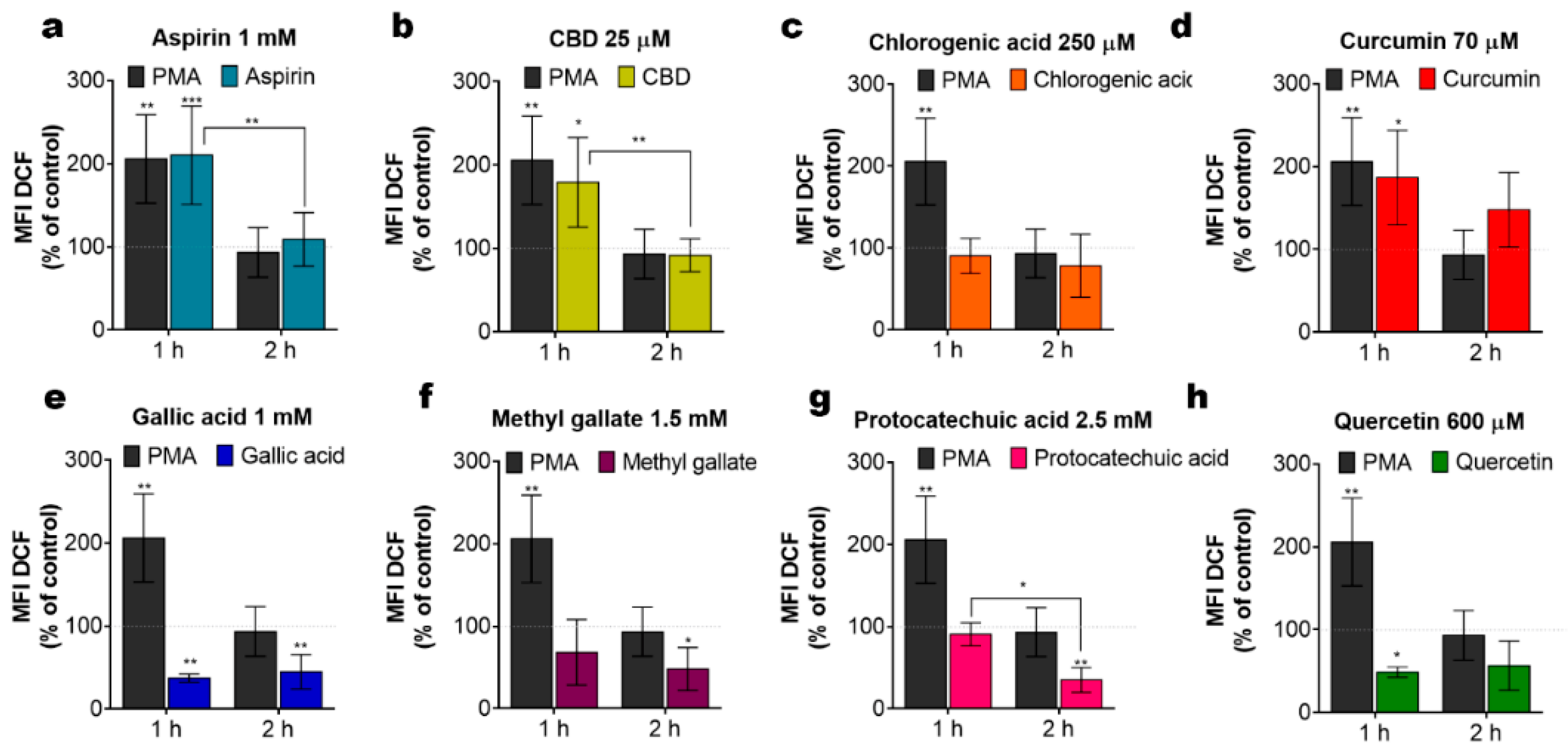
2.5. Phenols Differentially Regulate ROS Production
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. Cells and Culture Conditions
4.3. Viability Assay
4.4. Curcumin Uptake Monitoring
4.5. Mitochondrial Calcium [Ca2+]m Measurements
4.6. Evaluation of Mitochondrial Membrane Potential (∆Ψm)
4.7. Cell Death Analysis by DAPI Retention
4.8. Measurement of ROS by Flow Cytometry
4.9. Protein–Ligand Interaction Prediction
4.10. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ALL | Acute Lymphoblastic Leukemia |
| CBD | Cannabidiol |
| CsA | Cyclosporin A |
| m | Mitochondrial Membrane Potential |
| MCU | Mitochondrial Calcium Uniporter |
| ROS | Reactive Oxygen Species |
| T-ALL | T-lineage ALL |
| VDAC | Voltage-Dependent Anion Channel |
References
- Siegel, D.A.; Henley, S.J.; Li, J.; Pollack, L.A.; Van Dyne, E.A.; White, A. Rates and trends of pediatric acute lymphoblastic leukemia—United States, 2001–2014. MMWR Morb. Mortal. Wkly. Rep. 2017, 66, 950–954. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stankovic, T.; Marston, E. Molecular mechanisms involved in chemoresistance in paediatric acute lymphoblastic leukaemia. Srpski Arhiv za Celokupno Lekarstvo 2008, 136, 1887–1892. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.Y.; Cai, Y.Z.; Zhang, Y. Natural phenolic compounds from medicinal herbs and dietary plants: Potential use for cancer prevention. Nutr. Cancer 2010, 62, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Wang, F.; Trachootham, D.; Huang, P. Preferential killing of cancer cells with mitochondrial dysfunction by natural compounds. Mitochondrion 2010, 10, 614–625. [Google Scholar] [CrossRef] [Green Version]
- Mahbub, A.A.; Le Maitre, C.L.; Haywood-Small, S.L.; Cross, N.A.; Jordan-Mahy, N. Polyphenols act synergistically with doxorubicin and etoposide in leukemia cells. Cell Death Discov. 2015, 11, 5403. [Google Scholar]
- Wahle, K.W.L.; Brown, I.; Rotondo, D.; Heys, S.D. Plant Phenolics in the Prevention and Treatment of Cancer in Bio-Farms for Nutraceuticals. Advances in Experimental Medicine and Biology; Giardi, M.T., Rea, G., Berra, B., Eds.; Springer: Boston, MA, USA, 2010; pp. 36–51. [Google Scholar]
- Salimi, A.; Roudkenar, M.H.; Sadeghi, L. Ellagic acid, a polyphenolic compound, selectively induces ROS-mediated apoptosis in cancerous B-lymphocytes of CLL patients by directly targeting mitochondria. Redox Biol. 2015, 6, 461–471. [Google Scholar] [CrossRef] [Green Version]
- Tewari, D.; Ahmed, T.; Chirasani, V.R.; Singh, P.K.; Maji, S.K.; Senapati, S.; Kanti Bera, A. Modulation of the mitochondrial voltage dependent anion channel (VDAC) by curcumin. Biochimica et Biophysica Acta 2015, 1848, 151–158. [Google Scholar] [CrossRef] [Green Version]
