
Soluble Epoxide Hydrolase Blockade after Stroke Onset Protects Normal but Not Diabetic Mice

 ,
, 
 ,
,
Abstract
:1. Introduction
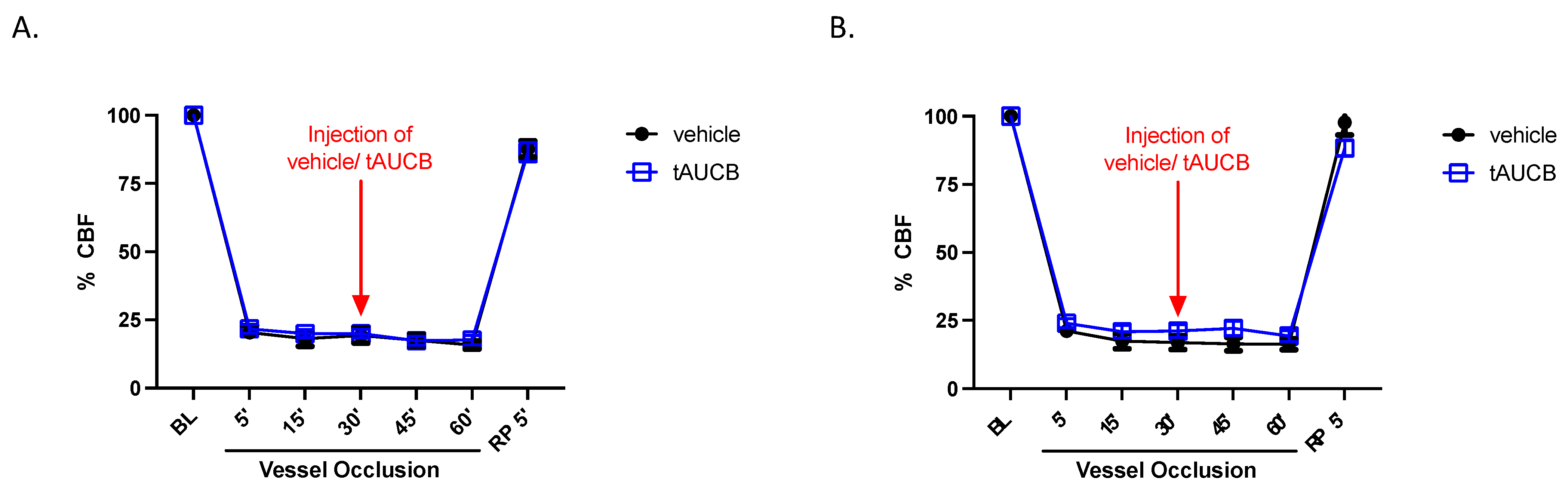
2. Results
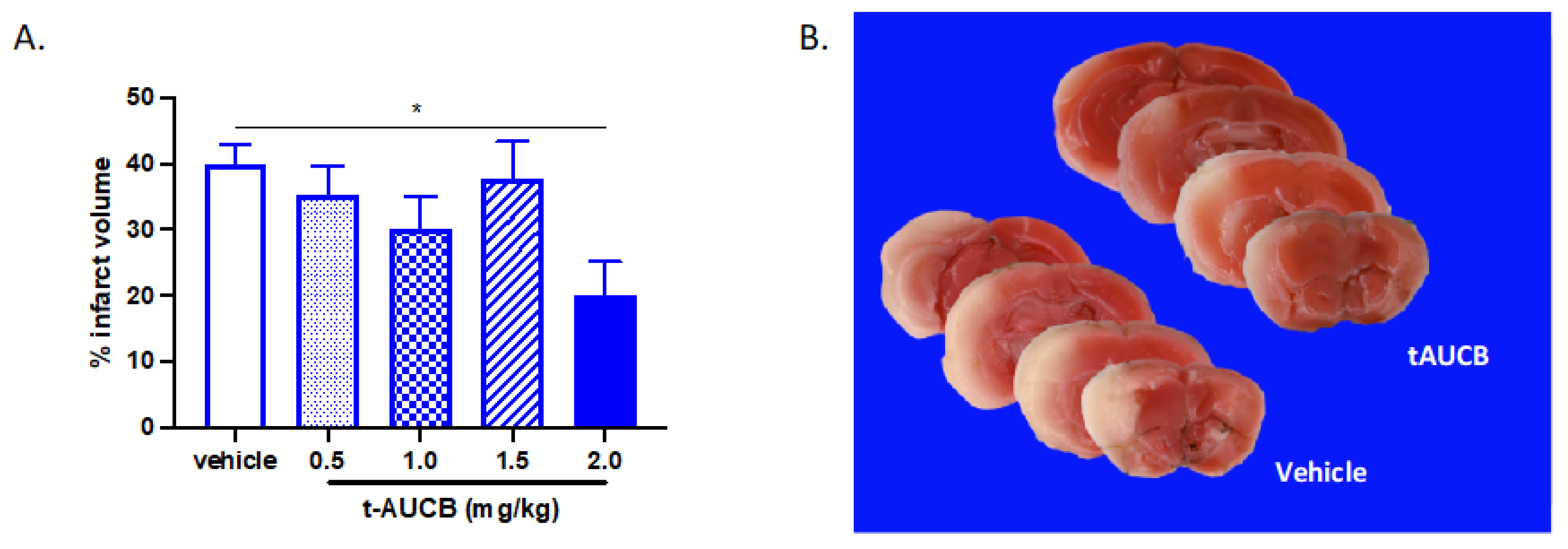
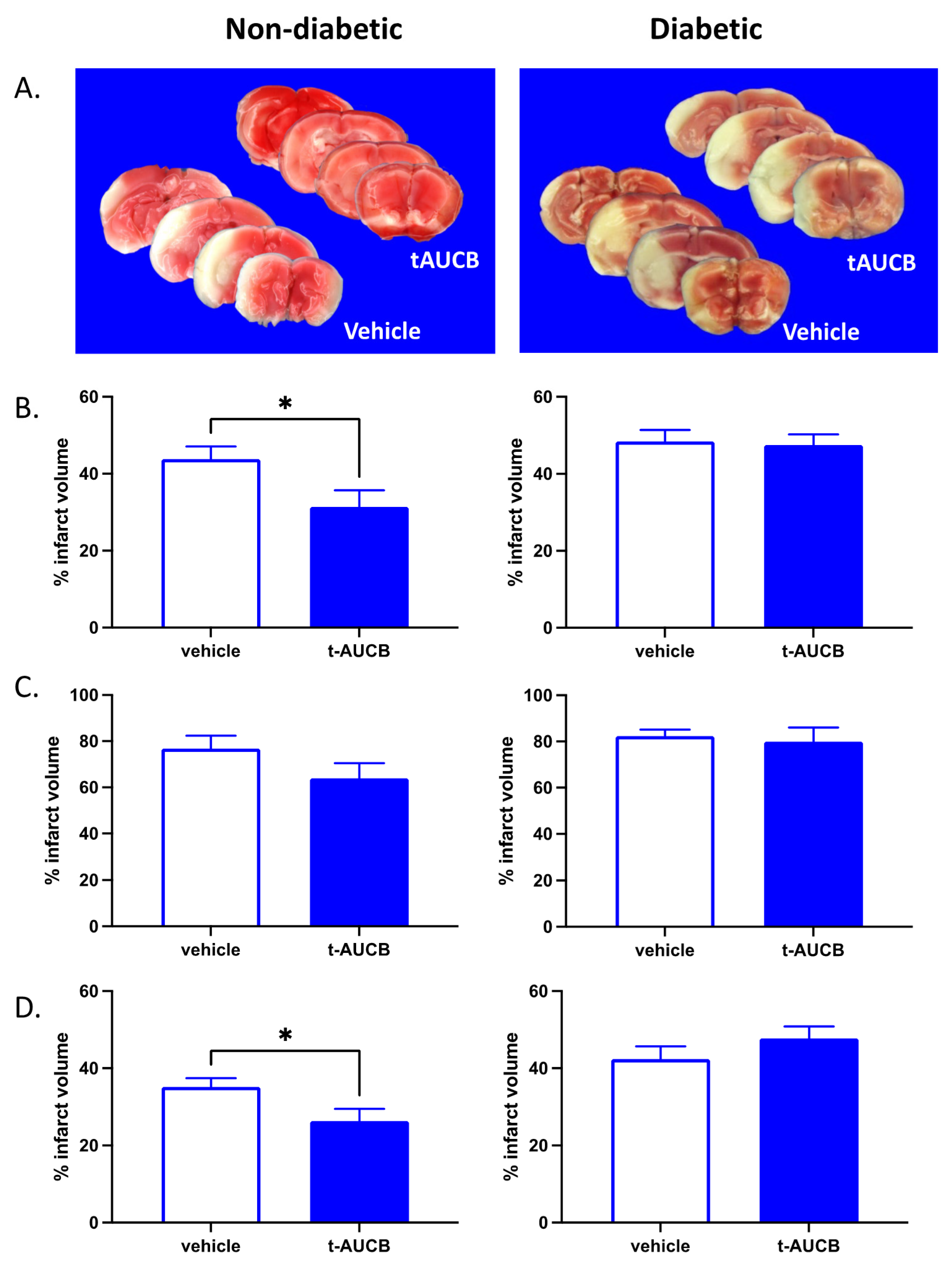
2.1. tAUCB Reduces Infarct Size in Non-Diabetic Mice
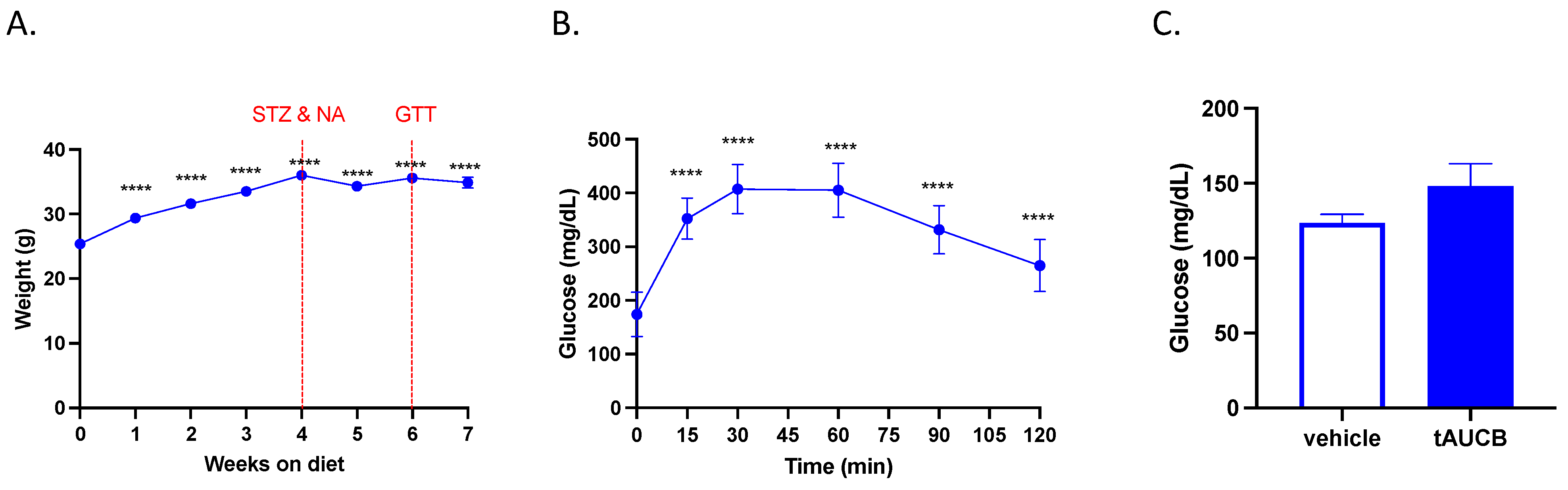
2.2. tAUCB Does Not Alter Blood Glucose Concentration in Diabetic Mice
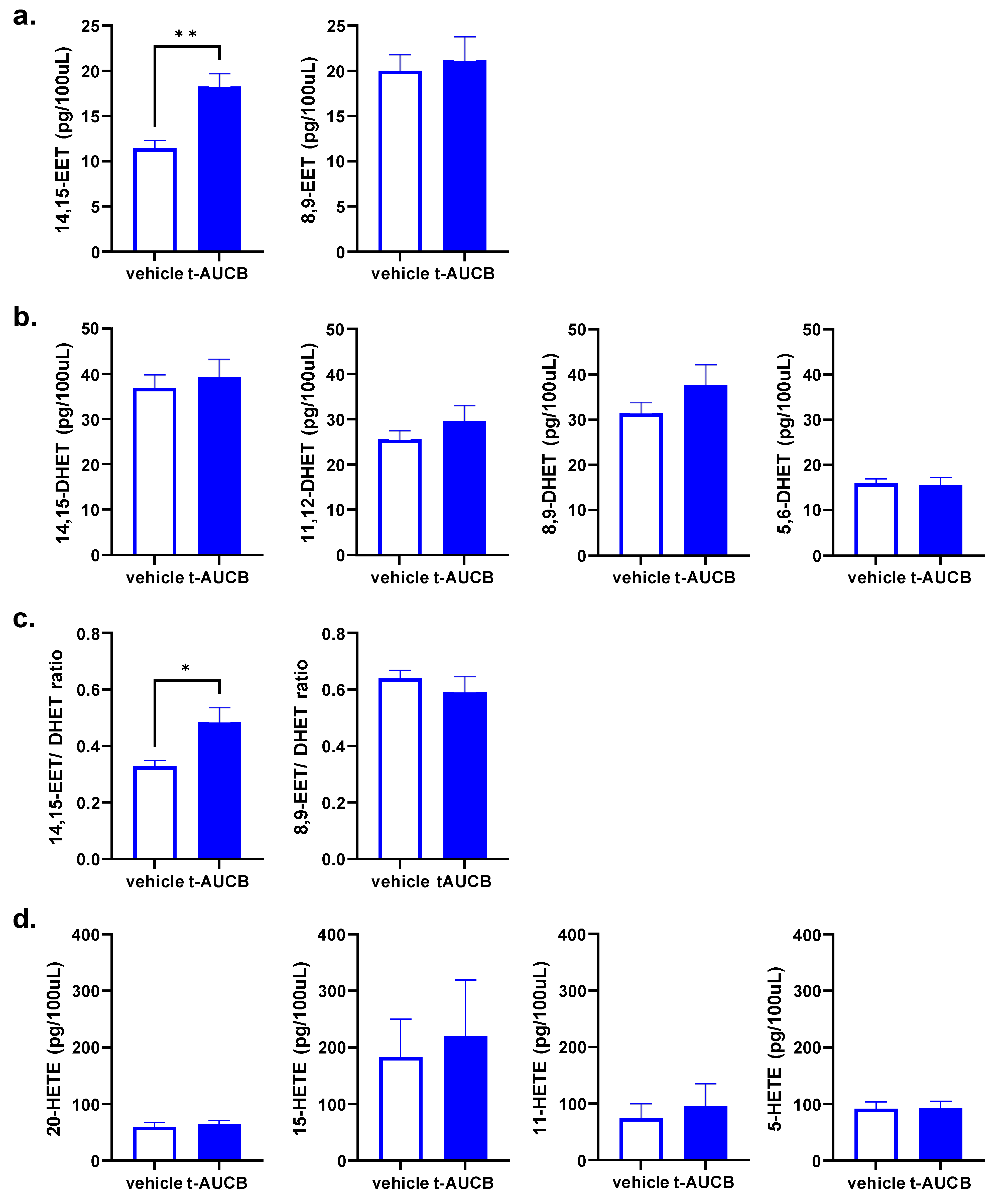
2.3. tAUCB Selectively Increases Plasma 14,15-EET Concentration
2.4. CBF Is Unaltered by tAUCB in Immediate Peri-Ischemia Period
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Induction of Type 2 Diabetes
4.3. Non-Diabetic Mice
4.4. Middle Cerebral Artery Occlusion (MCAO)
4.5. Brain Infarct Size
4.6. Plasma Eicosanoid Quantification
4.6.1. Chemicals and Reagents
4.6.2. Preparation of Eicosanoid Samples and Calibrators
4.6.3. LC-MS/MS Analysis for Eicosanoid Metabolites
4.7. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Spescha, R.D.; Klohs, J.; Semerano, A.; Giacalone, G.; Derungs, R.S.; Reiner, M.F.; Rodriguez Gutierrez, D.; Mendez-Carmona, N.; Glanzmann, M.; Savarese, G.; et al. Post-ischaemic silencing of p66Shc reduces ischaemia/reperfusion brain injury and its expression correlates to clinical outcome in stroke. Eur. Heart J. 2015, 36, 1590–1600. [Google Scholar] [CrossRef] [Green Version]
- Chamorro, Á.; Dirnagl, U.; Urra, X.; Planas, A.M. Neuroprotection in acute stroke: Targeting excitotoxicity, oxidative and nitrosative stress, and inflammation. Lancet Neurol. 2016, 15, 869–881. [Google Scholar] [CrossRef]
- Dinesh Shah, A.; Langenberg, C.; Rapsomaniki, E.; Denaxas, S.; Pujades-Rodriguez, M.; Gale, C.P.; Deanfield, J.; Smeeth, L.; Timmis, A.; Hemingway, H. Type 2 diabetes and incidence of a wide range of cardiovascular diseases: A cohort study in 1·9 million people. Lancet 2015, 385 (Suppl. S1), S86. [Google Scholar] [CrossRef] [Green Version]
