
Prolonged Transcriptional Consequences in Survivors of Sepsis



{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Survivorship and Morphological Changes in Survivors of the Sepsis
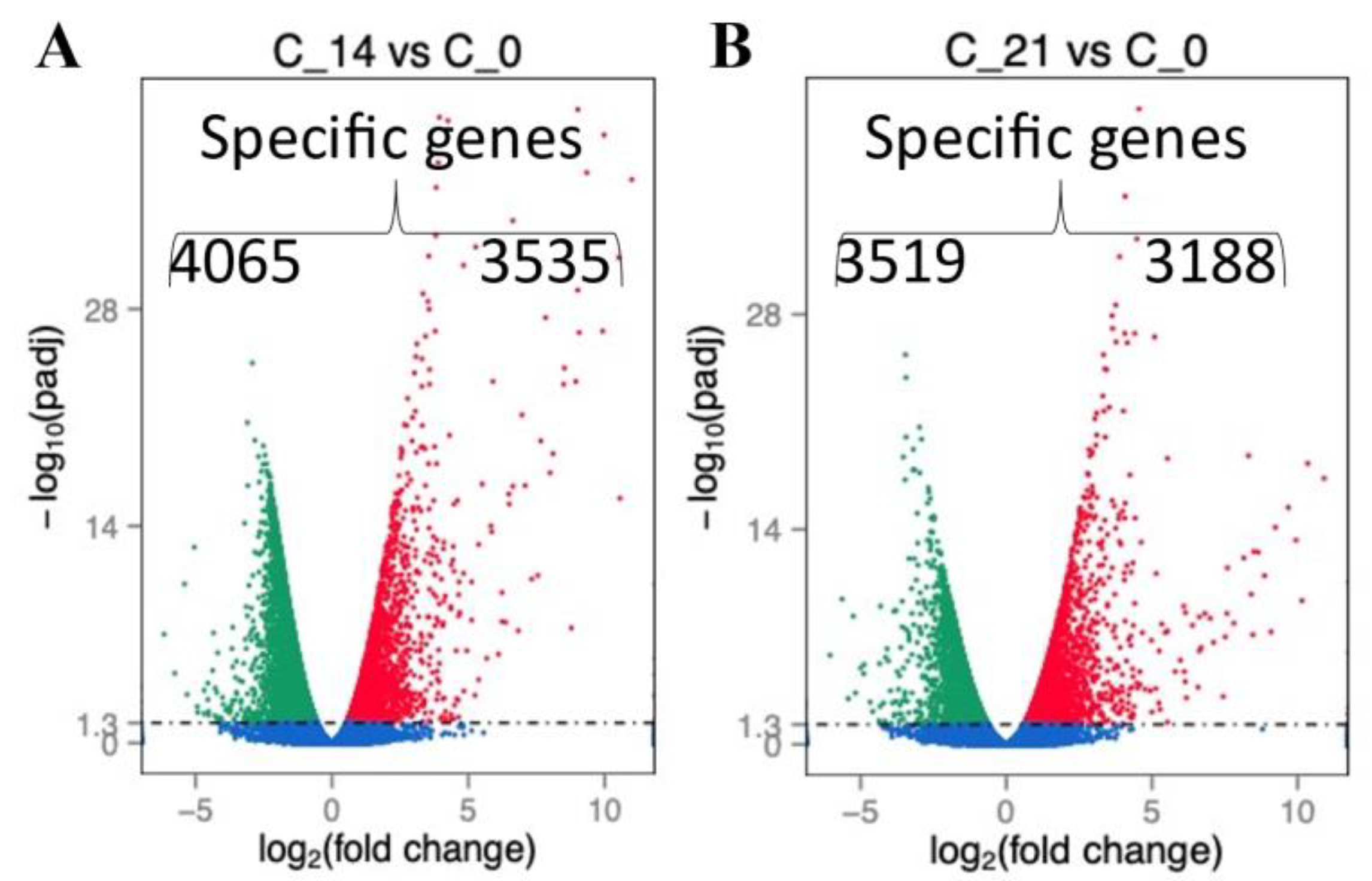
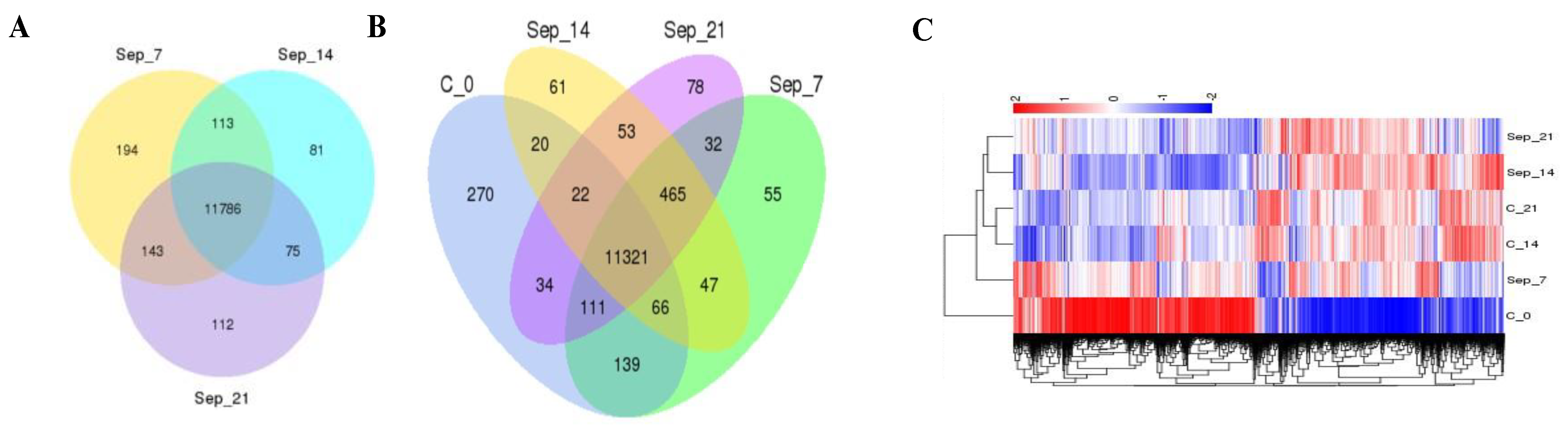
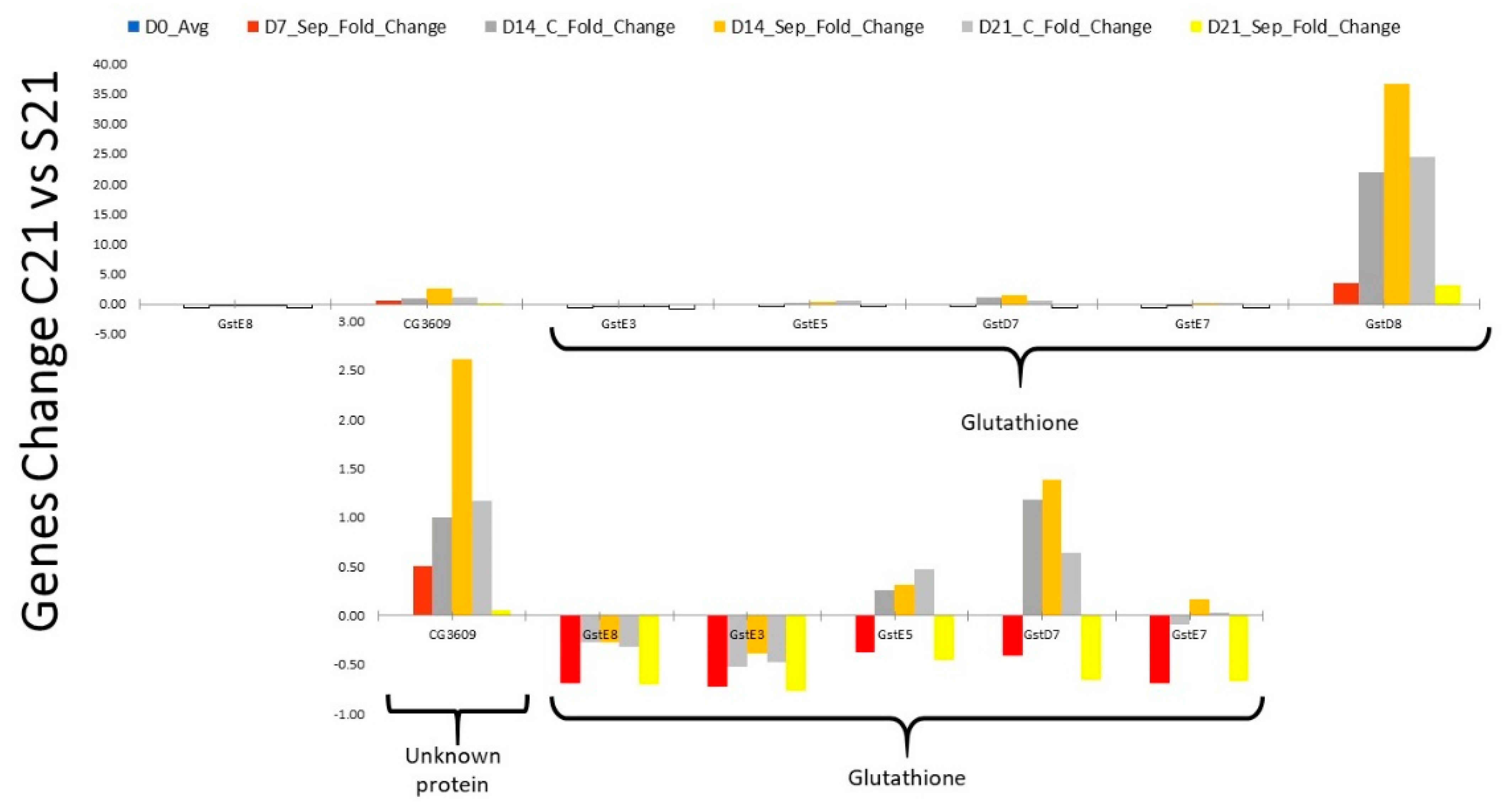
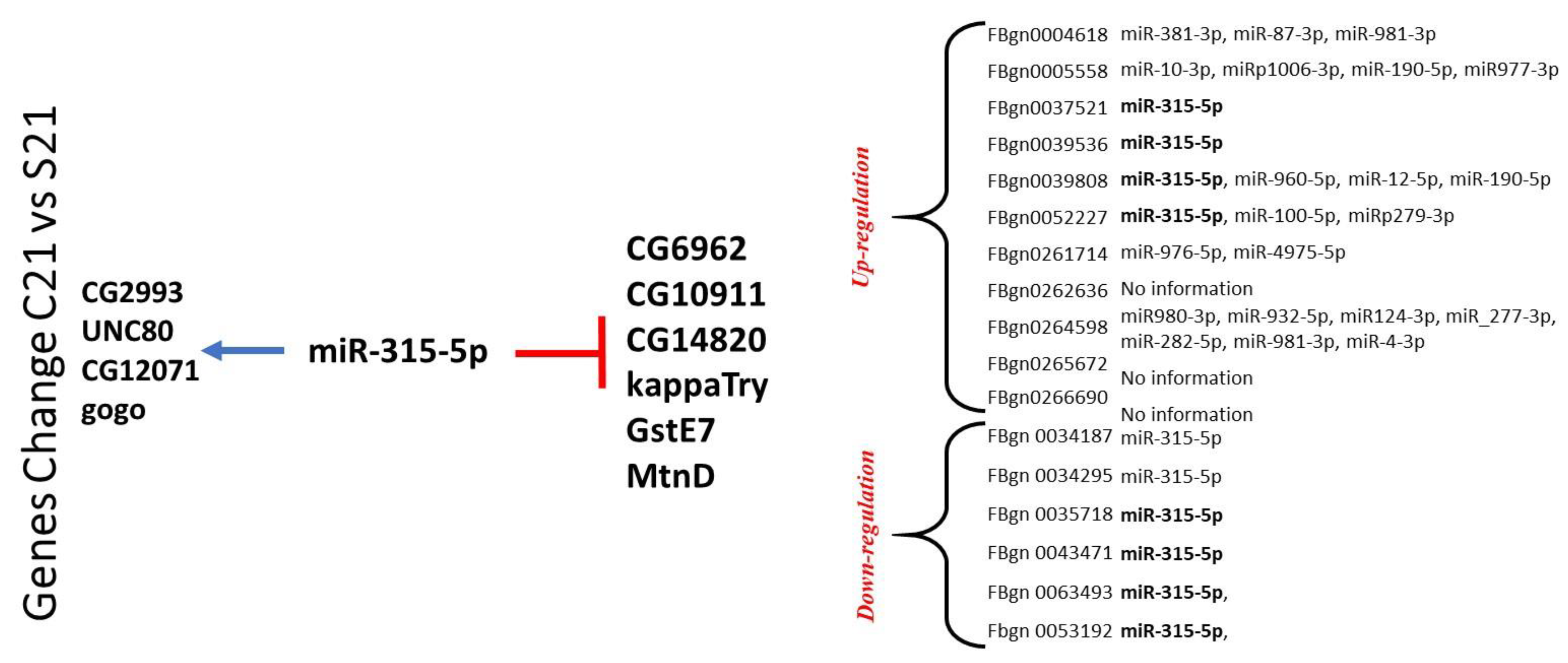
2.2. RNA Profile in the Sepsis Survivors at 7, 14, and 21 Days
3. Discussion
4. Materials and Methods
4.1. Fly Stock Maintenance and Experimental Procedure
4.2. RNA Extraction and Quantification
4.3. RNA Sequencing and Analysis
4.4. 3′ Untranslated Region and microRNA Analysis
4.5. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Vachharajani, V.; McCall, C.E. Epigenetic and metabolic programming of innate immunity in sepsis. Innate Immun. 2019, 25, 267–279. [Google Scholar] [CrossRef] [PubMed]
- Nowill, A.E.; Fornazin, M.C.; Spago, M.C.; Neto, V.D.; Pinheiro, V.R.P.; Alexandre, S.S.S.; Moraes, E.O.; Souza, G.H.M.F.; Eberlin, M.N.; Marques, L.A.; et al. Immune Response Resetting in Ongoing Sepsis. J. Immunol. 2019, 203, 1298–1312. [Google Scholar] [CrossRef] [PubMed]
- Mankowski, R.T.; Yende, S.; Angus, D.C. Long-term impact of sepsis on cardiovascular health. Intensiv. Care Med. 2019, 45, 78–81. [Google Scholar] [CrossRef]
- Abu-Kaf, H.; Mizrakli, Y.; Novack, V.; Dreiher, J. Long-Term Survival of Young Patients Surviving ICU Admission with Severe Sepsis. Crit. Care Med. 2018, 46, 1269–1275. [Google Scholar] [CrossRef]
- Shen, H.-N.; Lu, C.-L.; Yang, H.-H. Risk of Recurrence after Surviving Severe Sepsis. Crit. Care Med. 2016, 44, 1833–1841. [Google Scholar] [CrossRef]
