
Rhizomal Reclassification of Living Organisms

 ,
,  , and
, and 
Abstract
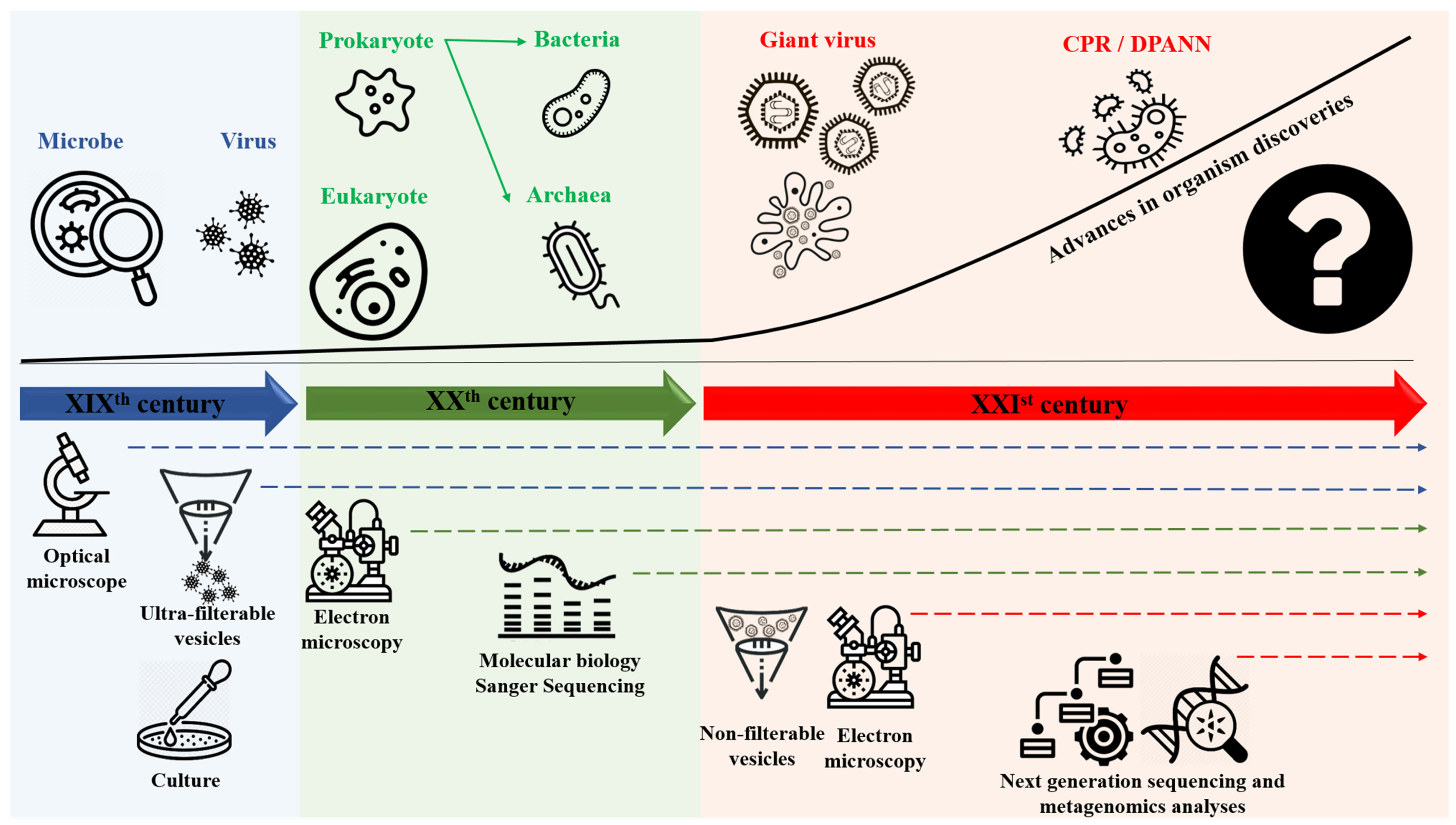
:1. Introduction
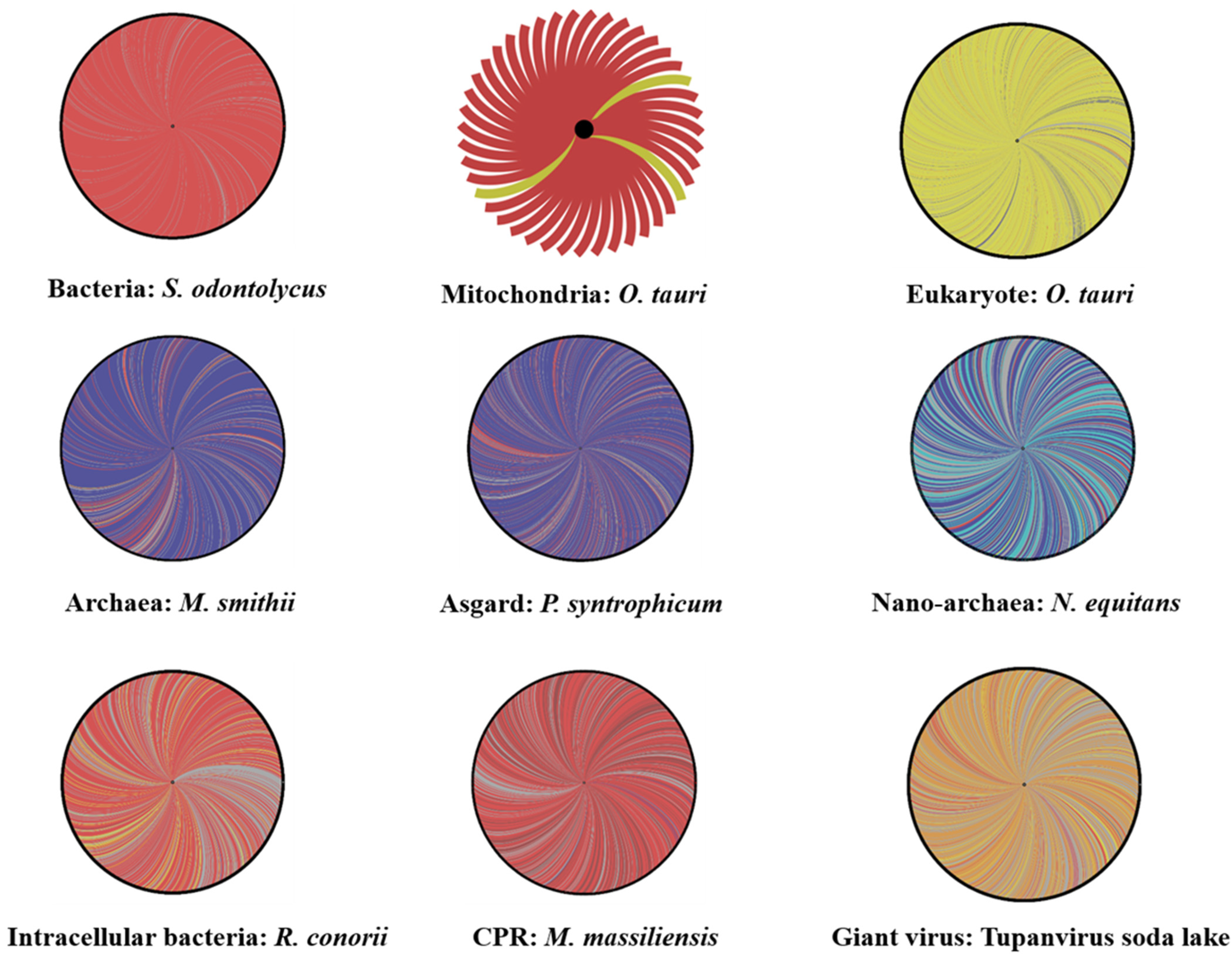
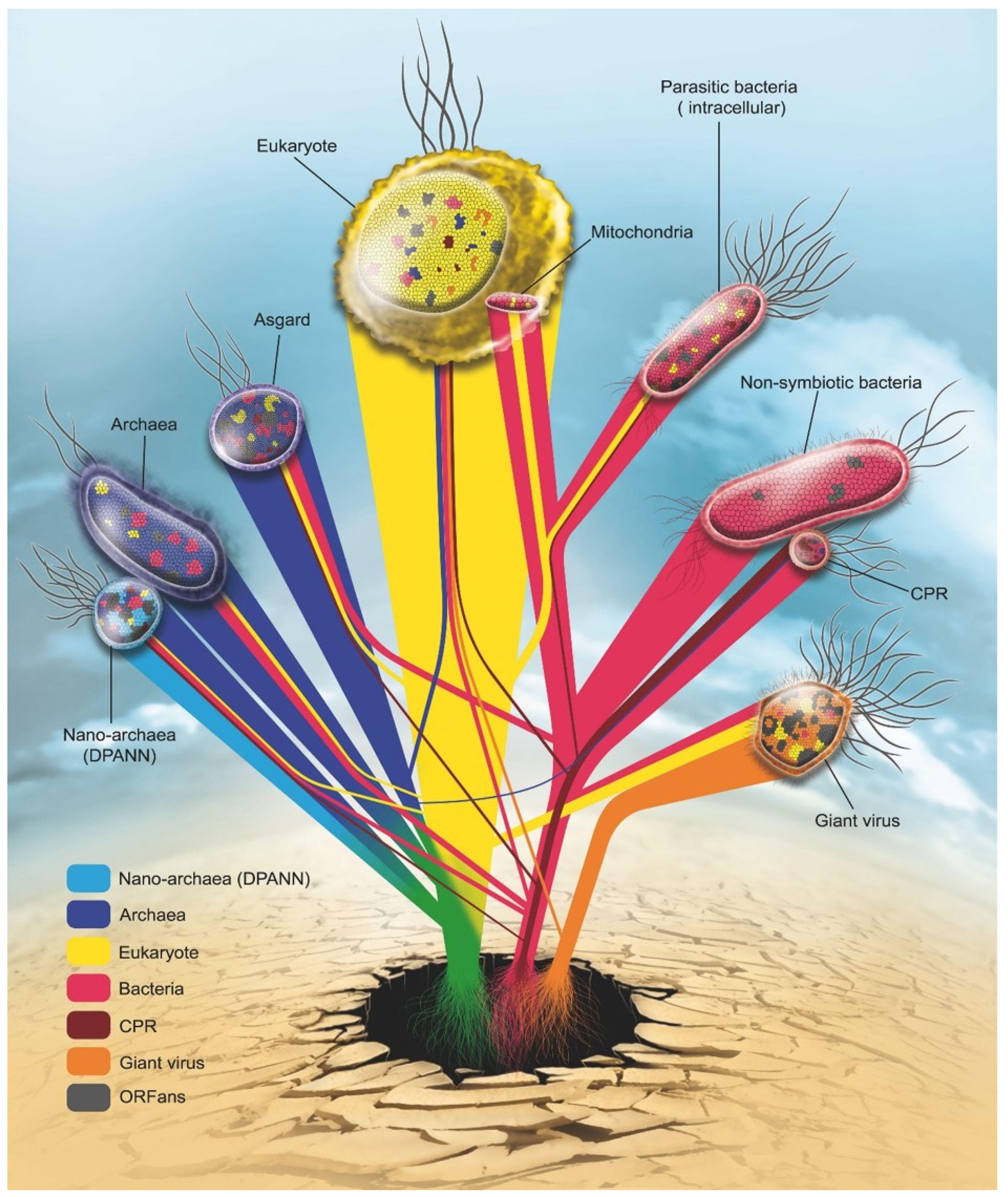
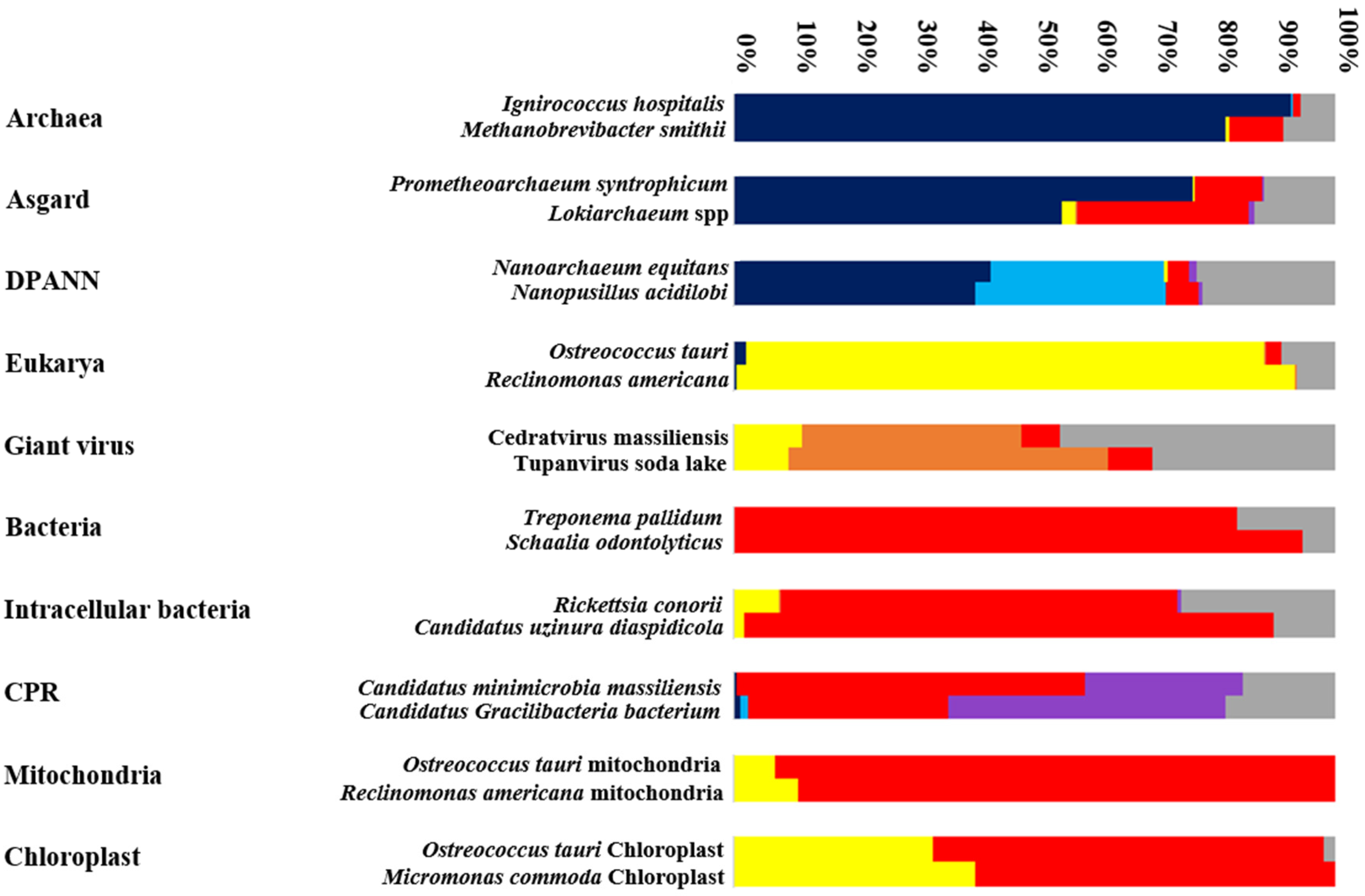
2. Genome Evolution and Mosaicism Representation: Rhizomes Construction
2.1. Rhizome of All Archaeal Members
2.2. Rhizome of Eukaryote
2.3. Giant Viruses: Rhizome of Living Viruses
2.4. Do CPR and Bacterial Species Have the Same Mosaic Profile?
2.5. Organelles’ Rhizomes: Mitochondria and Chloroplast
