
Chronic Low-Dose Alcohol Consumption Attenuates Post-Ischemic Inflammation via PPARγ in Mice


Abstract
:1. Introduction
2. Results
2.1. Control Conditions
2.2. Effect of Alcohol on PPARγ and MnSOD
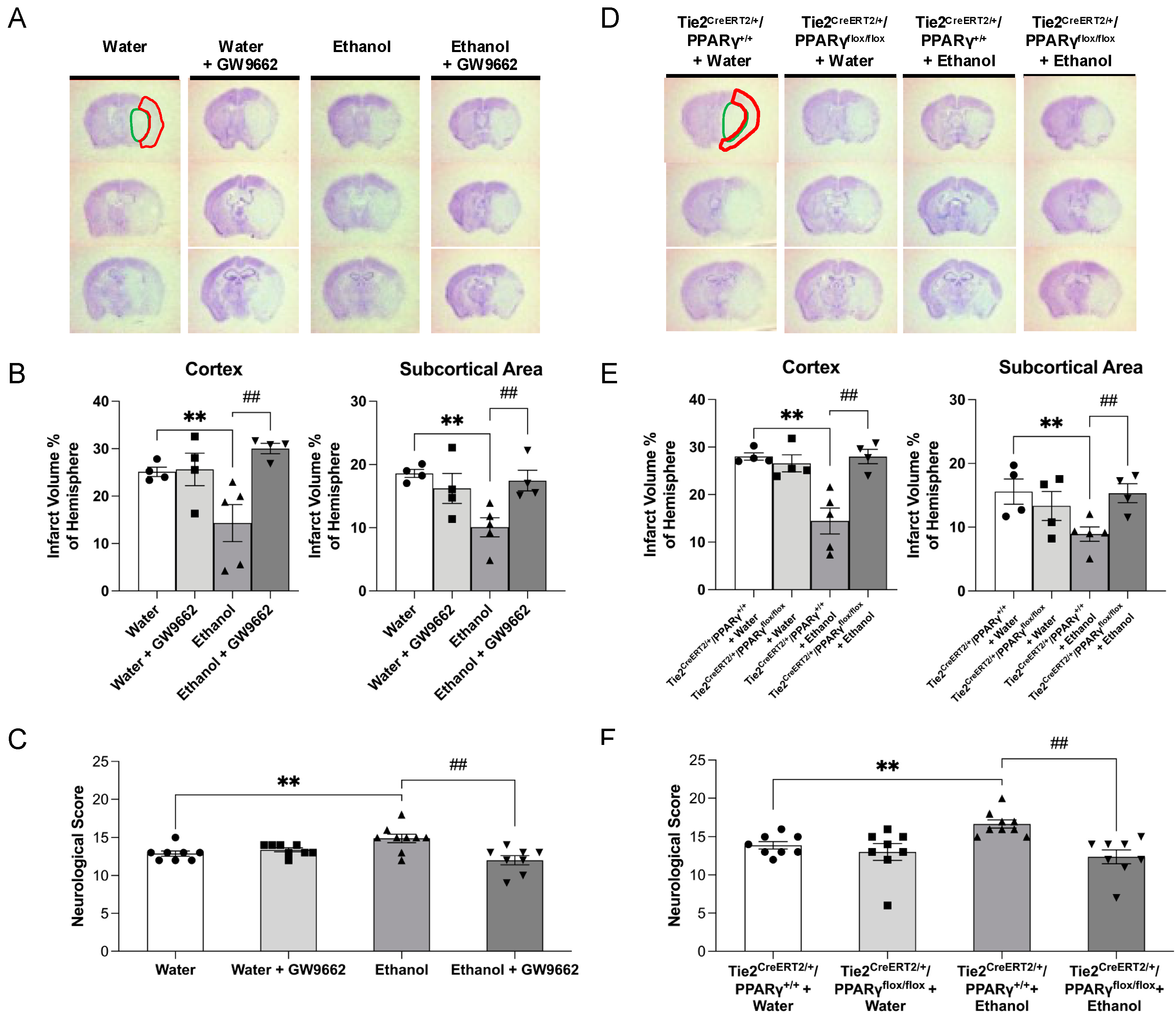
2.3. Effect of Chronic Alcohol Consumption on Cerebral I/R Injury
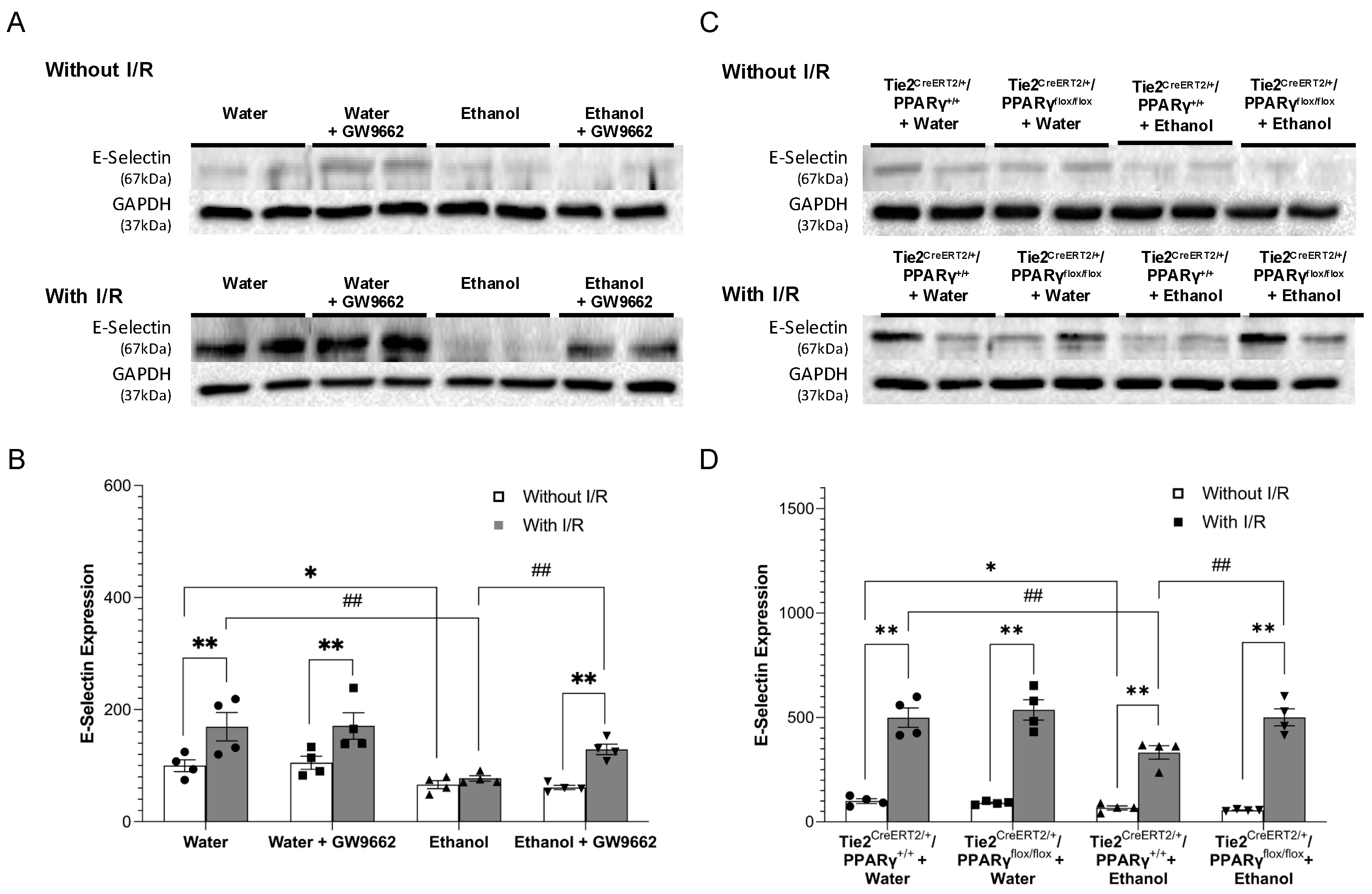
2.4. Effect of Chronic Alcohol Consumption on Protein Expression of Adhesion Molecules
2.5. Effect of Chronic Alcohol Consumption on Neutrophil Infiltration
2.6. Effect of Chronic Alcohol Consumption on Microglial Activation
3. Discussion
4. Materials and Methods
4.1. Mouse Brain Microvascular Endothelial Cells
4.2. Animal Models
4.3. Ethanol Preconditioning and Treatment
4.4. Nuclear PPARγ DNA-Binding Activity
4.5. Transient Focal Cerebral Ischemia
4.6. Brain Collection and Processing
4.7. Nissl Staining
4.8. Western Blot Analysis
4.9. Immunohistochemistry Staining
4.10. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Benjamin, E.J.; Blaha, M.J.; Chiuve, S.E.; Cushman, M.; Das, S.R.; Deo, R.; de Ferranti, S.D.; Floyd, J.; Fornage, M.; Gillespie, C.; et al. Heart Disease and Stroke Statistics-2017 Update: A Report From the American Heart Association. Circulation 2017, 135, e146–e603. [Google Scholar] [CrossRef] [PubMed]
- Favate, A.S.; Younger, D.S. Epidemiology of Ischemic Stroke. Neurol. Clin. 2016, 34, 967–980. [Google Scholar] [CrossRef]
- Jean, W.C.; Spellman, S.R.; Nussbaum, E.S.; Low, W.C. Reperfusion injury after focal cerebral ischemia: The role of inflammation and the therapeutic horizon. Neurosurgery 1998, 43, 1382–1396; Discussion 1396–1397. [Google Scholar] [CrossRef] [PubMed]
- Kalogeris, T.; Baines, C.P.; Krenz, M.; Korthuis, R.J. Cell biology of ischemia/reperfusion injury. Int. Rev. Cell Mol. Biol. 2012, 298, 229–317. [Google Scholar] [PubMed] [Green Version]
- Chen, W.; Sun, Y.; Liu, K.; Sun, X. Autophagy: A double-edged sword for neuronal survival after cerebral ischemia. Neural Regen. Res. 2014, 9, 1210–1216. [Google Scholar]
- Jin, R.; Liu, L.; Zhang, S.H.; Nanda, A.; Li, G.H. Role of Inflammation and Its Mediators in Acute Ischemic Stroke. J. Cardiovasc. Transl. 2013, 6, 834–851. [Google Scholar] [CrossRef] [Green Version]
- Weinstein, J.R.; Koerner, I.P.; Moller, T. Microglia in ischemic brain injury. Future Neurol. 2010, 5, 227–246. [Google Scholar] [CrossRef] [Green Version]