- Coopersmith, C.M.; De Backer, D.; Deutschman, C.S.; Ferrer, R.; Lat, I.; Machado, F.R.; Martin, G.S.; Martin-Loeches, I.; Nunnally, M.E.; Antonelli, M.; et al. Surviving sepsis campaign: Research priorities for sepsis and septic shock. Intensiv. Care Med. 2018, 44, 1400–1426. [Google Scholar] [CrossRef] [Green Version]
- Weiss, S.L.; Peters, M.J.; Alhazzani, W.; Agus, M.S.D.; Flori, H.R.; Inwald, D.P.; Nadel, S.; Schlapbach, L.J.; Tasker, R.C.; Argent, A.C.; et al. Surviving sepsis campaign international guidelines for the management of septic shock and sepsis-associated organ dysfunction in children. Intensiv. Care Med. 2020, 46, 10–67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nascimento, D.C.; Melo, P.H.; Piñeros, A.R.; Ferreira, R.G.; Colón, D.F.; Donate, P.B.; Castanheira, F.V.; Gozzi, A.; Czaikoski, P.G.; Niedbala, W.; et al. IL-33 contributes to sepsis-induced long-term immunosuppression by expanding the regulatory T cell population. Nat. Commun. 2017, 8, 14919. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lapko, N.; Zawadka, M.; Polosak, J.; Worthen, G.S.; Danet-Desnoyers, G.; Puzianowska-Kuźnicka, M.; Laudanski, K. Long-term Monocyte Dysfunction after Sepsis in Humanized Mice Is Related to Persisted Activation of Macrophage-Colony Stimulation Factor (M-CSF) and Demethylation of PU.1, and It Can Be Reversed by Blocking M-CSF In Vitro or by Transplanting Naïve Autologous Stem Cells In Vivo. Front. Immunol. 2017, 8, 401. [Google Scholar] [CrossRef]
- Kaynar, A.M.; Bakalov, V.; LaVerde, S.M.; Cambriel, A.I.F.; Lee, B.-H.; Towheed, A.; Gregory, A.D.; Webb, S.A.R.; Palladino, M.J.; Bozza, F.A.; et al. Cost of surviving sepsis: A novel model of recovery from sepsis in Drosophila melanogaster. Intensiv. Care Med. Exp. 2016, 4, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brakenridge, S.C.; Efron, P.A.; Cox, M.C.; Stortz, J.A.; Hawkins, R.B.; Ghita, G.; Gardner, A.; Mohr, A.M.; Anton, S.D.; Moldawer, L.L.; et al. Current Epidemiology of Surgical Sepsis. Ann. Surg. 2019, 270, 502–510. [Google Scholar] [CrossRef]