2.6. Genetic Network: The Representation of Interactions between Microbes
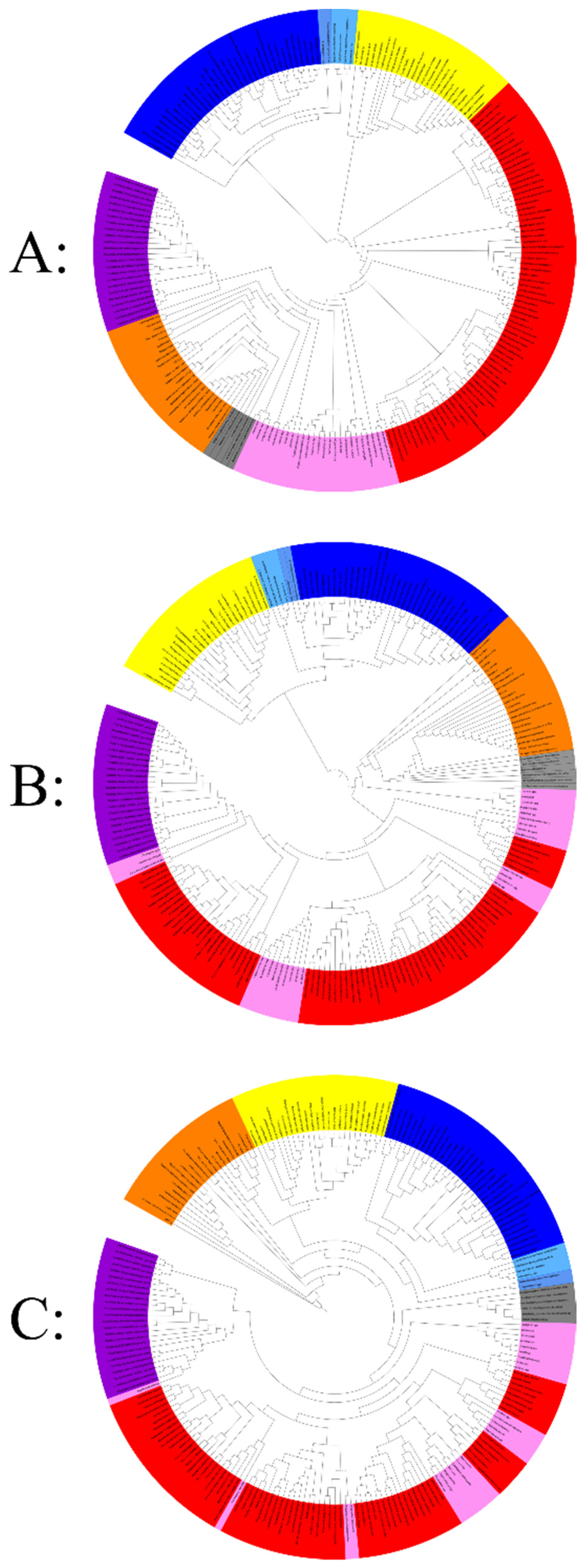
3. Hierarchical Clustering Based on Informational COGs and Fold Superfamily Domains: A Symbiotic Lifestyle Branch along with the Three Domains of Life
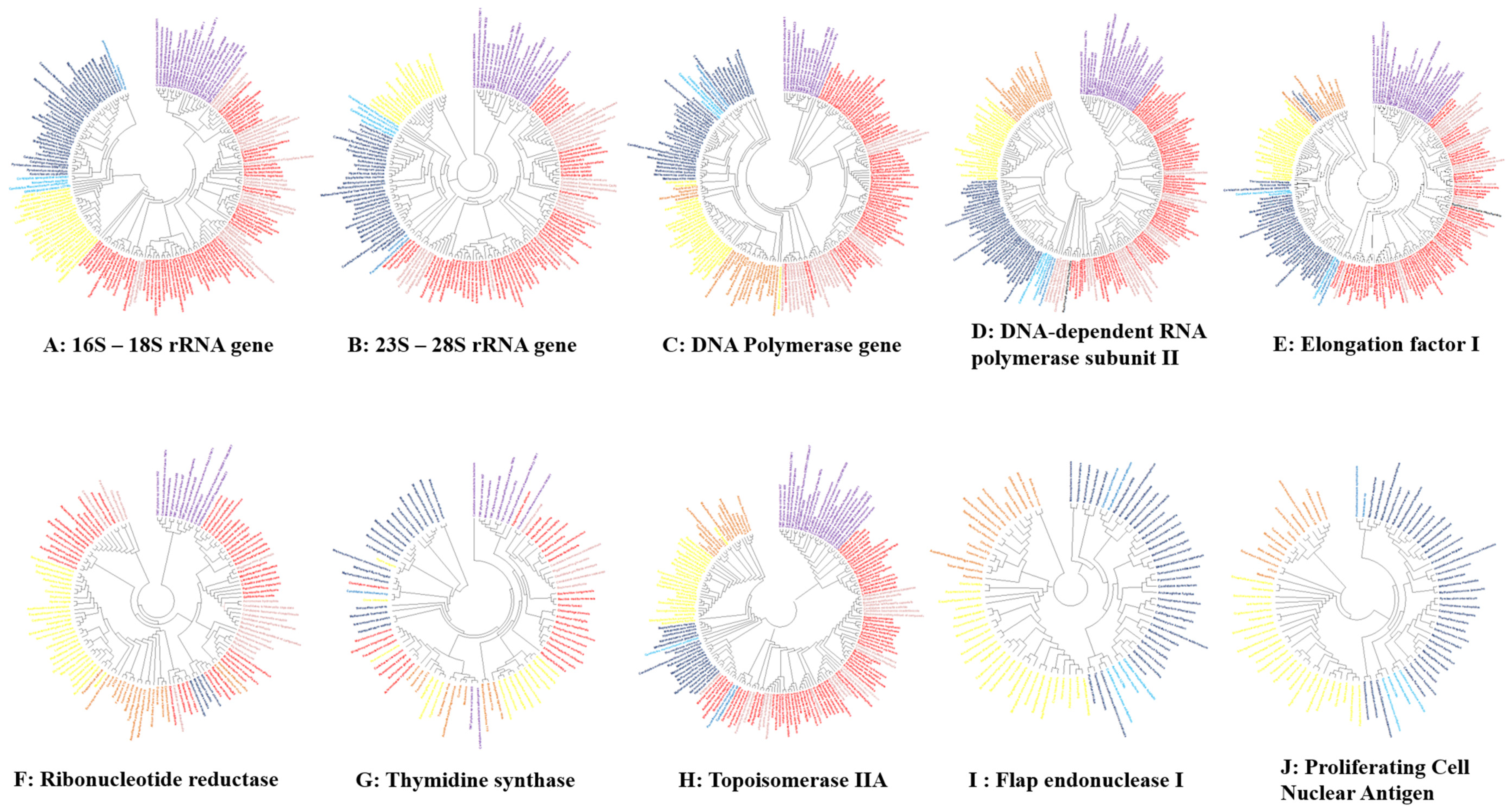
4. Phylogenetic Analysis of Ancestral Coding and Noncoding rRNA Genes
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- The Logic of Scientific Discovery—Karl Popper—Google Books. Available online: https://books.google.fr/books?hl=en&lr=&id=LWSBAgAAQBAJ&oi=fnd&pg=PP1&dq=popper+1934+the+logic+of+scientific+discovery&ots=pADe_00FdN&sig=rmw7bfCRO3OobPdGEpdjkLaMXBk&redir_esc=y#v=onepage&q=popper1934thelogicofscientificdiscovery&f=false (accessed on 24 January 2021).
- Raoult, D. TRUC or the need for a new microbial classification. Intervirology 2013, 56, 349–353. [Google Scholar] [CrossRef] [PubMed]
- Blevins, S.M.; Bronze, M.S. Robert Koch and the “golden age” of bacteriology. Int. J. Infect. Dis. 2010, 14, e744–e751. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coico, R. Gram staining. Curr. Protoc. Microbiol. 2005. [Google Scholar] [CrossRef]
- Ivanowski, D. Ueber die Mosaikkrankheit der Tabakspflanze. St. Petersb. Acad Imp. Sci. Bul. 1892, 35, 67–70. [Google Scholar]
- Bos, L. Beijerinck’s work on tobacco mosaic virus: Historical context and legacy. Philos. Trans. R. Soc. Lond. B Biol. Sci. 1999, 354, 675–685. [Google Scholar] [CrossRef] [Green Version]
- Wilkinson, L. 1. Beginnings At The Turn Of The Century. Med. Hist. 1974, 18, 211–221. [Google Scholar] [CrossRef] [Green Version]
- Lwoff, B.A. The Concept of Virus The Third Marjory Stephenson Memorial Lecture. J. Gen. Microbiol. 1957, 17, 239–253. [Google Scholar]
- Chatton, E. Pansporella Perplexa: Amœbien à Spores Protégées Parasite des Daphnies: Réflexions sur la Biologie et la Phylogénie des Protozoaires; Masson: Paris, France, 1925. [Google Scholar]
- Smith, K.C.A.; Wells, O.C.; Mcmullan, D. The fiftieth anniversary of the first applications of the scanning electron microscope in materials research. Phys. Procedia 2008, 1, 3–12. [Google Scholar] [CrossRef] [Green Version]
- History of Electron Microscopy, 1931–2000. Available online: https://authors.library.caltech.edu/5456/1/hrst.mit.edu/hrs/materials/public/ElectronMicroscope/EM_HistOverview.htm (accessed on 12 May 2021).