- Larsson, S.C.; Scott, R.A.; Traylor, M.; Langenberg, C.C.; Hindy, G.; Melander, O.; Orho-Melander, M.; Seshadri, S.; Wareham, N.J.; Markus, H.S. Type 2 diabetes, glucose, insulin, BMI, and ischemic stroke subtypes: Mendelian randomization study. Neurology 2017, 89, 454–460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rawshani, A.; Rawshani, A.; Franzén, S.; Sattar, N.; Eliasson, B.; Svensson, A.M.; Zethelius, B.; Miftaraj, M.; McGuire, D.K.; Rosengren, A.; et al. Risk Factors, Mortality, and Cardiovascular Outcomes in Patients with Type 2 Diabetes. N. Engl. J. Med. 2018, 379, 633–644. [Google Scholar] [CrossRef]
- Scott, J.F.; Robinson, G.M.; French, J.M.; O’Connell, J.E.; Alberti, K.G.; Gray, C.S. Prevalence of admission hyperglycaemia across clinical subtypes of acute stroke. Lancet 1999, 353, 376–377. [Google Scholar] [CrossRef]
- Baird, T.A.; Parsons, M.W.; Phan, T.; Butcher, K.S.; Desmond, P.M.; Tress, B.M.; Colman, P.G.; Chambers, B.R.; Davis, S.M. Persistent poststroke hyperglycemia is independently associated with infarct expansion and worse clinical outcome. Stroke 2003, 34, 2208–2214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lo, J.W.; Crawford, J.D.; Samaras, K.; Desmond, D.W.; Köhler, S.; Staals, J.; Verhey, F.R.J.; Bae, H.J.; Lee, K.J.; Kim, B.J.; et al. Association of Prediabetes and Type 2 Diabetes with Cognitive Function After Stroke: A STROKOG Collaboration Study. Stroke 2020, 51, 1640–1646. [Google Scholar] [CrossRef]
- Lau, L.H.; Lew, J.; Borschmann, K.; Thijs, V.; Ekinci, E.I. Prevalence of diabetes and its effects on stroke outcomes: A meta-analysis and literature review. J. Diabetes Investig. 2019, 10, 780–792. [Google Scholar] [CrossRef] [Green Version]
- Gangraram, U.; Dorasanamma, M.; Thiruvuru, I.; Krishna, K.S. Glycaemic levels as an independent predictor of outcome in acute ischemic stroke from a tertiary care hospital, Nellore, India. Int. J. Adv. Med. 2020, 7, 1232. [Google Scholar] [CrossRef]
- Forlivesi, S.; Micheletti, N.; Tomelleri, G.; Bovi, P.; Cappellari, M. Association of hyperglycemia, systolic and diastolic hypertension, and hyperthermia relative to baseline in the acute phase of stroke with poor outcome after intravenous thrombolysis. Blood Coagul. Fibrinolysis 2018, 29, 167–171. [Google Scholar] [CrossRef]
