CD85k Contributes to Regulatory T Cell Function in Chronic Viral Infections


 ,
,  ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
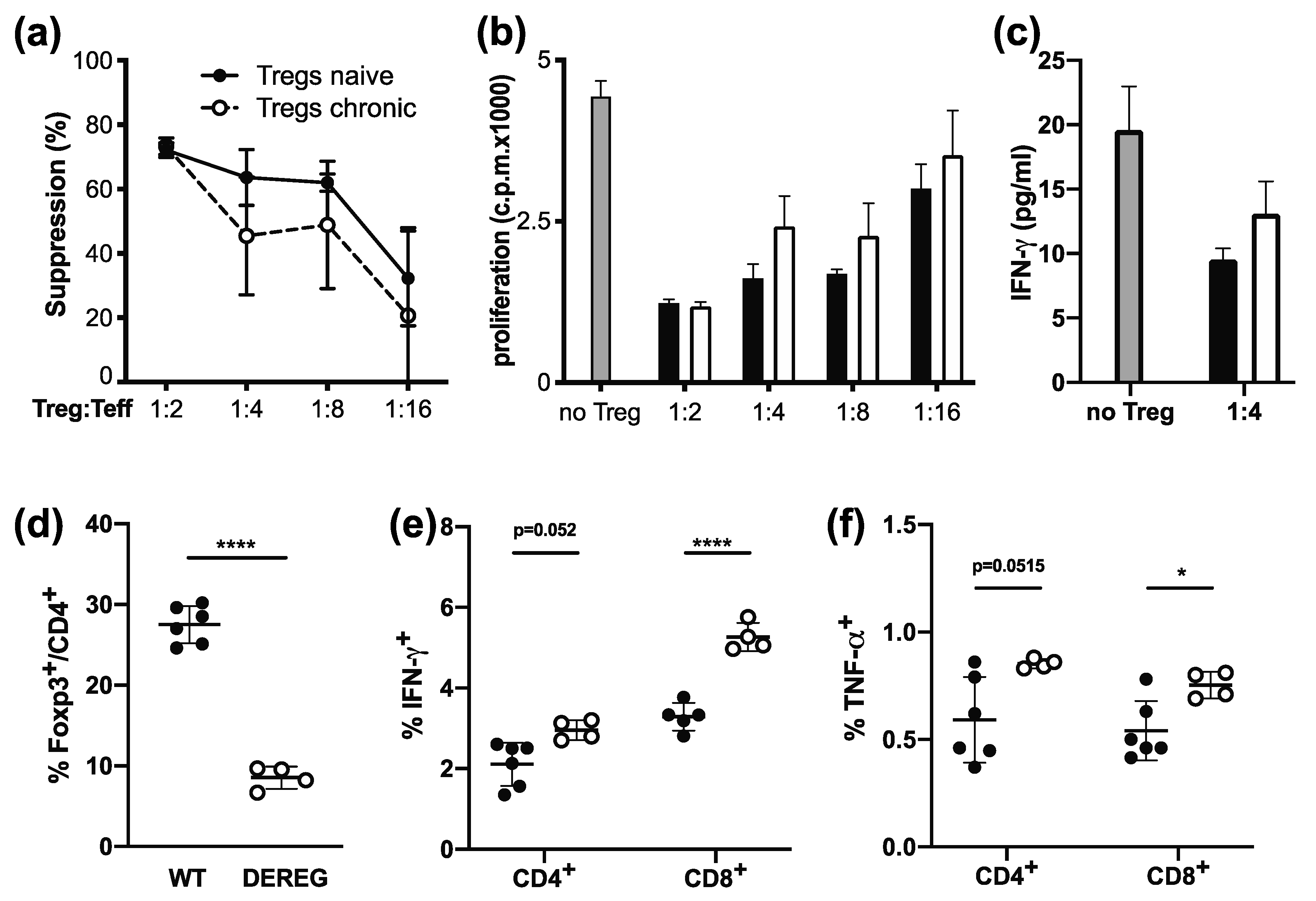
2.1. Tregs Maintain Suppressive Function during Chronic Infection
2.2. Tregs Undergo Classical Th1-Specific Specialization in Persistent Infection
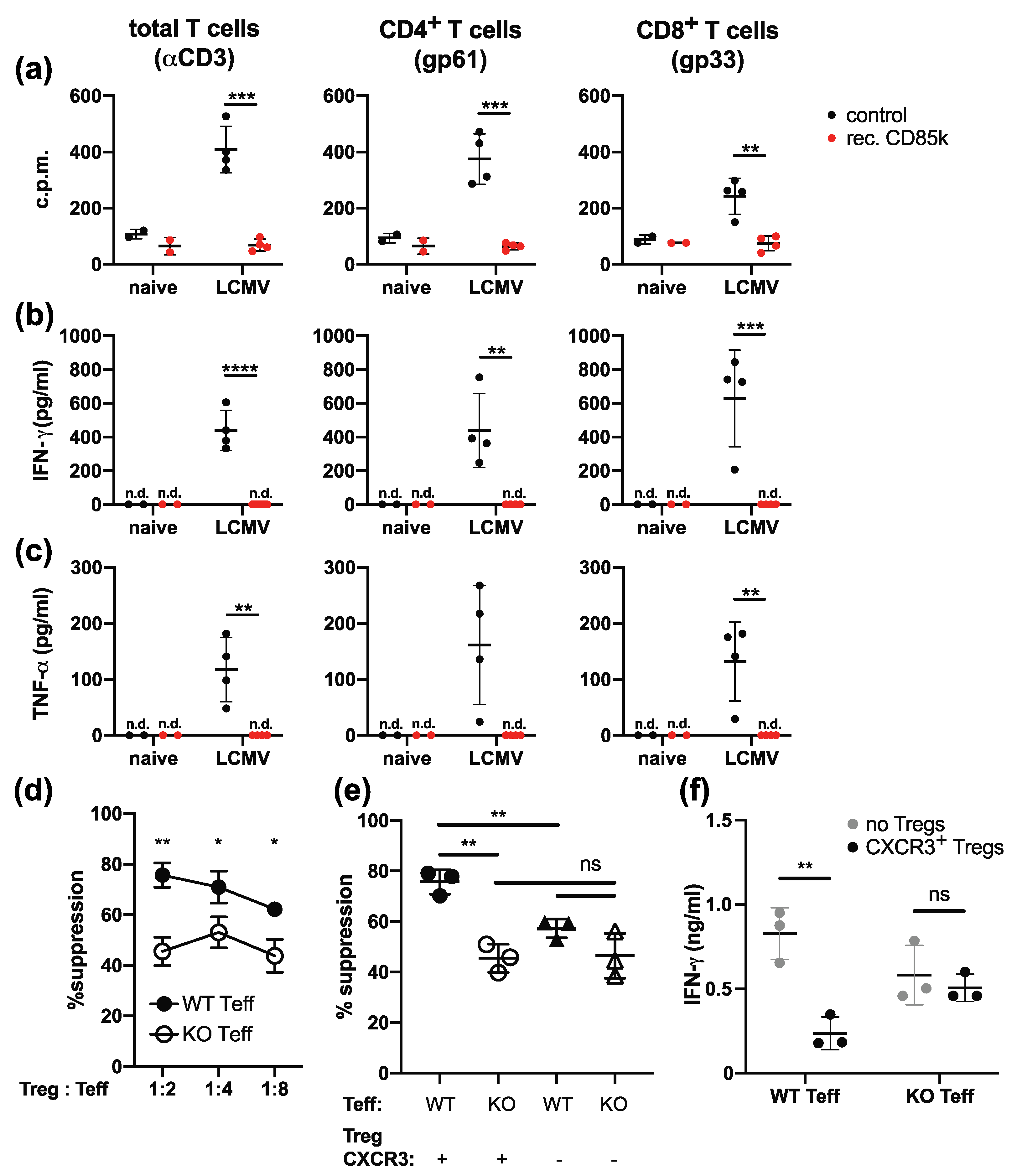
2.3. Effector T Cells Express the CD85k Ligand ALCAM during Chronic Infection
2.4. Type 1 Tregs Limit ALCAM-Expressing Effector T Cells
3. Discussion
4. Materials and Methods
4.1. Mice, Pathogens, and Infections
4.2. Isolation of Leukocytes
4.3. Flow Cytometry
4.4. In Vitro Co-Culture and Treg Suppression Assays
4.5. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| Treg | regulatory T cell |
| LCMV | Lymphocytic choriomeningitis virus |
| MCMV | Murine cytomegalovirus |
| APC | antigen presenting cell |
References
- Fontenot, J.D.; Gavin, M.A.; Rudensky, A.Y. Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nat. Immunol. 2003, 4, 330–336. [Google Scholar] [CrossRef] [PubMed]
- Wildin, R.S.; Ramsdell, F.; Peake, J.; Faravelli, F.; Casanova, J.L.; Buist, N.; Levy-Lahad, E.; Mazzella, M.; Goulet, O.; Perroni, L.; et al. X-linked neonatal diabetes mellitus, enteropathy and endocrinopathy syndrome is the human equivalent of mouse scurfy. Nat. Genet. 2001, 27, 18–20. [Google Scholar] [CrossRef] [PubMed]
- Hori, S.; Nomura, T.; Sakaguchi, S. Control of regulatory T cell development by the transcription factor Foxp3. Science 2003, 299, 1057–1061. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lund, J.M.; Hsing, L.; Pham, T.T.; Rudensky, A.Y. Coordination of early protective immunity to viral infection by regulatory T cells. Science 2008, 320, 1220–1224. [Google Scholar] [CrossRef] [Green Version]
- Belkaid, Y.; Piccirillo, C.A.; Mendez, S.; Shevach, E.M.; Sacks, D.L. CD4+CD25+ regulatory T cells control Leishmania major persistence and immunity. Nature 2002, 420, 502–507. [Google Scholar] [CrossRef]
- Suvas, S.; Kumaraguru, U.; Pack, C.D.; Lee, S.; Rouse, B.T. CD4+CD25+ T cells regulate virus-specific primary and memory CD8+ T cell responses. J. Exp. Med. 2003, 198, 889–901. [Google Scholar] [CrossRef]