- Woese, C.R.; Kandler, O.; Wheelis, M.L. Towards a natural system of organisms: Proposal for the domains Archaea, Bacteria, and Eucarya. Proc. Natl. Acad. Sci. USA 1990, 87, 4576–4579. [Google Scholar] [CrossRef] [Green Version]
- Lindsay, M.R.; Webb, R.I.; Strous, M.; Jetten, M.S.M.; Butler, M.K.; Forde, R.J.; Fuerst, J.A. Cell compartmentalisation in planctomycetes: Novel types of structural organisation for the bacterial cell. Arch. Microbiol. 2001, 175, 413–429. [Google Scholar] [CrossRef]
- Raoult, D.; Forterre, P. Redefining viruses: Lessons from Mimivirus. Nat. Rev. Microbiol. 2008, 6, 315–319. [Google Scholar] [CrossRef]
- Georgiades, K.; Raoult, D. The rhizome of Reclinomonas americana, Homo sapiens, Pediculus humanus and Saccharomyces cerevisiae mitochondria. Biol. Direct 2011, 6, 1–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- La Scola, B.; Audic, S.; Robert, C.; Jungang, L.; De Lamballerie, X.; Drancourt, M.; Birtles, R.; Claverie, J.M.; Raoult, D. A giant virus in amoebae. Science 2003, 299, 2033. [Google Scholar] [CrossRef] [PubMed]
- Raoult, D.; Audic, S.; Robert, C.; Abergel, C.; Renesto, P.; Ogata, H.; La Scola, B.; Suzan, M.; Claverie, J.M. The 1.2-megabase genome sequence of Mimivirus. Science 2004, 306, 1344–1350. [Google Scholar] [CrossRef] [PubMed]
- Zaremba-Niedzwiedzka, K.; Caceres, E.F.; Saw, J.H.; Bäckström, D.; Juzokaite, L.; Vancaester, E.; Seitz, K.W.; Anantharaman, K.; Starnawski, P.; Kjeldsen, K.U.; et al. Asgard archaea illuminate the origin of eukaryotic cellular complexity. Nature 2017, 541, 353–358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, C.T.; Hug, L.A.; Thomas, B.C.; Sharon, I.; Castelle, C.J.; Singh, A.; Wilkins, M.J.; Wrighton, K.C.; Williams, K.H.; Banfield, J.F. Unusual biology across a group comprising more than 15% of domain Bacteria. Nature 2015, 523, 208–211. [Google Scholar] [CrossRef] [PubMed]
- Hug, L.A.; Baker, B.J.; Anantharaman, K.; Brown, C.T.; Probst, A.J.; Castelle, C.J.; Butterfield, C.N.; Hernsdorf, A.W.; Amano, Y.; Ise, K.; et al. A new view of the tree of life. Nat. Microbiol. 2016, 1, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Soro, V.; Dutton, L.C.; Sprague, S.V.; Nobbs, A.H.; Ireland, A.J.; Sandy, J.R.; Jepson, M.A.; Micaroni, M.; Splatt, P.R.; Dymock, D.; et al. Axenic culture of a candidate division TM7 bacterium from the human oral cavity and biofilm interactions with other oral bacteria. Appl. Environ. Microbiol. 2014, 80, 6480–6489. [Google Scholar] [CrossRef] [Green Version]
- Dombrowski, N.; Lee, J.-H.; Williams, T.A.; Offre, P.; Spang, A. Genomic diversity, lifestyles and evolutionary origins of DPANN archaea. FEMS Microbiol. Lett. 2019, 366, fnz008. [Google Scholar] [CrossRef] [Green Version]
- Baker, B.J.; Comolli, L.R.; Dick, G.J.; Hauser, L.J.; Hyatt, D.; Dill, B.D.; Land, M.L.; VerBerkmoes, N.C.; Hettich, R.L.; Banfield, J.F. Enigmatic, ultrasmall, uncultivated Archaea. Proc. Natl. Acad. Sci. USA 2010, 107, 8806–8811. [Google Scholar] [CrossRef] [Green Version]
- Deschamps, P.; Zivanovic, Y.; Moreira, D.; Rodriguez-Valera, F.; Lopez-García, P. Pangenome evidence for extensive interdomain horizontal transfer affecting lineage coreandshell genes inuncultured planktonic thaumarchaeota and euryarchaeota. Genome Biol. Evol. 2014, 6, 1549–1563. [Google Scholar] [CrossRef] [Green Version]
- Merhej, V.; Raoult, D. Rhizome of life, catastrophes, sequence exchanges, gene creations, and giant viruses: How microbial genomics challenges Darwin. Front. Cell. Infect. Microbiol. 2012, 2, 113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raoult, D. The post-Darwinist rhizome of life. Lancet 2010, 375, 104–105. [Google Scholar] [CrossRef]