- Bomans, K.; Schenz, J.; Sztwiertnia, I.; Schaack, D.; Weigand, M.A.; Uhle, F. Sepsis Induces a Long-Lasting State of Trained Immunity in Bone Marrow Monocytes. Front. Immunol. 2018, 9, 2685. [Google Scholar] [CrossRef]
- Fouda, E.; Midan, D.A.E.; Ellaban, R.; El-Kousy, S.; Arafat, E. The diagnostic and prognostic role of MiRNA 15b and MiRNA 378a in neonatal sepsis. Biochem. Biophys. Rep. 2021, 26, 100988. [Google Scholar] [CrossRef]
- Tang, Y.; Yang, X.; Shu, H.; Yu, Y.; Pan, S.; Xu, J.; Shang, Y. Bioinformatic analysis identifies potential biomarkers and therapeutic targets of septic-shock-associated acute kidney injury. Hereditas 2021, 158, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Haas, L.E.M.; Boumendil, A.; Flaatten, H.; Guidet, B.; Ibarz, M.; Jung, C.; Moreno, R.; Morandi, A.; Andersen, F.H.; Zafeiridis, T.; et al. Frailty is associated with long-term outcome in patients with sepsis who are over 80 years old: Results from an observational study in 241 European ICUs. Age Ageing 2021. [Google Scholar] [CrossRef] [PubMed]
- Jeansonne, D.; Jeyaseelan, S. Role of an Anti-Aging Molecule in a Toxic Lifestyle: Relevance for Alcohol Effects on Sepsis. Alcohol. Clin. Exp. Res. 2021. [Google Scholar] [CrossRef] [PubMed]
- Popov, D.; Pavlov, G. Sepsis models in exprimental animals. Trakia J. Sci. 2013, 1, 13–23. [Google Scholar]
- Cho, S.-Y.; Kwon, Y.-K.; Nam, M.; Vaidya, B.; Kim, S.R.; Lee, S.; Kwon, J.; Kim, D.; Hwang, G.-S. Integrated profiling of global metabolomic and transcriptomic responses to viral hemorrhagic septicemia virus infection in olive flounder. Fish Shellfish Immunol. 2017, 71, 220–229. [Google Scholar] [CrossRef]