- Dittmer, U.; He, H.; Messer, R.J.; Schimmer, S.; Olbrich, A.R.; Ohlen, C.; Greenberg, P.D.; Stromnes, I.M.; Iwashiro, M.; Sakaguchi, S.; et al. Functional impairment of CD8+ T cells by regulatory T cells during persistent retroviral infection. Immunity 2004, 20, 293–303. [Google Scholar] [CrossRef] [Green Version]
- Penaloza-MacMaster, P.; Kamphorst, A.O.; Wieland, A.; Araki, K.; Iyer, S.S.; West, E.E.; O’Mara, L.; Yang, S.; Konieczny, B.T.; Sharpe, A.H.; et al. Interplay between regulatory T cells and PD-1 in modulating T cell exhaustion and viral control during chronic LCMV infection. J. Exp. Med. 2014, 211, 1905–1918. [Google Scholar] [CrossRef]
- Koch, M.A.; Tucker-Heard, G.; Perdue, N.R.; Killebrew, J.R.; Urdahl, K.B.; Campbell, D.J. The transcription factor T-bet controls regulatory T cell homeostasis and function during type 1 inflammation. Nat. Immunol. 2009, 10, 595–602. [Google Scholar] [CrossRef]
- Zheng, Y.; Chaudhry, A.; Kas, A.; deRoos, P.; Kim, J.M.; Chu, T.T.; Corcoran, L.; Treuting, P.; Klein, U.; Rudensky, A.Y. Regulatory T-cell suppressor program co-opts transcription factor IRF4 to control TH2 responses. Nature 2009, 458, 351–356. [Google Scholar] [CrossRef]
- Chaudhry, A.; Rudra, D.; Treuting, P.; Samstein, R.M.; Liang, Y.; Kas, A.; Rudensky, A.Y. CD4+ regulatory T cells control TH17 responses in a Stat3-dependent manner. Science 2009, 326, 986–991. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levine, A.G.; Medoza, A.; Hemmers, S.; Moltedo, B.; Niec, R.E.; Schizas, M.; Hoyos, B.E.; Putintseva, E.V.; Chaudhry, A.; Dikiy, S.; et al. Stability and function of regulatory T cells expressing the transcription factor T-bet. Nature 2017, 546, 421–425. [Google Scholar] [CrossRef] [PubMed]
- Hall, A.O.; Beiting, D.P.; Tato, C.; John, B.; Oldenhove, G.; Lombana, C.G.; Pritchard, G.H.; Silver, J.S.; Bouladoux, N.; Stumhofer, J.S.; et al. The cytokines interleukin 27 and interferon-gamma promote distinct Treg cell populations required to limit infection-induced pathology. Immunity 2012, 37, 511–523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koch, M.A.; Thomas, K.R.; Perdue, N.R.; Smigiel, K.S.; Srivastava, S.; Campbell, D.J. T-bet+ Treg cells undergo abortive Th1 cell differentiation due to impaired expression of IL-12 receptor beta2. Immunity 2012, 37, 501–510. [Google Scholar] [CrossRef] [Green Version]