- Caetano-Anollés, G.; Caetano-Anollés, D. An evolutionarily structural universe of protein architecture. Genome Res. 2003, 13, 1563–1571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dagan, T.; Martin, W. The tree of one percent. Genome Biol. 2006, 7, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Deleuze, G. Rhizome: Introduction; Editions de Minuit: Paris, France, 1976; ISBN 9782707301130. [Google Scholar]
- Merhej, V.; Notredame, C.; Royer-Carenzi, M.; Pontarotti, P.; Raoult, D. The rhizome of life: The sympatric Rickettsia felis paradigm demonstrates the random transfer of DNA sequences. Mol. Biol. Evol. 2011, 28, 3213–3223. [Google Scholar] [CrossRef]
- Levasseur, A.; Merhej, V.; Baptiste, E.; Sharma, V.; Pontarotti, P.; Raoult, D. The rhizome of lokiarchaeota illustrates the mosaicity of archaeal genomes. Genome Biol. Evol. 2017, 9, 2635–2639. [Google Scholar] [CrossRef] [Green Version]
- Darwin, C. CLASSICS On the Origin of Species by Means of Natural Selection, or the Preservation of Favoured Races in the Struggle for Life; John Murray: London, UK, 2009. [Google Scholar]
- Raoult, D.; Koonin, E.V. Microbial genomics challenge Darwin. Front. Cell. Infect. Microbiol. 2012, 2, 127. [Google Scholar] [CrossRef] [Green Version]
- Woese, C.R. Bacterial evolution. Microbiol. Rev. 1987, 51, 221–271. [Google Scholar] [CrossRef]
- Woese, C.R.; Fox, G.E. Phylogenetic structure of the prokaryotic domain: The primary kingdoms. Proc. Natl. Acad. Sci. USA 1977, 74, 5088–5090. [Google Scholar] [CrossRef] [Green Version]
- Bapteste, E.; Boucher, Y. Lateral gene transfer challenges principles of microbial systematics. Trends Microbiol. 2008, 16, 200–207. [Google Scholar] [CrossRef]
- Baumgartner, M.; Roffler, S.; Wicker, T.; Pernthaler, J. Letting go: Bacterial genome reduction solves the dilemma of adapting to predation mortality in a substrate-restricted environment. ISME J. 2017, 11, 2258–2266. [Google Scholar] [CrossRef] [Green Version]
- Georgiades, K.; Raoult, D. How microbiology helps define the rhizome of life. Front. Cell. Infect. Microbiol. 2012, 2, 60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gogarten, J.P.; Doolittle, W.F.; Lawrence, J.G. Prokaryotic evolution in light of gene transfer. Mol. Biol. Evol. 2002, 19, 2226–2238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koonin, E.V.; Puigbò, P.; Wolf, Y.I. Comparison of phylogenetic trees and search for a central trend in the “forest of life”. J. Comput. Biol. 2011, 18, 917–924. [Google Scholar] [CrossRef]
- Penny, D. Darwin’s Theory of Descent with Modification, versus the Biblical Tree of Life. PLoS Biol. 2011, 9, e1001096. [Google Scholar] [CrossRef] [PubMed]
- Robbens, S.; Derelle, E.; Ferraz, C.; Wuyts, J.; Moreau, H.; Van De Peer, Y. The Complete Chloroplast and Mitochondrial DNA Sequence of Ostreococcus tauri: Organelle Genomes of the Smallest Eukaryote Are Examples of Compaction. Mol. Biol. Evol. 2007, 24, 956–968. [Google Scholar] [CrossRef] [PubMed]
- Colson, P.; Levasseur, A.; La Scola, B.; Sharma, V.; Nasir, A.; Pontarotti, P.; Caetano-Anollés, G.; Raoult, D. Ancestrality and mosaicism of giant viruses supporting the definition of the fourth TRUC of microbes. Front. Microbiol. 2018, 9, 2668. [Google Scholar] [CrossRef] [Green Version]
- Nelson-Sathi, S.; Sousa, F.L.; Roettger, M.; Lozada-Chávez, N.; Thiergart, T.; Janssen, A.; Bryant, D.; Landan, G.; Schönheit, P.; Siebers, B.; et al. Origins of major archaeal clades correspond to gene acquisitions from bacteria. Nature 2015, 517, 77–80. [Google Scholar] [CrossRef] [Green Version]
- Da Cunha, V.; Gaia, M.; Nasir, A.; Forterre, P. Asgard archaea do not close the debate about the universal tree of life topology. PLOS Genet. 2018, 14, e1007215. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Makarova, K.S.; Huang, W.C.; Wolf, Y.I.; Nikolskaya, A.; Zhang, X.; Cai, M.; Zhang, C.J.; Xu, W.; Luo, Z.; et al. Expanding diversity of asgard archaea and the elusive ancestry of eukaryotes. bioRxiv 2020. [Google Scholar] [CrossRef]