- Douglas, A.E. The Drosophila model for microbiome research. Lab Anim. 2018, 47, 157–164. [Google Scholar] [CrossRef]
- Medzhitov, R.; Preston-Hurlburt, P.; Janewayr, C.A. A human homologue of the Drosophila Toll protein signals activation of adaptive immunity. Nat. Cell Biol. 1997, 388, 394–397. [Google Scholar] [CrossRef] [PubMed]
- Medvedev, A.E.; Murphy, M.P.; Zhou, H.; Li, X. E3 ubiquitin ligases Pellinos as regulators of pattern recognition receptor signaling and immune responses. Immunol. Rev. 2015, 266, 109–122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Istas, O.; Greenhalgh, A.; Cooper, R. The Effects of a Bacterial Endotoxin on Behavior and Sensory-CNS-Motor Circuits in Drosophila melanogaster. Insects 2019, 10, 115. [Google Scholar] [CrossRef] [Green Version]
- Limmer, S.; Haller, S.; Drenkard, E.; Lee, J.; Yu, S.; Kocks, C.; Ausubel, F.M.; Ferrandon, D. Pseudomonas aeruginosa RhlR is required to neutralize the cellular immune response in a Drosophila melanogaster oral infection model. Proc. Natl. Acad. Sci. USA 2011, 108, 17378–17383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramond, E.; Dudzic, J.P.; Lemaitre, B. Comparative RNA-Seq analyses of Drosophila plasmatocytes reveal gene specific signatures in response to clean injury and septic injury. PLoS ONE 2020, 15, e0235294. [Google Scholar] [CrossRef]
- Heo, Y.-J.; Lee, Y.-R.; Jung, H.-H.; Lee, J.; Ko, G.; Cho, Y.-H. Antibacterial Efficacy of Phages against Pseudomonas aeruginosa Infections in Mice and Drosophila melanogaster. Antimicrob. Agents Chemother. 2009, 53, 2469–2474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gotland, N.; Uhre, M.; Mejer, N.; Skov, R.; Petersen, A.; Larsen, A.; Benfield, T. Long-term mortality and causes of death associated with Staphylococcus aureus bacteremia. A matched cohort study. J. Infect. 2016, 73, 346–357. [Google Scholar] [CrossRef]