- Gronberg, N.V.; Johansen, F.F.; Kristiansen, U.; Hasseldam, H. Leukocyte infiltration in experimental stroke. J. Neuroinflamm. 2013, 10, 115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shukla, V.; Shakya, A.K.; Perez-Pinzon, M.A.; Dave, K.R. Cerebral ischemic damage in diabetes: An inflammatory perspective. J. Neuroinflamm. 2017, 14, 21. [Google Scholar] [CrossRef] [Green Version]
- Herz, J.; Sabellek, P.; Lane, T.E.; Gunzer, M.; Hermann, D.M.; Doeppner, T.R. Role of Neutrophils in Exacerbation of Brain Injury After Focal Cerebral Ischemia in Hyperlipidemic Mice. Stroke 2015, 46, 2916–2925. [Google Scholar] [CrossRef] [Green Version]
- Supanc, V.; Biloglav, Z.; Kes, V.B.; Demarin, V. Role of cell adhesion molecules in acute ischemic stroke. Ann. Saudi Med. 2011, 31, 365–370. [Google Scholar] [CrossRef] [PubMed]
- Hansagi, H.; Romelsjo, A.; de Verdier, M.G.; Andreasson, S.; Leifman, A. Alcohol consumption and stroke mortality. Stroke 1995, 26, 1768–1773. [Google Scholar] [CrossRef]
- Ikehara, S.; Iso, H.; Toyoshima, H.; Date, C.; Yamamoto, A.; Kikuchi, S.; Kondo, T.; Watanabe, Y.; Koizumi, A.; Wada, Y.; et al. Alcohol consumption and mortality from stroke and coronary heart disease among Japanese men and women: The Japan collaborative cohort study. Stroke 2008, 39, 2936–2942. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ronksley, P.E.; Brien, S.E.; Turner, B.J.; Mukamal, K.J.; Ghali, W.A. Association of alcohol consumption with selected cardiovascular disease outcomes: A systematic review and meta-analysis. BMJ 2011, 342, d671. [Google Scholar] [CrossRef] [Green Version]
- Patra, J.; Taylor, B.; Irving, H.; Roerecke, M.; Baliunas, D.; Mohapatra, S.; Rehm, J. Alcohol consumption and the risk of morbidity and mortality for different stroke types--a systematic review and meta-analysis. BMC Public Health 2010, 10, 258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ducroquet, A.; Leys, D.; Al Saabi, A.; Richard, F.; Cordonnier, C.; Girot, M.; Deplanque, D.; Casolla, B.; Allorge, D.; Bordet, R. Influence of chronic ethanol consumption on the neurological severity in patients with acute cerebral ischemia. Stroke 2013, 44, 2324–2326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, C.; Qin, Y.Y.; Chen, Q.; Jiang, H.; Chen, X.Z.; Xu, C.L.; Mao, P.J.; He, J.; Zhou, Y.H. Alcohol intake and risk of stroke: A dose-response meta-analysis of prospective studies. Int. J. Cardiol. 2014, 174, 669–677. [Google Scholar] [CrossRef] [PubMed]
- Christensen, A.I.; Nordestgaard, B.G.; Tolstrup, J.S. Alcohol Intake and Risk of Ischemic and Haemorrhagic Stroke: Results from a Mendelian Randomisation Study. J. Stroke 2018, 20, 218–227. [Google Scholar] [CrossRef]