- Osei, E.; Fonville, S.; Zandbergen, A.A.M.; Koudstaal, P.J.; Dippel, D.W.J.; den Hertog, H.M. Impaired fasting glucose is associated with unfavorable outcome in ischemic stroke patients treated with intravenous alteplase. J. Neurol. 2018, 265, 1426–1431. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.T.; Jahan, R.; Saver, J.L. Impact of Glucose on Outcomes in Patients Treated with Mechanical Thrombectomy: A Post Hoc Analysis of the Solitaire Flow Restoration with the Intention for Thrombectomy Study. Stroke 2016, 47, 120–127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.T.; Liebeskind, D.S.; Jahan, R.; Menon, B.K.; Goyal, M.; Nogueira, R.G.; Pereira, V.M.; Gralla, J.; Saver, J.L. Impact of Hyperglycemia According to the Collateral Status on Outcomes in Mechanical Thrombectomy. Stroke 2018, 49, 2706–2714. [Google Scholar] [CrossRef]
- Chamorro, Á.; Brown, S.; Amaro, S.; Hill, M.D.; Muir, K.W.; Dippel, D.W.J.; van Zwam, W.; Butcher, K.; Ford, G.A.; den Hertog, H.M.; et al. Glucose Modifies the Effect of Endovascular Thrombectomy in Patients with Acute Stroke. Stroke 2019, 50, 690–696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Capes, S.E.; Hunt, D.; Malmberg, K.; Pathak, P.; Gerstein, H.C. Stress hyperglycemia and prognosis of stroke in nondiabetic and diabetic patients: A systematic overview. Stroke 2001, 32, 2426–2432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ben Assayag, E.; Eldor, R.; Korczyn, A.D.; Kliper, E.; Shenhar-Tsarfaty, S.; Tene, O.; Molad, J.; Shapira, I.; Berliner, S.; Volfson, V.; et al. Type 2 Diabetes Mellitus and Impaired Renal Function Are Associated with Brain Alterations and Poststroke Cognitive Decline. Stroke 2017, 48, 2368–2374. [Google Scholar] [CrossRef]
- Zhang, W.; Koerner, I.P.; Noppens, R.; Grafe, M.; Tsai, H.J.; Morisseau, C.; Luria, A.; Hammock, B.D.; Falck, J.R.; Alkayed, N.J. Soluble epoxide hydrolase: A novel therapeutic target in stroke. J. Cereb. Blood Flow Metab. 2007, 27, 1931–1940. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marowsky, A.; Burgener, J.; Falck, J.R.; Fritschy, J.M.; Arand, M. Distribution of soluble and microsomal epoxide hydrolase in the mouse brain and its contribution to cerebral epoxyeicosatrienoic acid metabolism. Neuroscience 2009, 163, 646–661. [Google Scholar] [CrossRef]
- Revermann, M. Pharmacological inhibition of the soluble epoxide hydrolase-from mouse to man. Curr. Opin. Pharmacol. 2010, 10, 173–178. [Google Scholar] [CrossRef]