- Artero, A.; Inglada, L.; Gómez-Belda, A.; Capdevila, J.A.; Diez, L.F.; Arca, A.; Romero, J.M.; Domínguez-Gil, M.; Serra-Centelles, C.; De La Fuente, J. The clinical impact of bacteremia on outcomes in elderly patients with pyelonephritis or urinary sepsis: A prospective multicenter study. PLoS ONE 2018, 13, e0191066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matova, N.; Anderson, K.V. Rel/NF- B double mutants reveal that cellular immunity is central to Drosophila host defense. Proc. Natl. Acad. Sci. USA 2006, 103, 16424–16429. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Xie, C.; Tian, L.; Hong, L.; Wu, X.; Han, J. Participation of the p38 pathway in Drosophila host defense against pathogenic bacteria and fungi. Proc. Natl. Acad. Sci. USA 2010, 107, 20774–20779. [Google Scholar] [CrossRef] [Green Version]
- Avet-Rochex, A.; Perrin, J.; Bergeret, E.; Fauvarque, M.-O. Rac2 is a major actor of Drosophila resistance to Pseudomonas aeruginosa acting in phagocytic cells. Genes Cells 2007, 12, 1193–1204. [Google Scholar] [CrossRef]
- Bajgar, A.; Krejčová, G.; Doležal, T. Polarization of Macrophages in Insects: Opening Gates for Immuno-Metabolic Research. Front. Cell Dev. Biol. 2021, 9, 629238. [Google Scholar] [CrossRef]
- Bakalov, V.; Amathieu, R.; Triba, M.N.; Clément, M.-J.; Uribe, L.R.; Le Moyec, L.; Kaynar, A.M. Metabolomics with Nuclear Magnetic Resonance Spectroscopy in a Drosophila melanogaster Model of Surviving Sepsis. Metabolites 2016, 6, 47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Irving, P.; Ubeda, J.-M.; Doucet, D.; Troxler, L.; Lagueux, M.; Zachary, D.; Hoffmann, J.A.; Hetru, C.; Meister, M. New insights into Drosophila larval haemocyte functions through genome-wide analysis. Cell. Microbiol. 2005, 7, 335–350. [Google Scholar] [CrossRef]