- Tewari, D.; Majumdar, D.; Vallabhaneni, S.; Bera, A.K. Aspirin induces cell death by directly modulating mitochondrial voltage-dependent anion channel (VDAC). Sci. Rep. 2017, 7, 45184. [Google Scholar] [CrossRef] [Green Version]
- Olivas-Aguirre, M.; Torres-López, L.; Valle-Reyes, J.S.; Hernandez-Cruz, A.; Pottosin, I.; Dobrovinskaya, O. Cannabidiol directly targets mitochondria and disturbs calcium homeostasis in acute lymphoblastic leukemia. Cell Death Dis. 2019, 10, 779. [Google Scholar] [CrossRef] [Green Version]
- Stevens, J.F.; Revel, J.S.; Maier, C.S. Mitochondria-centric review of polyphenol bioactivity in cancer models. Antioxid. Redox Signal. 2018, 29, 1589–1611. [Google Scholar] [CrossRef]
- Ralph, S.J.; Low, P.; Lawen, A.; Neuzil, J. Mitocans: Mitochondrial targeted anti-cancer drugs as improved therapies and related patent documents. Recent Pat. Anticancer Drug Discov. 2006, 1, 327–346. [Google Scholar] [CrossRef]
- Olivas-Aguirre, M.; Pottosin, I.; Dobrovinskaya, O. Mitochondria as emerging targets for therapies against T cell acute lymphoblastic leukemia. J. Leukoc. Biol. 2019, 105, 935–946. [Google Scholar] [CrossRef]
- Rimmerman, N.; Ben-Hail, D.; Porat, Z.; Juknat, A.; Kozela, E.; Daniels, M.P.; Connelly, P.S.; Leishman, E.; Bradshaw, H.B.; Shoshan-Barmatz, V.; et al. Direct modulation of the outer mitochondrial membrane channel, voltage-dependent anion channel 1 (VDAC1) by cannabidiol: A novel mechanism for cannabinoid-induced cell death. Cell Death Dis. 2013, 4, e949. [Google Scholar] [CrossRef] [PubMed]
- Colombini, M. VDAC structure, selectivity and dynamics. Biochimica et Biophysica Acta 2012, 1818, 1457–1465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Magri, A.; Reina, S.; De Pinto, V. VDAC1 as pharmacological target in cancer and neurodegeneration: Focus on its role in apoptosis. Front. Chem. 2018, 6, 108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shoshan-Barmatz, V.; Shteinfer-Kuzmine, A.; Verma, A. VDAC1 at the intersection of cell metabolism, apoptosis, and diseases. Biomolecules 2020, 10, 1485. [Google Scholar] [CrossRef] [PubMed]
- Chignell, C.F.; Bilski, P.; Reszka, K.J.; Motten, A.G.; Sik, R.H.; Dahl, T.A. Spectral and photochemical properties of curcumin. Photochem. Photobiol. 1994, 59, 295–302. [Google Scholar] [CrossRef]
- Gorlach, S.; Fichna, J.; Lewandowska, U. Polyphenols as mitochondria-targeted anticancer drugs. Cancer Lett. 2015, 366, 141–149. [Google Scholar] [CrossRef]