- Klatsky, A.L.; Armstrong, M.A.; Friedman, G.D.; Sidney, S. Alcohol drinking and risk of hospitalization for ischemic stroke. Am. J. Cardiol. 2001, 88, 703–706. [Google Scholar] [CrossRef]
- Zhao, H.; Mayhan, W.G.; Arrick, D.M.; Xiong, W.; Sun, H. Dose-related influence of chronic alcohol consumption on cerebral ischemia/reperfusion injury. Alcohol. Clin. Exp. Res. 2011, 35, 1265–1269. [Google Scholar] [CrossRef] [Green Version]
- Xu, G.; Li, C.; Parsiola, A.L.; Li, J.; McCarter, K.D.; Shi, R.; Mayhan, W.G.; Sun, H. Dose-Dependent Influences of Ethanol on Ischemic Stroke: Role of Inflammation. Front. Cell Neurosci. 2019, 13, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, C.; Li, J.Y.; Xu, G.D.; Sun, H. Influence of Chronic Ethanol Consumption on Apoptosis and Autophagy Following Transient Focal Cerebral Ischemia in Male Mice. Sci. Rep. 2020, 10, 6164. [Google Scholar] [CrossRef] [Green Version]
- Sun, H.; Xiong, W.; Arrick, D.M.; Mayhan, W.G. Low-dose alcohol consumption protects against transient focal cerebral ischemia in mice: Possible role of PPARgamma. PLoS ONE 2012, 7, e41716. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shibuya, A.; Wada, K.; Nakajima, A.; Saeki, M.; Katayama, K.; Mayumi, T.; Kadowaki, T.; Niwa, H.; Kamisaki, Y. Nitration of PPARgamma inhibits ligand-dependent translocation into the nucleus in a macrophage-like cell line, RAW 264. FEBS Lett. 2002, 525, 43–47. [Google Scholar] [CrossRef] [Green Version]
- Ketsawatsomkron, P.; Pelham, C.J.; Groh, S.; Keen, H.L.; Faraci, F.M.; Sigmund, C.D. Does Peroxisome Proliferator-activated Receptor-gamma (PPAR gamma) Protect from Hypertension Directly through Effects in the Vasculature? J. Biol. Chem. 2010, 285, 9311–9316. [Google Scholar] [CrossRef] [Green Version]
- Cai, W.; Yang, T.; Liu, H.; Han, L.J.; Zhang, K.; Hu, X.M.; Zhang, X.J.; Yin, K.J.; Gao, Y.Q.; Bennett, M.V.L.; et al. Peroxisome proliferator-activated receptor gamma (PPAR gamma): A master gatekeeper in CNS injury and repair. Prog. Neurobiol. 2018, 163, 27–58. [Google Scholar] [CrossRef]
- Shimazu, T.; Inoue, I.; Araki, N.; Asano, Y.; Sawada, M.; Furuya, D.; Nagoya, H.; Greenberg, J.H. A peroxisome proliferator-activated receptor-gamma agonist reduces infarct size in transient but not in permanent ischemia. Stroke. J. Cereb. Circ. 2005, 36, 353–359. [Google Scholar] [CrossRef]