- Horti, A.G.; Wang, Y.; Minn, I.; Lan, X.; Wang, J.; Koehler, R.C.; Alkayed, N.J.; Dannals, R.F.; Pomper, M.G. 18F-FNDP for PET Imaging of Soluble Epoxide Hydrolase. J. Nucl. Med. 2016, 57, 1817–1822. [Google Scholar] [CrossRef] [Green Version]
- Coughlin, J.M.; Slania, S.; Du, Y.; Shinehouse, L.K.; Brosnan, M.K.; Azad, B.B.; Holt, D.P.; Fan, H.; Lesniak, W.G.; Minn, I.; et al. First-in-human neuroimaging of soluble epoxide hydrolase using [(18)F]FNDP PET. Eur. J. Nucl. Med. Mol. Imaging 2021. [Google Scholar] [CrossRef]
- Jouihan, S.A.; Zuloaga, K.L.; Zhang, W.; Shangraw, R.E.; Krasnow, S.M.; Marks, D.L.; Alkayed, N.J. Role of soluble epoxide hydrolase in exacerbation of stroke by streptozotocin-induced type 1 diabetes mellitus. J. Cereb. Blood Flow Metab. 2013, 33, 1650–1656. [Google Scholar] [CrossRef]
- Zuloaga, K.L.; Krasnow, S.M.; Zhu, X.; Zhang, W.; Jouihan, S.A.; Shangraw, R.E.; Alkayed, N.J.; Marks, D.L. Mechanism of protection by soluble epoxide hydrolase inhibition in type 2 diabetic stroke. PLoS ONE 2014, 9, e97529. [Google Scholar] [CrossRef] [Green Version]
- Van Leyen, K.; Wang, X.; Selim, M.; Lo, E.H. Opening the time window. J. Cereb. Blood Flow Metab. 2019, 39, 2539–2540. [Google Scholar] [CrossRef]
- Parsons, M.W.; Barber, P.A.; Desmond, P.M.; Baird, T.A.; Darby, D.G.; Byrnes, G.; Tress, B.M.; Davis, S.M. Acute hyperglycemia adversely affects stroke outcome: A magnetic resonance imaging and spectroscopy study. Ann. Neurol. 2002, 52, 20–28. [Google Scholar] [CrossRef]
- Kersten, J.R.; Toller, W.G.; Gross, E.R.; Pagel, P.S.; Warltier, D.C. Diabetes abolishes ischemic preconditioning: Role of glucose, insulin, and osmolality. Am. J. Physiol. Heart Circ. Physiol. 2000, 278, H1218–H1224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Z.; Tian, Y.; Liu, Y.; Hennessy, S.; Kron, I.L.; French, B.A. Acute hyperglycemia abolishes ischemic preconditioning by inhibiting Akt phosphorylation: Normalizing blood glucose before ischemia restores ischemic preconditioning. Oxidative Med. Cell. Longev. 2013, 2013, 329183. [Google Scholar] [CrossRef]
- Schenning, K.J.; Anderson, S.; Alkayed, N.J.; Hutchens, M.P. Hyperglycemia abolishes the protective effect of ischemic preconditioning in glomerular endothelial cells in vitro. Physiol. Rep. 2015, 3, e12346. [Google Scholar] [CrossRef]