- Littringer, K.; Moresi, C.; Rakebrandt, N.; Zhou, X.; Schorer, M.; Dolowschiak, T.; Kirchner, F.; Rost, F.; Keller, C.W.; McHugh, D.; et al. Common features of regulatory T cell specialization during Th1 responses. Front. Immunol. 2018, 9. [Google Scholar] [CrossRef] [Green Version]
- Cella, M.; Dohring, C.; Samaridis, J.; Dessing, M.; Brockhaus, M.; Lanzavecchia, A.; Colonna, M. A novel inhibitory receptor (ILT3) expressed on monocytes, macrophages, and dendritic cells involved in antigen processing. J. Exp. Med. 1997, 185, 1743–1751. [Google Scholar] [CrossRef]
- Chang, C.C.; Ciubotariu, R.; Manavalan, J.S.; Yuan, J.; Colovai, A.I.; Piazza, F.; Lederman, S.; Colonna, M.; Cortesini, R.; Dalla-Favera, R.; et al. Tolerization of dendritic cells by T(S) cells: The crucial role of inhibitory receptors ILT3 and ILT4. Nat. Immunol. 2002, 3, 237–243. [Google Scholar] [CrossRef]
- Kim-Schulze, S.; Scotto, L.; Vlad, G.; Piazza, F.; Lin, H.; Liu, Z.; Cortesini, R.; Suciu-Foca, N. Recombinant Ig-Like Transcript 3-Fc Modulates T Cell Responses via Induction of Th Anergy and Differentiation of CD8 + T Suppressor Cells. J. Immunol. 2006. [Google Scholar] [CrossRef] [Green Version]
- Xu, Z.; Chang, C.-C.; Li, M.; Zhang, Q.-Y.; Vasilescu, E.-R.M.; D′Agati, V.; Floratos, A.; Vlad, G.; Suciu-Foca, N. ILT3.Fc–CD166 Interaction Induces Inactivation of p70 S6 Kinase and Inhibits Tumor Cell Growth. J. Immunol. 2018, 200, 1207–1219. [Google Scholar] [CrossRef] [Green Version]
- Ulges, A.; Klein, M.; Reuter, S.; Gerlitzki, B.; Hoffmann, M.; Grebe, N.; Staudt, V.; Stergiou, N.; Bohn, T.; Bruhl, T.J.; et al. Protein kinase CK2 enables regulatory T cells to suppress excessive TH2 responses in vivo. Nat. Immunol. 2015, 16, 267–275. [Google Scholar] [CrossRef]
- Nilsson, J.; Boasso, A.; Velilla, P.A.; Zhang, R.; Vaccari, M.; Franchini, G.; Shearer, G.M.; Andersson, J.; Chougnet, C. HIV-1-driven regulatory T-cell accumulation in lymphoid tissues is associated with disease progression in HIV/AIDS. Blood 2006, 108, 3808–3817. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cabrera, R.; Tu, Z.; Xu, Y.; Firpi, R.J.; Rosen, H.R.; Liu, C.; Nelson, D.R. An immunomodulatory role for CD4+CD25+ regulatory T lymphocytes in hepatitis C virus infection. Hepatology 2004, 40, 1062–1071. [Google Scholar] [CrossRef] [PubMed]
