The Role of Bone in Muscle Wasting


Abstract
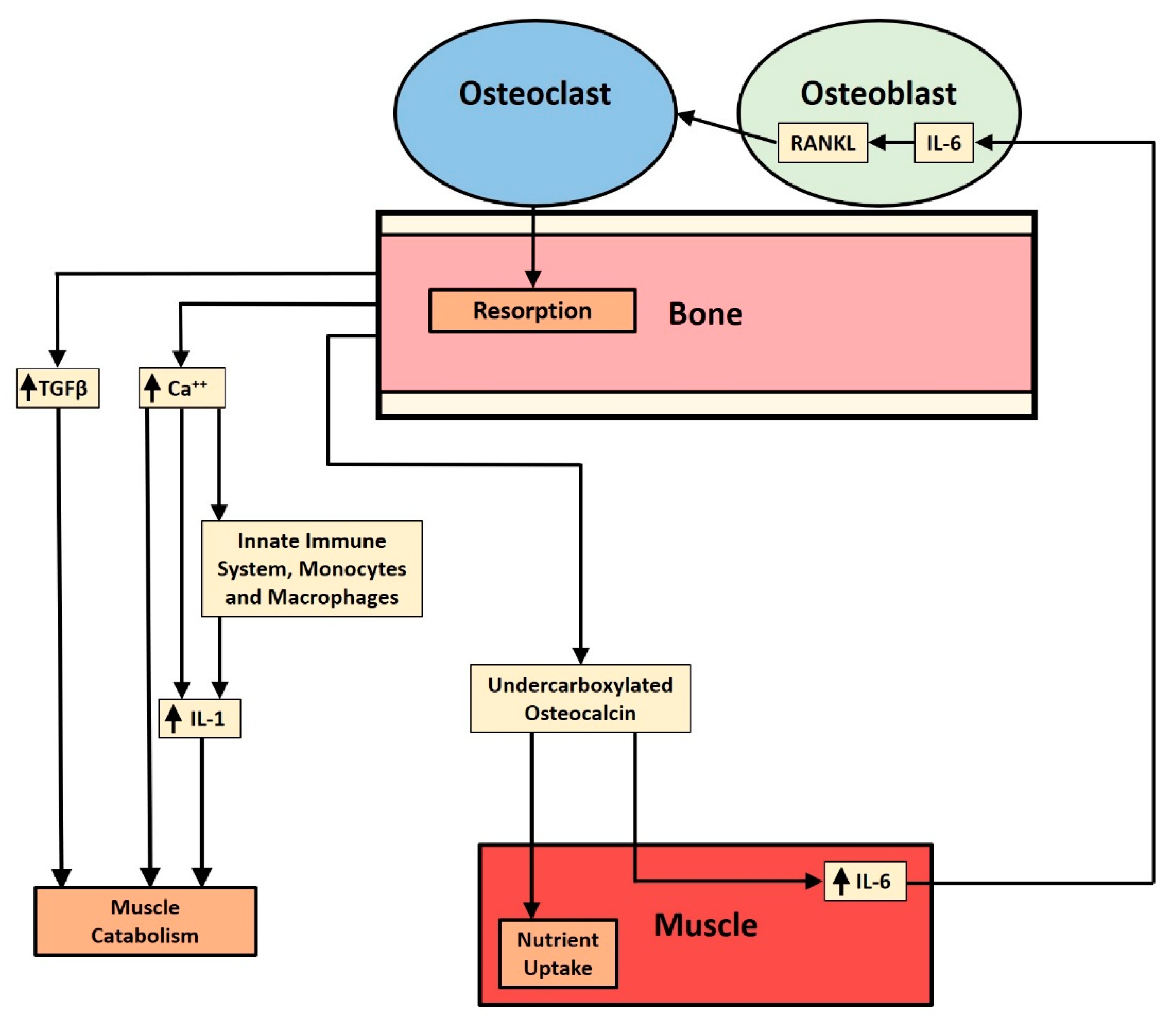
:1. Introduction: Bone Resorption
1.1. Clinical Conditions That Can Cause Resorptive Bone Loss
1.2. Factors Liberated by Bone Resorption: What We Know and Do Not Know
1.3. Bone Factor Release and Muscle Catabolism
1.4. Muscle Factors Influencing Atrophy
2. Discussion and Summary
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Shiwaku, Y.; Neff, L.; Nagano, K.; Takayama, K.-I.; de Bruijn, J.; Dard, M.; Gori, F.; Baron, R. The crosstalk between osteoclasts and osteoblasts is dependent upon the structure of biphasic calcium phosphates. PLoS ONE 2015, 10, e0132903. [Google Scholar] [CrossRef]
- Ikebuchi, Y.; Aoki, S.; Honma, M.; Hayashi, M.; Sugamori, Y.; Khan, M.; Kariya, Y.; Kato, G.; Tabata, Y.; Penninger, J.M.; et al. Coupling of bone resorption and formation by RANKL reverse signaling. Nature 2018, 561, 195–200. [Google Scholar] [CrossRef]
- Waning, D.L.; Mohammad, K.S.; Reiken, S.; Xie, W.; Andersson, D.C.; John, S.K.; Chiechi, A.; Wright, L.E.; Umanskaya, A.; Niewolna, M.; et al. Excess TGF-β mediates muscle weakness associated with bone metastases in mice. Nat. Med. 2015, 21, 1262–1271. [Google Scholar] [CrossRef]
- Klein, G.L.; Herndon, D.; Goodman, W.; Langman, C.; Phillips, W.; Dickson, I.; Eastell, R.; Naylor, K.; Maloney, N.; Desai, M.; et al. Histomorphometric and biochemical characterization of bone following acute severe burns in children. Bone 1995, 17, 455–460. [Google Scholar] [CrossRef]
- Klein, G.L.; Bi, L.X.; Sherrard, D.J.; Beavan, S.R.; Ireland, D.; Compston, J.E.; Williams, E.G.; Herndon, D.N. Evidence supporting a role of glucocorticoids in short-term bone loss in burned children. Osteoporos. Int. 2004, 15, 468–474. [Google Scholar] [CrossRef]
- Klein, G.L. The role of the musculoskeletal system in post-burn hypermetabolism. Metabolism 2019, 97, 81–86. [Google Scholar] [CrossRef] [PubMed]
- Rossol, M.; Pierer, M.; Raulien, N.; Quandt, D.; Meusch, U.; Rothe, K.; Schubert, K.; Schöneberg, T.; Schaefer, M.; Krügel, U.; et al. Extracellular Ca2+ is a danger signal activating the NLRP3 inflammasome through G protein-coupled calcium sensing receptors. Nat. Commun. 2012, 3, 1329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klein, G.L.; Castro, S.M.; Garofalo, R.P. The calcium-sensing receptor as a mediator of inflammation. Semin. Cell Dev. Biol. 2016, 49, 52–56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nielsen, P.K.; Rasmussen, A.K.; Butters, R.; Feldt-Rasmussen, U.; Bendtzen, K.; Diaz, R.; Brown, E.M.; Olgaard, K. Inhibition of PTH secretion by interleukin-1 beta in bovine parathyroid glands in vitro is associated with upregulation of the calcium sensing receptor mRNA. Biochem. Biophys. Res. Commun. 1997, 238, 880–885. [Google Scholar] [CrossRef]