- Yao, M.; Ni, J.; Zhou, L.; Peng, B.; Zhu, Y.; Cui, L. Elevated Fasting Blood Glucose Is Predictive of Poor Outcome in Non-Diabetic Stroke Patients: A Sub-Group Analysis of SMART. PLoS ONE 2016, 11, e0160674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van den Berghe, G. How does blood glucose control with insulin save lives in intensive care? J. Clin. Investig. 2004, 114, 1187–1195. [Google Scholar] [CrossRef] [Green Version]
- Luitse, M.J.; Biessels, G.J.; Rutten, G.E.; Kappelle, L.J. Diabetes, hyperglycaemia, and acute ischaemic stroke. Lancet Neurol. 2012, 11, 261–271. [Google Scholar] [CrossRef]
- Carvalho, C.; Santos, M.S.; Oliveira, C.R.; Moreira, P.I. Alzheimer’s disease and type 2 diabetes-related alterations in brain mitochondria, autophagy and synaptic markers. Biochim. Biophys. Acta 2015, 1852, 1665–1675. [Google Scholar] [CrossRef] [Green Version]
- Lee, R.H.; Bergmeier, W. Sugar makes neutrophils RAGE: Linking diabetes-associated hyperglycemia to thrombocytosis and platelet reactivity. J. Clin. Investig. 2017, 127, 2040–2043. [Google Scholar] [CrossRef]
- Ferris, J.K.; Peters, S.; Brown, K.E.; Tourigny, K.; Boyd, L.A. Type-2 diabetes mellitus reduces cortical thickness and decreases oxidative metabolism in sensorimotor regions after stroke. J. Cereb. Blood Flow Metab. 2018, 38, 823–834. [Google Scholar] [CrossRef]
- Gray, C.S.; Hildreth, A.J.; Sandercock, P.A.; O’Connell, J.E.; Johnston, D.E.; Cartlidge, N.E.; Bamford, J.M.; James, O.F.; Alberti, K.G. Glucose-potassium-insulin infusions in the management of post-stroke hyperglycaemia: The UK Glucose Insulin in Stroke Trial (GIST-UK). Lancet Neurol. 2007, 6, 397–406. [Google Scholar] [CrossRef]
- Johnston, K.C.; Bruno, A.; Pauls, Q.; Hall, C.E.; Barrett, K.M.; Barsan, W.; Fansler, A.; Van de Bruinhorst, K.; Janis, S.; Durkalski-Mauldin, V.L. Intensive vs Standard Treatment of Hyperglycemia and Functional Outcome in Patients with Acute Ischemic Stroke: The SHINE Randomized Clinical Trial. JAMA 2019, 322, 326–335. [Google Scholar] [CrossRef]
- Liu, R.; Wang, H.; Xu, B.; Chen, W.; Turlova, E.; Dong, N.; Sun, C.L.; Lu, Y.; Fu, H.; Shi, R.; et al. Cerebrovascular Safety of Sulfonylureas: The Role of KATP Channels in Neuroprotection and the Risk of Stroke in Patients with Type 2 Diabetes. Diabetes 2016, 65, 2795–2809. [Google Scholar] [CrossRef] [Green Version]