- Gamboa, J.; Blankenship, D.A.; Niemi, J.P.; Landreth, G.E.; Karl, M.; Hilow, E.; Sundararajan, S. Extension of the neuroprotective time window for thiazolidinediones in ischemic stroke is dependent on time of reperfusion. Neuroscience 2010, 170, 846–857. [Google Scholar] [CrossRef] [Green Version]
- Fisher, H.R.; Simpson, R.I.; Kapur, B.M. Calculation of blood alcohol concentration (BAC) by sex, weight, number of drinks and time. Can. J. Public Health 1987, 78, 300–304. [Google Scholar]
- Sun, H.; Zhao, H.; Sharpe, G.M.; Arrick, D.M.; Mayhan, W.G. Effect of chronic alcohol consumption on brain damage following transient focal ischemia. Brain Res. 2008, 1194, 73–80. [Google Scholar] [CrossRef] [Green Version]
- Mitra, S.K.; Varma, S.R.; Godavarthi, A.; Nandakumar, K.S. Liv.52 regulates ethanol induced PPARgamma and TNF alpha expression in HepG2 cells. Mol. Cell Biochem. 2008, 315, 9–15. [Google Scholar] [CrossRef]
- Yeligar, S.M.; Mehta, A.J.; Harris, F.L.; Brown, L.A.S.; Hart, C.M. Peroxisome Proliferator-Activated Receptor gamma Regulates Chronic Alcohol-Induced Alveolar Macrophage Dysfunction. Am. J. Resp. Cell Mol. 2016, 55, 35–46. [Google Scholar] [CrossRef] [Green Version]
- Petersen, R.K.; Larsen, S.B.; Jensen, D.M.; Christensen, J.; Olsen, A.; Loft, S.; Nellemann, C.; Overvad, K.; Kristiansen, K.; Tjonneland, A.; et al. PPARgamma-PGC-1alpha activity is determinant of alcohol related breast cancer. Cancer Lett. 2012, 315, 59–68. [Google Scholar] [CrossRef]
- Chavez, P.R.; Lian, F.; Chung, J.; Liu, C.; Paiva, S.A.; Seitz, H.K.; Wang, X.D. Long-term ethanol consumption promotes hepatic tumorigenesis but impairs normal hepatocyte proliferation in rats. J. Nutr. 2011, 141, 1049–1055. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.H.; Song, S.J.; Kim, A.; Choi, Y.; Seok, J.W.; Kim, H.J.; Lee, Y.J.; Lee, K.S.; Kim, J.W. Suppression of PPAR gamma-mediated monoacylglycerol O-acyltransferase 1 expression ameliorates alcoholic hepatic steatosis. Sci. Rep. 2016, 6, 29352. [Google Scholar] [CrossRef] [Green Version]
- Fortunato, F.; Berger, I.; Gross, M.L.; Rieger, P.; Buechler, M.W.; Werner, J. Immune-compromised state in the rat pancreas after chronic alcohol exposure: The role of peroxisome proliferator-activated receptor gamma. J. Pathol. 2007, 213, 441–452. [Google Scholar] [CrossRef] [PubMed]
- Yilmaz, G.; Granger, D.N. Cell adhesion molecules and ischemic stroke. Neurol. Res. 2008, 30, 783–793. [Google Scholar] [CrossRef] [PubMed]