- Silver, S.J.; Hagen, J.W.; Okamura, K.; Perrimon, N.; Lai, E.C. Functional screening identifies miR-315 as a potent activator of Wingless signaling. Proc. Natl. Acad. Sci. USA 2007, 104, 18151–18156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, S.P.; Coronella, J.A.; Beneš, H.; Cochrane, B.J.; Zimniak, P. Catalytic function ofDrosophila melanogasterglutathioneS-transferase DmGSTS1-1 (GST-2) in conjugation of lipid peroxidation end products. JBIC J. Biol. Inorg. Chem. 2001, 268, 2912–2923. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.; Ralston, M.M.; Meng, X.; Bongiovanni, K.D.; Jones, A.L.; Benndorf, R.; Nelin, L.D.; Frazier, W.J.; Rogers, L.K.; Smith, C.V.; et al. Glutathione reductase is essential for host defense against bacterial infection. Free Radic. Biol. Med. 2013, 61, 320–332. [Google Scholar] [CrossRef] [Green Version]
- Udomsinprasert, R.; Bogoyevitch, M.A.; Ketterman, A.J. Reciprocal regulation of glutathione S-transferase spliceforms and the Drosophila c-Jun N-terminal kinase pathway components. Biochem. J. 2004, 383, 483–490. [Google Scholar] [CrossRef] [Green Version]
- Wongtrakul, J.; Sukittikul, S.; Saisawang, C.; Ketterman, A.J. Mitogen-Activated Protein Kinase p38b Interaction with Delta Class Glutathione Transferases from the Fruit Fly, Drosophila melanogaster. J. Insect Sci. 2012, 12, 1–12. [Google Scholar] [CrossRef]
- Ekas, L.A.; Baeg, G.-H.; Flaherty, M.S.; Ayala-Camargo, A.; Bach, E.A. JAK/STAT signaling promotes regional specification by negatively regulating wingless expression in Drosophila. Development 2006, 133, 4721–4729. [Google Scholar] [CrossRef] [Green Version]