- Canaff, L.; Zhou, X.; Hendy, G.N. The pro-inflammatory cytokine, interleukin-6, up-regulates calcium-sensing receptor gene transcription via Stat 1/3 and Sp 1/3. J. Biol. Chem. 2008, 283, 13586–13600. [Google Scholar] [CrossRef] [Green Version]
- Murphey, E.D.; Chattopadhyay, N.; Bai, M.; Kifor, O.; Harper, D.; Traber, D.L.; Hawkins, H.K.; Brown, E.M.; Klein, G.L. Up-regulation of the parathyroid calcium-sensing receptor after burn injury in sheep: A potential contributing factor to post-burn hypocalcemia. Crit. Care Med. 2000, 28, 3885–3890. [Google Scholar] [CrossRef] [PubMed]
- Klein, G.L.; Nicolai, M.; Langman, C.B.; Cuneo, B.F.; Sailer, D.E.; Herndon, D.N. Dysregulation of calcium metabolism after severe burn injury in children: Possible role of magnesium depletion. J. Pediatr. 1997, 131, 246–251. [Google Scholar] [CrossRef]
- Klein, G.L.; Herndon, D.N.; Rutan, T.C.; Sherrard, D.J.; Coburn, J.W.; Langman, C.B.; Thomas, M.L.; Cooper, C.W.; Miller, N.C. Bone disease in burn patients. J. Bone Miner. Res. 1993, 8, 337–345. [Google Scholar] [CrossRef] [PubMed]
- Rousseau, A.-F.; Damas, P.; LeDoux, D.; Cavalier, E. Effect of cholecalciferol recommended daily allowances on Vitamin D status and fibroblast growth factor-23: An observational study in acute burn patients. Burns 2014, 40, 865–870. [Google Scholar] [CrossRef] [PubMed]
- Finnerty, C.C.; Jeschke, M.B.; Herndon, D.N.; Gamelli, R.; Gibran, N.; Klein, M.; Silver, G.; Arnoldo, B.; Remick, D.; Tompkins, B.G. Temporal cytokine profiles in severely burned patients: A comparison between adults and children. Mol. Med. 2008, 14, 553–560. [Google Scholar] [CrossRef] [PubMed]
- Borsheim, E.; Herndon, D.N.; Hawkins, H.K.; Suman, O.E.; Cotter, M.; Klein, G.L. Pamidronate attenuates post-burn muscle loss after pediatric burn injury. J. Bone Miner. Res. 2014, 29, 1369–1372. [Google Scholar] [CrossRef] [Green Version]
- Morikawa, M.; Derynck, R.; Miyazono, K. TGF-β and the TGF-β family: Context dependent roles in cell and tissue physiology. Cold Spring Harb. Perspect. Biol. 2016, 8, a021873. [Google Scholar] [CrossRef] [Green Version]
- Dallas, S.L.; Rosser, J.L.; Mundy, G.R.; Bonewald, L.F. Proteolysis of latent transforming growth factor (TGF-β) binding protein 1 by osteoclasts. A cellular mechanism for the release of TGF-β from bone matrix. J. Biol. Chem. 2002, 277, 21352–21360. [Google Scholar] [CrossRef] [Green Version]