- Moustapha, A.; Pérétout, P.A.; Rainey, N.E.; Sureau, F.; Geze, M.; Petit, J.-M.; Dewailly, E.; Slomianny, C.; Petit, P.X. Curcumin induces crosstalk between autophagy and apoptosis mediated by calcium release from endoplasmic reticulum, lysosomal destabilization and mitochondrial events. Cell Death Discov. 2015, 1, 15017. [Google Scholar] [CrossRef]
- Zhang, L.; Cheng, X.; Xu, S.; Bao, J.; Yu, H. Curcumin induces endoplasmic reticulum stress-associated apoptosis in human papillary thyroid carcinoma BCPAP cells via disruption of intracellular calcium homeostasis. Medicine 2018, 97, e11095. [Google Scholar] [CrossRef]
- Sanderson, T.H.; Reynolds, C.A.; Kumar, R.; Przyklenk, K.; Huttemann, M. Molecular mechanisms of ischemia–reperfusion injury in brain: Pivotal role of the mitochondrial membrane potential in reactive oxygen species generation. Mol. Neurobiol. 2013, 47, 9–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rasola, A.; Paolo, B. The mitochondrial permeability transition pore and its adaptive responses in tumor cells. Cell Calcium 2014, 56, 437–445. [Google Scholar] [CrossRef] [PubMed]
- Zorov, D.B.; Juhashzova, M.; Sollott, S.J. Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS-release. Physiol. Rev. 2014, 94, 909–950. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Denton, R.M. Regulation of mitochondrial dehydrogenases by calcium ions. Biochimica et Biophysica Acta 2009, 1787, 1309–1316. [Google Scholar] [CrossRef] [Green Version]
- Tan, W.; Colombini, M. VDAC closure increases calcium ion flux. Biochimica et Biophysica Acta 2007, 1768, 2510–2515. [Google Scholar] [CrossRef] [Green Version]
- Böhm, R.; Amodeo, G.F.; Murlidaran, S.; Chavali, S.; Wagner, G.; Winterhalter, M.; Brannigan, G.; Hiller, S. The Structural Basis for Low Conductance in the Membrane Protein VDAC upon β-NADH Binding and Voltage Gating. Structure 2020, 28, 206–214. [Google Scholar] [CrossRef]
- Mertins, B.; Psakis, G.; Grosse, W.; Back, K.C.; Salisowski, A.; Reiss, P.; Koert, U.; Essen, L.-O. Flexibility of the N-Terminal mVDAC1 segment controls the channels gating behavior. PLoS ONE 2012, 7, e47938. [Google Scholar] [CrossRef]
- Ben-Hail, D.; Begas-Shvartz, R.; Shalev, M.; Shteinfer-Kuzmine, A.; Gruzman, A.; Reina, S.; De Pinto, V.; Shoshan-Barmatz, V. Novel compounds targeting the mitochondrial protein VDAC1 inhibit apoptosis and protect against mitochondrial dysfunction. J. Biol. Chem. 2016, 291, 24986–25003. [Google Scholar] [CrossRef] [Green Version]
- Reuter, S.; Eifes, S.; Dicato, M.; Aggarwal, B.B.; Diederich, M. Modulation of antiapoptotic and survival pathways by curcumin as a strategy to induce apoptosis in cancer cells. Biochem. Pharmacol. 2008, 76, 1340–1351. [Google Scholar] [CrossRef]