- Zinman, B.; Wanner, C.; Lachin, J.M.; Fitchett, D.; Bluhmki, E.; Hantel, S.; Mattheus, M.; Devins, T.; Johansen, O.E.; Woerle, H.J.; et al. Empagliflozin, Cardiovascular Outcomes, and Mortality in Type 2 Diabetes. N. Engl. J. Med. 2015, 373, 2117–2128. [Google Scholar] [CrossRef]
- Marso, S.P.; Daniels, G.H.; Brown-Frandsen, K.; Kristensen, P.; Mann, J.F.; Nauck, M.A.; Nissen, S.E.; Pocock, S.; Poulter, N.R.; Ravn, L.S.; et al. Liraglutide and Cardiovascular Outcomes in Type 2 Diabetes. N. Engl. J. Med. 2016, 375, 311–322. [Google Scholar] [CrossRef] [Green Version]
- Gerstein, H.C.; Colhoun, H.M.; Dagenais, G.R.; Diaz, R.; Lakshmanan, M.; Pais, P.; Probstfield, J.; Riesmeyer, J.S.; Riddle, M.C.; Rydén, L.; et al. Dulaglutide and cardiovascular outcomes in type 2 diabetes (REWIND): A double-blind, randomised placebo-controlled trial. Lancet 2019, 394, 121–130. [Google Scholar] [CrossRef]
- Kernan, W.N.; Viscoli, C.M.; Furie, K.L.; Young, L.H.; Inzucchi, S.E.; Gorman, M.; Guarino, P.D.; Lovejoy, A.M.; Peduzzi, P.N.; Conwit, R.; et al. Pioglitazone after Ischemic Stroke or Transient Ischemic Attack. N. Engl. J. Med. 2016, 374, 1321–1331. [Google Scholar] [CrossRef]
- Augestad, I.L.; Pintana, H.; Larsson, M.; Krizhanovskii, C.; Nyström, T.; Klein, T.; Darsalia, V.; Patrone, C. Regulation of Glycemia in the Recovery Phase After Stroke Counteracts the Detrimental Effect of Obesity-Induced Type 2 Diabetes on Neurological Recovery. Diabetes 2020, 69, 1961–1973. [Google Scholar] [CrossRef]
- Davis, C.M.; Liu, X.; Alkayed, N.J. Cytochrome P450 eicosanoids in cerebrovascular function and disease. Pharmacol. Ther. 2017, 179, 31–46. [Google Scholar] [CrossRef]
- Iliff, J.J.; Jia, J.; Nelson, J.; Goyagi, T.; Klaus, J.; Alkayed, N.J. Epoxyeicosanoid signaling in CNS function and disease. Prostaglandins Other Lipid Mediat. 2010, 91, 68–84. [Google Scholar] [CrossRef] [Green Version]
- Simpkins, A.N.; Rudic, R.D.; Schreihofer, D.A.; Roy, S.; Manhiani, M.; Tsai, H.J.; Hammock, B.D.; Imig, J.D. Soluble epoxide inhibition is protective against cerebral ischemia via vascular and neural protection. Am. J. Pathol. 2009, 174, 2086–2095. [Google Scholar] [CrossRef] [Green Version]