- Karsenty, G.; Mera, P. Molecular basis of crosstalk between bone and muscle. Bone 2018, 115, 43–49. [Google Scholar] [CrossRef]
- Londhe, P.; Guttridge, D.C. Inflammation induced loss of skeletal muscle. Bone 2015, 60, 131–142. [Google Scholar] [CrossRef] [Green Version]
- Pin, F.; Bonetto, A.; Bonewald, L.F.; Klein, G.L. Molecular mechanisms responsible for the rescue effects of pamidronate on muscle atrophy in pediatric burn patients. Front. Endocrinol. (Lausanne) 2019, 10, 543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Essex, A.L.; Pin, F.; Huot, J.R.; Bonewald, L.F.; Plotkin, L.J.; Bonetto, A. A bisphosphonate treatment ameliorates chemotherapy induced bone and muscle abnormalities in young mice. Front. Endocrinol. (Lausanne) 2019, 40, 809. [Google Scholar] [CrossRef] [PubMed]
- Hain, B.; Xu, H.; Wilcox, J.R.; Mutua, D.; Waning, D.L. Chemotherapy-induced loss of bone and muscle mass in a mouse model of bone metastases and cachexia. JCSM Rapid Commun. 2019, 2, e00075. [Google Scholar] [CrossRef] [PubMed]
- Bonnet, N.; Bourgoin, L.; Biver, E.; Douni, E.; Ferrari, S. RANKL inhibition improves muscle strength and insulin sensitivity and restores bone mass. J. Clin. Investig. 2019, 129, 3214–3223. [Google Scholar] [CrossRef]
- Mera, P.; Laue, K.; Ferron, M.; Confraveux, C.; Wei, J.; Galan-Diez, M.; Lacampagne, A.; Mitchell, S.J.; Mattison, J.A.; Chen, Y.; et al. Osteocalcin signaling in myofibers is necessary and sufficient for optimum adaptation to exercise. Cell Metab. 2016, 23, 1078–1092. [Google Scholar] [CrossRef] [Green Version]
- Chowdhury, S.; Schulz, L.; Palmisano, B.; Singh, P.; Berger, J.M.; Yadav, V.K.; Mera, P.; Ellingsgaard, H.; Hidalgo, J.; Brüning, J.; et al. Muscle-derived interleukin-6 increases exercise capacity by signaling in osteoblasts. J. Clin. Investig. 2020, 130, 2888–2902. [Google Scholar] [CrossRef] [Green Version]
- Walker, R.G.; Doggioli, T.; Katsimpardi, L.; Buchanan, S.M.; Oh, J.; Wattrus, S. Biochemistry and biology of GDF11 and myostatin. Differences and questions for future intervention. Circ. Res. 2016, 11, 1125–1142. [Google Scholar] [CrossRef] [Green Version]
- Dankbar, B.; Fennen, M.; Brunert, D.; Hayer, S.; Frank, S.; Wehmeyer, C.; Beckmann, D.; Paruzel, P.; Bertrand, J.; Redlich, K.; et al. Myostatin is a direct regulator of osteoclast differentiation and its inhibition reduces inflammatory joint destruction in mice. Nat. Med. 2015, 21, 1085–1090. [Google Scholar] [CrossRef]
- Assyov, Y.S.; Velikova, T.V.; Kamenov, Z.A. Myostatin and carbohydrate disturbances. Endocr. Res. 2017, 42, 102–109. [Google Scholar] [CrossRef]