- Aalaei-Andabili, S.H.; Alavian, S.M. Regulatory T cells are the most important determinant factor of hepatitis B infection prognosis: A systematic review and meta-analysis. Vaccine 2012, 30, 5595–5602. [Google Scholar] [CrossRef] [PubMed]
- Tovar-Salazar, A.; Weinberg, A. Cytomegalovirus infection in HIV-infected and uninfected individuals is characterized by circulating regulatory T cells of unconstrained antigenic specificity. PLoS ONE 2017, 12, e0180691. [Google Scholar] [CrossRef]
- Almanan, M.; Raynor, J.; Sholl, A.; Wang, M.; Chougnet, C.; Cardin, R.D.; Hildeman, D.A. Tissue-specific control of latent CMV reactivation by regulatory T cells. PLoS Pathog. 2017, 13, e1006507. [Google Scholar] [CrossRef] [Green Version]
- Popovic, B.; Golemac, M.; Podlech, J.; Zeleznjak, J.; Bilic-Zulle, L.; Lukic, M.L.; Cicin-Sain, L.; Reddehase, M.J.; Sparwasser, T.; Krmpotic, A.; et al. IL-33/ST2 pathway drives regulatory T cell dependent suppression of liver damage upon cytomegalovirus infection. PLoS Pathog. 2017, 13, e1006345. [Google Scholar] [CrossRef] [Green Version]
- Moskophidis, D.; Lechner, F.; Pircher, H.; Zinkernagel, R.M. Virus persistence in acutely infected immunocompetent mice by exhaustion of antiviral cytotoxic effector T cells. Nature 1993, 362, 758–761. [Google Scholar] [CrossRef]
- Lahl, K.; Loddenkemper, C.; Drouin, C.; Freyer, J.; Arnason, J.; Eberl, G.; Hamann, A.; Wagner, H.; Huehn, J.; Sparwasser, T. Selective depletion of Foxp3+ regulatory T cells induces a scurfy-like disease. J. Exp. Med. 2007, 204, 57–63. [Google Scholar] [CrossRef] [Green Version]
- Christiaansen, A.F.; Boggiatto, P.M.; Varga, S.M. Limitations of Foxp3+ Treg depletion following viral infection in DEREG mice. J. Immunol. Methods 2014, 406, 58–65. [Google Scholar] [CrossRef] [Green Version]
- Schönherr, F.A.; Sparber, F.; Kirchner, F.R.; Guiducci, E.; Trautwein-Weidner, K.; Gladiator, A.; Sertour, N.; Hetzel, U.; Le, G.T.T.; Pavelka, N.; et al. The intraspecies diversity of C. albicans triggers qualitatively and temporally distinct host responses that determine the balance between commensalism and pathogenicity. Mucosal Immunol. 2017, 10, 1335–1350. [Google Scholar] [CrossRef] [PubMed]
- Wing, K.; Onishi, Y.; Prieto-Martin, P.; Yamaguchi, T.; Miyara, M.; Fehervari, Z.; Nomura, T.; Sakaguchi, S. CTLA-4 control over Foxp3+ regulatory T cell function. Science 2008, 322, 271–275. [Google Scholar] [CrossRef] [PubMed]