- Heizer, M.L.; McKinney, J.S.; Ellis, E.F. 14,15-Epoxyeicosatrienoic acid inhibits platelet aggregation in mouse cerebral arterioles. Stroke 1991, 22, 1389–1393. [Google Scholar] [CrossRef] [Green Version]
- Spector, A.A.; Norris, A.W. Action of epoxyeicosatrienoic acids on cellular function. Am. J. Physiol. Cell Physiol. 2007, 292, C996–C1012. [Google Scholar] [CrossRef]
- Yang, B.; Graham, L.; Dikalov, S.; Mason, R.P.; Falck, J.R.; Liao, J.K.; Zeldin, D.C. Overexpression of cytochrome P450 CYP2J2 protects against hypoxia-reoxygenation injury in cultured bovine aortic endothelial cells. Mol. Pharmacol. 2001, 60, 310–320. [Google Scholar] [CrossRef] [Green Version]
- Merkel, M.J.; Liu, L.; Cao, Z.; Packwood, W.; Young, J.; Alkayed, N.J.; Van Winkle, D.M. Inhibition of soluble epoxide hydrolase preserves cardiomyocytes: Role of STAT3 signaling. Am. J. Physiol. Heart Circ. Physiol. 2010, 298, H679–H687. [Google Scholar] [CrossRef] [Green Version]
- Percie du Sert, N.; Hurst, V.; Ahluwalia, A.; Alam, S.; Avey, M.T.; Baker, M.; Browne, W.J.; Clark, A.; Cuthill, I.C.; Dirnagl, U.; et al. The ARRIVE guidelines 2.0: Updated guidelines for reporting animal research. J. Cereb. Blood Flow Metab. 2020, 40, 1769–1777. [Google Scholar] [CrossRef] [PubMed]
- Obrosova, I.G.; Ilnytska, O.; Lyzogubov, V.V.; Pavlov, I.A.; Mashtalir, N.; Nadler, J.L.; Drel, V.R. High-fat diet induced neuropathy of pre-diabetes and obesity: Effects of “healthy” diet and aldose reductase inhibition. Diabetes 2007, 56, 2598–2608. [Google Scholar] [CrossRef] [Green Version]
- Nakamura, T.; Terajima, T.; Ogata, T.; Ueno, K.; Hashimoto, N.; Ono, K.; Yano, S. Establishment and pathophysiological characterization of type 2 diabetic mouse model produced by streptozotocin and nicotinamide. Biol. Pharm. Bull. 2006, 29, 1167–1174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zuloaga, K.L.; Johnson, L.A.; Roese, N.E.; Marzulla, T.; Zhang, W.; Nie, X.; Alkayed, F.N.; Hong, C.; Grafe, M.R.; Pike, M.M.; et al. High fat diet-induced diabetes in mice exacerbates cognitive deficit due to chronic hypoperfusion. J. Cereb. Blood Flow Metab. 2016, 36, 1257–1270. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | RetentionTime (min) | Q1 Mass | Q3 Mass | DP | EP | CE | CXP |
|---|---|---|---|---|---|---|---|