- Mouisel, E.; Relizani, E.; Mille Hamard, L.; Denis, R.; Hourde, C.; Agbulut, O.; Patel, K.; Arandel, L.; Morales-Gonzalez, S.; Vignaud, A.; et al. Myostatin is a key regulator between energy metabolism and endurance capacity of skeletal muscle. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2014, 307, R444–R454. [Google Scholar] [CrossRef]
- Perikakis, N.; Triantafyllou, C.A.; Fernandez-Real, J.M.; Huh, J.Y.; Seufert, J.; Mantzoros, C.S. Physiology and role of irisin in glucose homeostasis. Nat. Rev. Endocrinol. 2017, 13, 324–337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, H.; Wrann, C.D.; Jedrychowski, M.; Vidoni, S.; Kitase, Y.; Nagano, K.; Zhou, C.; Chou, J.; Parkman, V.-J.A.; Novick, S.J.; et al. Irisin Mediates Effects on Bone and Fat via αV Integrin Receptors. Cell 2018, 175, 1756–1768.e17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klein, G.L.; Wolf, S.E.; Langman, C.B.; Rosen, C.J.; Mohan, S.; Keenan, B.S.; Matin, S.; Steffen, C.; Nicolai, M.; Sailer, D.E.; et al. Effect of therapy with recombinant human growth hormone on insulin-like growth factor system components and serum levels of biochemical markers of bone formation in children after severe burn injury. J. Clin. Endocrinol. Metab. 1998, 83, 21–24. [Google Scholar] [PubMed]
- Klein, G.L.; Herndon, D.N.; Langman, C.B.; Rutan, T.C.; Young, W.E.; Pembleton, G.; Nusynowitz, M.; Barnett, J.L.; Broemeling, L.D.; Sailer, D.E. Long-term reduction in bone mass after severe burn injury in children. J. Pediatr. 1995, 126, 252–256. [Google Scholar] [CrossRef]


{kind=link}
| Factor | Source | Function |
|---|---|---|
| Calcium | bone | prolongs or intensifies inflammation, leading to increased bone and muscle wasting |
| TGF-β | bone | promotes muscle protein catabolism and suppresses anabolism |
| Osteocalcin | bone | promotes muscle fiber glucose and nutrient uptake, anabolic |
| Myostatin | muscle | inhibits muscle fiber uptake of nutrients, inhibits fiber growth |
| Irisin | muscle | stimulates muscle fiber nutrient uptake, decreases bone turnover, anabolic |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Klein, G.L. The Role of Bone in Muscle Wasting. Int. J. Mol. Sci. 2021, 22, 392. https://doi.org/10.3390/ijms22010392
Klein GL. The Role of Bone in Muscle Wasting. International Journal of Molecular Sciences. 2021; 22(1):392. https://doi.org/10.3390/ijms22010392
Chicago/Turabian StyleKlein, Gordon L. 2021. "The Role of Bone in Muscle Wasting" International Journal of Molecular Sciences 22, no. 1: 392. https://doi.org/10.3390/ijms22010392
APA StyleKlein, G. L. (2021). The Role of Bone in Muscle Wasting. International Journal of Molecular Sciences, 22(1), 392. https://doi.org/10.3390/ijms22010392