- Kitagawa, K.; Matsumoto, M.; Mabuchi, T.; Yagita, Y.; Ohtsuki, T.; Hori, M.; Yanagihara, T. Deficiency of intercellular adhesion molecule 1 attenuates microcirculatory disturbance and infarction size in focal cerebral ischemia. J. Cereb. Blood Flow Metab. 1998, 18, 1336–1345. [Google Scholar] [CrossRef] [Green Version]
- Huang, J.; Choudhri, T.F.; Winfree, C.J.; McTaggart, R.A.; Kiss, S.; Mocco, J.; Kim, L.J.; Protopsaltis, T.S.; Zhang, Y.; Pinsky, D.J.; et al. Postischemic cerebrovascular E-selectin expression mediates tissue injury in murine stroke. Stroke 2000, 31, 3047–3053. [Google Scholar] [CrossRef] [Green Version]
- Connolly, E.S.; Winfree, C.J.; Prestigiacomo, C.J.; Kim, S.C.; Choudhri, T.F.; Hoh, B.L.; Naka, Y.; Solomon, R.A.; Pinsky, D.J. Exacerbation of cerebral injury in mice that express the P-selectin gene—Identification of P-selectin blockade as a new target for the treatment of stroke. Circ. Res. 1997, 81, 304–310. [Google Scholar] [CrossRef]
- Justicia, C.; Martin, A.; Rojas, S.; Gironella, M.; Cervera, A.; Panes, J.; Chamorro, A.; Planas, A.M. Anti-VCAM-1 antibodies did not protect against ischemic damage either in rats or in mice. J. Cerebr. Blood F Met. 2006, 26, 421–432. [Google Scholar] [CrossRef] [Green Version]
- Pasceri, V.; Wu, H.D.; Willerson, J.T.; Yeh, E.T.H. Modulation of vascular inflammation in vitro and in vivo by peroxisome proliferator-activated receptor-gamma activators. Circulation 2000, 101, 235–238. [Google Scholar] [CrossRef] [Green Version]
- Wang, N.P.; Verna, L.; Chen, N.G.; Chen, J.; Li, H.L.; Forman, B.M.; Stemerman, M.B. Constitutive activation of peroxisome proliferator-activated receptor-gamma suppresses pro-inflammatory adhesion molecules in human vascular endothelial cells. J. Biol. Chem. 2002, 277, 34176–34181. [Google Scholar] [CrossRef] [Green Version]
- Verrier, E.; Wang, L.J.; Wadham, C.; Albanese, N.; Hahn, C.; Gamble, J.R.; Krishna, V.; Chatterjee, K.; Vadas, M.A.; Xia, P. PPAR gamma agonists ameliorate endothelial cell activation via inhibition of diacylglycerol-protein kinase C signaling pathway—Role of diacylglycerol kinase. Circ. Res. 2004, 94, 1515–1522. [Google Scholar] [CrossRef] [Green Version]
- Tureyen, K.; Kapadia, R.; Bowen, K.K.; Satriotomo, I.; Liang, J.; Feinstein, D.L.; Vemuganti, R. Peroxisome proliferator-activated receptor-gamma agonists induce neuroprotection following transient focal ischemia in normotensive, normoglycemic as well as hypertensive and type-2 diabetic rodents. J. Neurochem. 2007, 101, 41–56. [Google Scholar] [CrossRef]