| 14,15-EET | 7.45 | 319 | 219 | −120 | −10 | −18 | −11 |
| 11,12-EET | 8.14 | 319 | 208 | −80 | −10 | −18 | −9 |
| 8,9-EET | 8.50 | 319 | 68.9 | −90 | −10 | −26 | −11 |
| 5,6-EET | 8.83 | 319 | 191 | −85 | −10 | −16 | −7 |
| d8 14,15-EET | 7.30 | 327 | 226 | −95 | −10 | −18 | −9 |
| 5-HETE | 6.90 | 319.2 | 115 | −100 | −10 | −20 | −9 |
| 11-HETE | 6.08 | 319.2 | 167 | −75 | −10 | −24 | −3 |
| 12-HETE | 6.31 | 319.2 | 179 | −105 | −10 | −20 | −3 |
| 15-HETE | 5.74 | 319.2 | 219 | −115 | −10 | −20 | −9 |
| 18-HETE | 4.96 | 319.2 | 261 | −65 | −10 | −22 | −9 |
| 19-HETE | 4.62 | 319.2 | 231 | −100 | −10 | −22 | −7 |
| 20-HETE | 4.80 | 319.1 | 289 | −130 | −10 | −26 | −11 |
| d8 15-HETE | 5.6 | 327 | 226 | −85 | −10 | −20 | −9 |
| d6 20-HETE | 4.7 | 325 | 281 | −75 | −10 | −24 | −9 |
| 14,15-DHET | 3.6 | 337 | 207 | −100 | −10 | −26 | −3 |
| 11,12-DHET | 4.00 | 337 | 167 | −90 | −10 | −28 | −11 |
| 8,9-DHET | 4.40 | 337 | 127 | −75 | −10 | −30 | −9 |
| 5,6-DHET | 4.90 | 337 | 145 | −115 | −10 | −24 | −9 |
| d11 14,15-DHET | 3.60 | 348 | 207 | −120 | −10 | −40 | −15 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Davis, C.M.; Zhang, W.H.; Allen, E.M.; Bah, T.M.; Shangraw, R.E.; Alkayed, N.J. Soluble Epoxide Hydrolase Blockade after Stroke Onset Protects Normal but Not Diabetic Mice. Int. J. Mol. Sci. 2021, 22, 5419. https://doi.org/10.3390/ijms22115419
Davis CM, Zhang WH, Allen EM, Bah TM, Shangraw RE, Alkayed NJ. Soluble Epoxide Hydrolase Blockade after Stroke Onset Protects Normal but Not Diabetic Mice. International Journal of Molecular Sciences. 2021; 22(11):5419. https://doi.org/10.3390/ijms22115419
Chicago/Turabian StyleDavis, Catherine M., Wenri H. Zhang, Elyse M. Allen, Thierno M. Bah, Robert E. Shangraw, and Nabil J. Alkayed. 2021. "Soluble Epoxide Hydrolase Blockade after Stroke Onset Protects Normal but Not Diabetic Mice" International Journal of Molecular Sciences 22, no. 11: 5419. https://doi.org/10.3390/ijms22115419
APA StyleDavis, C. M., Zhang, W. H., Allen, E. M., Bah, T. M., Shangraw, R. E., & Alkayed, N. J. (2021). Soluble Epoxide Hydrolase Blockade after Stroke Onset Protects Normal but Not Diabetic Mice. International Journal of Molecular Sciences, 22(11), 5419. https://doi.org/10.3390/ijms22115419