- Mockett, R.J.; Sohal, R.S.; Orr, W.C. Overexpression of glutathione reductase extends survival in transgenic Drosophila melanogaster under hyperoxia but not normoxia. FASEB J. 1999, 13, 1733–1742. [Google Scholar] [CrossRef] [PubMed]
- Missirlis, F.; Rahlfs, S.; Dimopoulos, N.; Bauer, H.; Becker, K.; Hilliker, A.; Phillips, J.P.; Jäckle, H. A Putative Glutathione Peroxidase of Drosophila Encodes a Thioredoxin Peroxidase That Provides Resistance against Oxidative Stress But Fails to Complement a Lack of Catalase Activity. Biol. Chem. 2003, 384, 463–472. [Google Scholar] [CrossRef]
- Silveira-Lessa, A.L.; Quinello, C.; Lima, L.; Redondo, A.C.C.; Ceccon, M.E.J.R.; Carneiro-Sampaio, M.; Palmeira, P. TLR expression, phagocytosis and oxidative burst in healthy and septic newborns in response to Gram-negative and Gram-positive rods. Hum. Immunol. 2016, 77, 972–980. [Google Scholar] [CrossRef]
- Kennell, J.A.; Gerin, I.; MacDougald, O.A.; Cadigan, K.M. The microRNA miR-8 is a conserved negative regulator of Wnt signaling. Proc. Natl. Acad. Sci. USA 2008, 105, 15417–15422. [Google Scholar] [CrossRef] [Green Version]
- Jridi, I.; Canté-Barrett, K.; Pike-Overzet, K.; Staal, F.J.T. Inflammation and Wnt Signaling: Target for Immunomodulatory Therapy? Front. Cell Dev. Biol. 2021, 8, 615131. [Google Scholar] [CrossRef]
- Kwee, S.A.; Tiirikainen, M. Beta-catenin activation and immunotherapy resistance in hepatocellular carcinoma: Mechanisms and biomarkers. Hepatoma Res. 2021, 7. [Google Scholar] [CrossRef]
- Claudel, M.; Jouzeau, J.; Cailotto, F. Secreted Frizzled-related proteins (sFRPs) in osteo-articular diseases: Much more than simple antagonists of Wnt signaling? FEBS J. 2019, 286, 4832–4851. [Google Scholar] [CrossRef] [PubMed]