- Sasaki, M.; Jordan, P.; Welbourne, T.; Minagar, A.; Joh, T.; Itoh, M.; Elrod, J.W.; Alexander, J.S. Troglitazone, a PPAR-gamma activator prevents endothelial cell adhesion molecule expression and lymphocyte adhesion mediated by TNF-alpha. BMC Physiol. 2005, 5, 3. [Google Scholar] [CrossRef] [Green Version]
- Reddy, A.T.; Lakshmi, S.P.; Kleinhenz, J.M.; Sutliff, R.L.; Hart, C.M.; Reddy, R.C. Endothelial cell peroxisome proliferator-activated receptor gamma reduces endotoxemic pulmonary inflammation and injury. J. Immunol. 2012, 189, 5411–5420. [Google Scholar] [CrossRef] [Green Version]
- Rupalla, K.; Allegrini, P.R.; Sauer, D.; Wiessner, C. Time course of microglia activation and apoptosis in various brain regions after permanent focal cerebral ischemia in mice. Acta Neuropathol. 1998, 96, 172–178. [Google Scholar] [CrossRef]
- Kim, J.Y.; Park, J.; Chang, J.Y.; Kim, S.H.; Lee, J.E. Inflammation after Ischemic Stroke: The Role of Leukocytes and Glial Cells. Exp. Neurobiol. 2016, 25, 241–251. [Google Scholar] [CrossRef] [Green Version]
- Emmrich, J.V.; Ejaz, S.; Neher, J.J.; Williamson, D.J.; Baron, J.C. Regional distribution of selective neuronal loss and microglial activation across the MCA territory after transient focal ischemia: Quantitative versus semiquantitative systematic immunohistochemical assessment. J. Cereb. Blood Flow Metab. 2015, 35, 20–27. [Google Scholar] [CrossRef] [Green Version]
- Morrison, H.W.; Filosa, J.A. A quantitative spatiotemporal analysis of microglia morphology during ischemic stroke and reperfusion. J. Neuroinflamm. 2013, 10, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jayaraj, R.L.; Azimullah, S.; Beiram, R.; Jalal, F.Y.; Rosenberg, G.A. Neuroinflammation: Friend and foe for ischemic stroke. J. Neuroinflamm. 2019, 16, 142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bernardo, A.; Minghetti, L. PPAR-gamma agonists as regulators of microglial activation and brain inflammation. Curr. Pharm. Des. 2006, 12, 93–109. [Google Scholar] [CrossRef]
- Ding, G.; Fu, M.; Qin, Q.; Lewis, W.; Kim, H.W.; Fukai, T.; Bacanamwo, M.; Chen, Y.E.; Schneider, M.D.; Mangelsdorf, D.J.; et al. Cardiac peroxisome proliferator-activated receptor gamma is essential in protecting cardiomyocytes from oxidative damage. Cardiovasc. Res. 2007, 76, 269–279. [Google Scholar] [CrossRef] [Green Version]