- Joller, N.; Lozano, E.; Burkett, P.R.; Patel, B.; Xiao, S.; Zhu, C.; Xia, J.; Tan, T.G.; Sefik, E.; Yajnik, V.; et al. Treg cells expressing the coinhibitory molecule TIGIT selectively inhibit proinflammatory Th1 and Th17 cell responses. Immunity 2014, 40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Francisco, L.M.; Salinas, V.H.; Brown, K.E.; Vanguri, V.K.; Freeman, G.J.; Kuchroo, V.K.; Sharpe, A.H. PD-L1 regulates the development, maintenance, and function of induced regulatory T cells. J. Exp. Med. 2009, 206, 3015–3029. [Google Scholar] [CrossRef] [PubMed]
- Bergthaler, A.; Flatz, L.; Verschoor, A.; Hegazy, A.N.; Holdener, M.; Fink, K.; Eschli, B.; Merkler, D.; Sommerstein, R.; Horvath, E.; et al. Impaired antibody response causes persistence of prototypic T cell-contained virus. PLoS Biol. 2009, 7, 789–799. [Google Scholar] [CrossRef]
- Zajac, A.J.; Blattman, J.N.; Murali-Krishna, K.; Sourdive, D.J.D.; Suresh, M.; Altman, J.D.; Ahmed, R. Viral immune evasion due to persistence of activated T cells without effector function. J. Exp. Med. 1998, 188, 2205–2213. [Google Scholar] [CrossRef]
- Kirchner, F.R.; Littringer, K.; Altmeier, S.; Van Du, T.T.; Schönherr, F.; Lemberg, C.; Pagni, M.; Sanglard, D.; Joller, N.; LeibundGut-Landmann, S. Persistence of Candida albicans in the Oral Mucosa Induces a Curbed Inflammatory Host Response That Is Independent of Immunosuppression. Front. Immunol. 2019, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kirchner, F.R.; Leibundgut-landmann, S. Tissue-resident memory Th17 cells maintain stable fungal commensalism in the oral mucosa. Mucosal Immunol. 2020, 1–13. [Google Scholar] [CrossRef]
- McLane, L.M.; Abdel-Hakeem, M.S.; Wherry, E.J. CD8 T Cell Exhaustion During Chronic Viral Infection and Cancer. Annu. Rev. Immunol. 2019, 37, 457–495. [Google Scholar] [CrossRef] [Green Version]
- Munks, M.W.; Cho, K.S.; Pinto, A.K.; Sierro, S.; Klenerman, P.; Hill, A.B. Four Distinct Patterns of Memory CD8 T Cell Responses to Chronic Murine Cytomegalovirus Infection. J. Immunol. 2006. [Google Scholar] [CrossRef]
- Huang, C.T.; Workman, C.J.; Flies, D.; Pan, X.; Marson, A.L.; Zhou, G.; Hipkiss, E.L.; Ravi, S.; Kowalski, J.; Levitsky, H.I.; et al. Role of LAG-3 in regulatory T cells. Immunity 2004, 21, 503–513. [Google Scholar] [CrossRef] [Green Version]