- Rastogi, M.; Srivastava, N.; Singh, S.K. Exploitation of microRNAs by Japanese Encephalitis virus in human microglial cells. J. Med. Virol. 2018, 90, 648–654. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Dong, X.; Zhao, N.; Su, X.; Wang, Y.; Li, Y.; Wen, M.; Li, Z.; Wang, C.; Chen, J.; et al. Schisandrin B attenuates bleomycin-induced pulmonary fibrosis in mice through the wingless/integrase-1 signaling pathway. Exp. Lung Res. 2020, 46, 185–194. [Google Scholar] [CrossRef]
- Blankesteijn, W.M. Interventions in WNT Signaling to Induce Cardiomyocyte Proliferation: Crosstalk with Other Pathways. Mol. Pharmacol. 2019, 97, 90–101. [Google Scholar] [CrossRef]
- Grealy, R.; White, M.; Stordeur, P.; Kelleher, D.; Doherty, D.G.; McManus, R.; Ryan, T. Characterising Cytokine Gene Expression Signatures in Patients with Severe Sepsis. Mediat. Inflamm. 2013, 2013, 164246. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Laudanski, K.; Soh, J.; DiMeglio, M.; Sullivan, K.E. Prolonged Transcriptional Consequences in Survivors of Sepsis. Int. J. Mol. Sci. 2021, 22, 5422. https://doi.org/10.3390/ijms22115422
Laudanski K, Soh J, DiMeglio M, Sullivan KE. Prolonged Transcriptional Consequences in Survivors of Sepsis. International Journal of Molecular Sciences. 2021; 22(11):5422. https://doi.org/10.3390/ijms22115422
Chicago/Turabian StyleLaudanski, Krzysztof, James Soh, Matthew DiMeglio, and Kathleen E. Sullivan. 2021. "Prolonged Transcriptional Consequences in Survivors of Sepsis" International Journal of Molecular Sciences 22, no. 11: 5422. https://doi.org/10.3390/ijms22115422
APA StyleLaudanski, K., Soh, J., DiMeglio, M., & Sullivan, K. E. (2021). Prolonged Transcriptional Consequences in Survivors of Sepsis. International Journal of Molecular Sciences, 22(11), 5422. https://doi.org/10.3390/ijms22115422