- Dikalov, S.I.; Ungvari, Z. Role of mitochondrial oxidative stress in hypertension. Am. J. Physiol. Heart Circ. Physiol. 2013, 305, H1417–H1427. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Tabas, I. Emerging roles of mitochondria ROS in atherosclerotic lesions: Causation or association? J. Atheroscler. Thromb. 2014, 21, 381–390. [Google Scholar] [CrossRef] [Green Version]
- Dai, D.F.; Chiao, Y.A.; Marcinek, D.J.; Szeto, H.H.; Rabinovitch, P.S. Mitochondrial oxidative stress in aging and healthspan. Longev. Healthspan 2014, 3, 6. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.L.; Chen, C.L.; Yeh, S.T.; Zweier, J.L.; Chen, Y.R. Biphasic modulation of the mitochondrial electron transport chain in myocardial ischemia and reperfusion. Am. J. Physiol. Heart Circ. Physiol. 2012, 302, H1410–H1422. [Google Scholar] [CrossRef] [Green Version]
- Kalogeris, T.; Bao, Y.; Korthuis, R.J. Mitochondrial reactive oxygen species: A double edged sword in ischemia/reperfusion vs. preconditioning. Redox Biol. 2014, 2, 702–714. [Google Scholar] [CrossRef] [Green Version]
- Zinkevich, N.S.; Gutterman, D.D. ROS-induced ROS release in vascular biology: Redox-redox signaling. Am. J. Physiol. Heart Circ. Physiol. 2011, 301, H647–H653. [Google Scholar] [CrossRef] [Green Version]
- Lakhan, S.E.; Kirchgessner, A.; Hofer, M. Inflammatory mechanisms in ischemic stroke: Therapeutic approaches. J. Transl. Med. 2009, 7, 97. [Google Scholar] [CrossRef] [Green Version]
- Rodrigues, S.F.; Granger, D.N. Cerebral microvascular inflammation in DOCA salt-induced hypertension: Role of angiotensin II and mitochondrial superoxide. J. Cereb. Blood Flow Metab. Off. J. Int. Soc. Cereb. Blood Flow Metab. 2012, 32, 368–375. [Google Scholar] [CrossRef] [Green Version]
- Jung, Y.; Song, S.; Choi, C. Peroxisome proliferator activated receptor gamma agonists suppress TNFalpha-induced ICAM-1 expression by endothelial cells in a manner potentially dependent on inhibition of reactive oxygen species. Immunol. Lett. 2008, 117, 63–69. [Google Scholar] [CrossRef] [PubMed]
- Choi, D.H.; Kim, J.H.; Lee, K.H.; Kim, H.Y.; Kim, Y.S.; Choi, W.S.; Lee, J. Role of neuronal NADPH oxidase 1 in the peri-infarct regions after stroke. PLoS ONE 2015, 10, e0116814. [Google Scholar] [CrossRef] [Green Version]
- McCarter, K.D.; Li, C.; Jiang, Z.; Lu, W.; Smith, H.C.; Xu, G.; Mayhan, W.G.; Sun, H. Effect of Low-Dose Alcohol Consumption on Inflammation Following Transient Focal Cerebral Ischemia in Rats. Sci. Rep. 2017, 7, 12547. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Salayandia, V.M.; Thompson, J.F.; Yang, L.Y.; Estrada, E.Y.; Yang, Y. Attenuation of acute stroke injury in rat brain by minocycline promotes blood-brain barrier remodeling and alternative microglia/macrophage activation during recovery. J. Neuroinflamm. 