- Zhang, X.; Chen, Q.; Wang, Y.; Peng, W.; Cai, H. Effects of curcumin on ion channels and transporters. Front. Physiol. 2014, 5, 94. [Google Scholar] [CrossRef] [Green Version]
- Shukla, S.; Zaher, H.; Hartz, A.; Ware, J.A.; Ambudkar, S.V. Curcumin inhibits the activity of ABCG2/BCRP1, a multidrug resistance-linked ABC drug transporter in mice. Pharm. Res. 2009, 26, 480–487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, S.; Yang, X.; Morris, M.E. Flavonoids are inhibitors of breast cancer resistance protein (ABCG2)-mediated transport. Mol. Pharmacol. 2004, 65, 1208–1216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, S.; Chopra, K.; Kulkarni, S.K.; Agrewala, J.N. Resveratrol and curcumin suppress immune response through CD28/CTLA-4 and CD80 co-stimulatory pathway. Clin. Exp. Immunol. 2007, 147, 155–163. [Google Scholar] [CrossRef] [PubMed]
- Millar, S.A.; Stone, N.L.; Yates, A.S.; O’Sullivan, S.E. A systematic pharmacokinetics of cannabidiol in humans. Front. Pharmacol. 2018, 9, 1365. [Google Scholar] [CrossRef]
- Shoba, G.; Joy, D.; Joseph, T.; Majeed, M.; Rajendran, R.; Srinivas, P.S. Influence of piperine on the pharmacokinetics of curcumin in animals and human volunteers. Planta Medica 1998, 64, 353–356. [Google Scholar] [CrossRef] [Green Version]
- Neuzil, J.; Dong, L.; Rohlena, J.; Truksa, J.; Ralph, S.J. Classification of mitocans, anti-cancer drugs acting on mitochondria. Mitochondrion 2013, 13, 199–208. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Olivas-Aguirre, M.; Torres-López, L.; Pottosin, I.; Dobrovinskaya, O. Phenolic Compounds Cannabidiol, Curcumin and Quercetin Cause Mitochondrial Dysfunction and Suppress Acute Lymphoblastic Leukemia Cells. Int. J. Mol. Sci. 2021, 22, 204. https://doi.org/10.3390/ijms22010204
Olivas-Aguirre M, Torres-López L, Pottosin I, Dobrovinskaya O. Phenolic Compounds Cannabidiol, Curcumin and Quercetin Cause Mitochondrial Dysfunction and Suppress Acute Lymphoblastic Leukemia Cells. International Journal of Molecular Sciences. 2021; 22(1):204. https://doi.org/10.3390/ijms22010204
Chicago/Turabian StyleOlivas-Aguirre, Miguel, Liliana Torres-López, Igor Pottosin, and Oxana Dobrovinskaya. 2021. "Phenolic Compounds Cannabidiol, Curcumin and Quercetin Cause Mitochondrial Dysfunction and Suppress Acute Lymphoblastic Leukemia Cells" International Journal of Molecular Sciences 22, no. 1: 204. https://doi.org/10.3390/ijms22010204
APA StyleOlivas-Aguirre, M., Torres-López, L., Pottosin, I., & Dobrovinskaya, O. (2021). Phenolic Compounds Cannabidiol, Curcumin and Quercetin Cause Mitochondrial Dysfunction and Suppress Acute Lymphoblastic Leukemia Cells. International Journal of Molecular Sciences, 22(1), 204. https://doi.org/10.3390/ijms22010204