- Bettelli, E.; Carrier, Y.; Gao, W.; Korn, T.; Strom, T.B.; Oukka, M.; Weiner, H.L.; Kuchroo, V.K. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature 2006, 441, 235–238. [Google Scholar] [CrossRef] [PubMed]
- Weiner, J.A.; Koo, S.J.; Nicolas, S.; Fraboulet, S.; Pfaff, S.L.; Pourquié, O.; Sanes, J.R. Axon fasciculation defects and retinal dysplasias in mice lacking the immunoglobulin superfamily adhesion molecule BEN/ALCAM/SC1. Mol. Cell. Neurosci. 2004, 27, 59–69. [Google Scholar] [CrossRef] [PubMed]
- Jordan, S.; Krause, J.; Prager, A.; Mitrovic, M.; Jonjic, S.; Koszinowski, U.H.; Adler, B. Virus Progeny of Murine Cytomegalovirus Bacterial Artificial Chromosome pSM3fr Show Reduced Growth in Salivary Glands due to a Fixed Mutation of MCK-2. J. Virol. 2011, 85, 10346–10353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Altman, J.D.; Moss, P.A.H.; Goulder, P.J.R.; Barouch, D.H.; McHeyzer-Williams, M.G.; Bell, J.I.; McMichael, A.J.; Davis, M.M. Phenotypic analysis of antigen-specific T lymphocytes. Science 1996, 274, 94–96. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Estrada Brull, A.; Rost, F.; Oderbolz, J.; Kirchner, F.R.; Leibundgut-Landmann, S.; Oxenius, A.; Joller, N. CD85k Contributes to Regulatory T Cell Function in Chronic Viral Infections. Int. J. Mol. Sci. 2021, 22, 31. https://doi.org/10.3390/ijms22010031
Estrada Brull A, Rost F, Oderbolz J, Kirchner FR, Leibundgut-Landmann S, Oxenius A, Joller N. CD85k Contributes to Regulatory T Cell Function in Chronic Viral Infections. International Journal of Molecular Sciences. 2021; 22(1):31. https://doi.org/10.3390/ijms22010031
Chicago/Turabian StyleEstrada Brull, Anna, Felix Rost, Josua Oderbolz, Florian R. Kirchner, Salomé Leibundgut-Landmann, Annette Oxenius, and Nicole Joller. 2021. "CD85k Contributes to Regulatory T Cell Function in Chronic Viral Infections" International Journal of Molecular Sciences 22, no. 1: 31. https://doi.org/10.3390/ijms22010031
APA StyleEstrada Brull, A., Rost, F., Oderbolz, J., Kirchner, F. R., Leibundgut-Landmann, S., Oxenius, A., & Joller, N. (2021). CD85k Contributes to Regulatory T Cell Function in Chronic Viral Infections. International Journal of Molecular Sciences, 22(1), 31. https://doi.org/10.3390/ijms22010031