2015, 12, 26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taylor, R.A.; Sansing, L.H. Microglial responses after ischemic stroke and intracerebral hemorrhage. Clin. Dev. Immunol. 2013, 2013, 746068. [Google Scholar] [CrossRef]
- Ahmad, S.; Elsherbiny, N.M.; Haque, R.; Khan, M.B.; Ishrat, T.; Shah, Z.A.; Khan, M.M.; Ali, M.; Jamal, A.; Katare, D.P.; et al. Sesamin attenuates neurotoxicity in mouse model of ischemic brain stroke. Neurotoxicology 2014, 45, 100–110. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Body Weight (g) | MABP (mmHg) | Heart Rate (bpm) | Fasting Blood Glucose (mg/dL) | |
|---|---|---|---|---|
| Water | 27 ± 0.6 | 85 ± 3 | 656 ± 22 | 142 ± 7 |
| Water + GW9662 | 28 ± 0.6 | 82 ± 3 | 680 ± 32 | 167 ± 12 |
| Ethanol | 28 ± 0.5 | 85 ± 3 | 663 ± 41 | 134 ± 9 |
| Ethanol + GW9662 | 28 ± 0.55 | 86 ± 1 | 699 ± 39 | 162 ± 22 |
| Tie2CreERT2/+/PPARγ+/+ + Water | 27 ± 1.3 | 84 ± 2 | 605 ± 23 | 131 ± 18 |
| Tie2CreERT2/+/PPARγflox/flox + Water | 27 ± 1.1 | 87 ± 2 | 664 ± 26 | 147 ± 8 |
| Tie2CreERT2/+/PPARγ+/+ + Ethanol | 28 ± 1.6 | 84 ± 9 | 666 ± 24 | 124 ± 10 |
| Tie2CreERT2/+/PPARγflox/flox + Ethanol | 28 ± 1.0 | 81 ± 5 | 607 ± 27 | 139 ± 11 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, C.; Li, J.; Loreno, E.G.; Miriyala, S.; Panchatcharam, M.; Lu, X.; Sun, H. Chronic Low-Dose Alcohol Consumption Attenuates Post-Ischemic Inflammation via PPARγ in Mice. Int. J. Mol. Sci. 2021, 22, 5121. https://doi.org/10.3390/ijms22105121
Li C, Li J, Loreno EG, Miriyala S, Panchatcharam M, Lu X, Sun H. Chronic Low-Dose Alcohol Consumption Attenuates Post-Ischemic Inflammation via PPARγ in Mice. International Journal of Molecular Sciences. 2021; 22(10):5121. https://doi.org/10.3390/ijms22105121
Chicago/Turabian StyleLi, Chun, Jiyu Li, Ethyn G. Loreno, Sumitra Miriyala, Manikandan Panchatcharam, Xiaohong Lu, and Hong Sun. 2021. "Chronic Low-Dose Alcohol Consumption Attenuates Post-Ischemic Inflammation via PPARγ in Mice" International Journal of Molecular Sciences 22, no. 10: 5121. https://doi.org/10.3390/ijms22105121
APA StyleLi, C., Li, J., Loreno, E. G., Miriyala, S., Panchatcharam, M., Lu, X., & Sun, H. (2021). Chronic Low-Dose Alcohol Consumption Attenuates Post-Ischemic Inflammation via PPARγ in Mice. International Journal of Molecular Sciences, 22(10), 5121. https://doi.org/10.3390/ijms22105121