- Macleod, F.; Kindler, G.S.; Wong, H.L.; Chen, R.; Burns, B.P. Asgard archaea: Diversity, function, and evolutionary implications in a range of microbiomes. AIMS Microbiol. 2019, 5, 48–61. [Google Scholar] [CrossRef] [PubMed]
- Koonin, E.V.; Yutin, N. The dispersed archaeal eukaryome and the complex archaeal ancestor of eukaryotes. Cold Spring Harb. Perspect. Biol. 2014, 6. [Google Scholar] [CrossRef] [Green Version]
- López-García, P.; Moreira, D. Cultured Asgard Archaea Shed Light on Eukaryogenesis. Cell 2020, 181, 232–235. [Google Scholar] [CrossRef]
- Karnkowska, A.; Vacek, V.; Zubáčová, Z.; Treitli, S.C.; Petrželková, R.; Eme, L.; Novák, L.; Žárský, V.; Barlow, L.D.; Herman, E.K.; et al. A eukaryote without a mitochondrial organelle. Curr. Biol. 2016, 26, 1274–1284. [Google Scholar] [CrossRef]
- López-García, P.; Moreira, D. The Syntrophy hypothesis for the origin of eukaryotes revisited. Nat. Microbiol. 2020, 5, 655–667. [Google Scholar] [CrossRef] [PubMed]
- Falcón, L.I.; Magallón, S.; Castillo, A. Dating the cyanobacterial ancestor of the chloroplast. ISME J. 2010, 4, 777–783. [Google Scholar] [CrossRef] [Green Version]
- Doolittle, W.F.; Bapteste, E. Pattern pluralism and the Tree of Life hypothesis. Proc. Natl. Acad. Sci. USA 2007, 104, 2043–2049. [Google Scholar] [CrossRef] [Green Version]
- Eme, L.; Spang, A.; Lombard, J.; Stairs, C.W.; Ettema, T.J.G. Erratum: Archaea and the origin of eukaryotes (Nature reviews. Microbiology (2017) 15 12 (711–723)). Nat. Rev. Microbiol. 2018, 16, 120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eme, L.; Spang, A.; Lombard, J.; Stairs, C.W.; Ettema, T.J.G. Archaea and the origin of eukaryotes. Nat. Rev. Microbiol. 2017, 15, 711–723. [Google Scholar] [CrossRef] [PubMed]
- Forterre, P.; Gaïa, M. Giant viruses and the origin of modern eukaryotes. Curr. Opin. Microbiol. 2016, 31, 44–49. [Google Scholar] [CrossRef]
- Feschotte, C.; Gilbert, C. Endogenous viruses: Insights into viral evolution and impact on host biology. Nat. Rev. Genet. 2012, 13, 283–296. [Google Scholar] [CrossRef] [Green Version]
- Nikoh, N.; Tanaka, K.; Shibata, F.; Kondo, N.; Hizume, M.; Shimada, M.; Fukatsu, T. Wolbachia genome integrated in an insect chromosome: Evolution and fate of laterally transferred endosymbiont genes. Genome Res. 2008, 18, 272–280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Callaway, E. Genomes within genomes. Nature 2007, 449, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arbuckle, J.H.; Medveczky, M.M.; Luka, J.; Hadley, S.H.; Luegmayr, A.; Ablashi, D.; Lund, T.C.; Tolar, J.; De Meirleir, K.; Montoya, J.G.; et al. The latent human herpesvirus-6A genome specifically integrates in telomeres of human chromosomes in vivo and in vitro. Proc. Natl. Acad. Sci. USA 2010, 107, 5563–5568. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abi-Rached, L.; Jobin, M.J.; Kulkarni, S.; McWhinnie, A.; Dalva, K.; Gragert, L.; Babrzadeh, F.; Gharizadeh, B.; Luo, M.; Plummer, F.A.; et al. The shaping of modern human immune systems by multiregional admixture with archaic humans. Science 2011, 334, 89–94. [Google Scholar] [CrossRef] [Green Version]
- Lacroix, B.; Citovsky, V. Transfer of DNA from bacteria to eukaryotes. mBio 2016, 7. [Google Scholar] [CrossRef] [Green Version]
- Brueckner, J.; Martin, W.F. Bacterial Genes Outnumber Archaeal Genes in Eukaryotic Genomes. Genome Biol. Evol. 2020, 12, 282–292. [Google Scholar] [CrossRef] [Green Version]
- Stoye, J.P. Koala retrovirus: A genome invasion in real time. Genome Biol. 2006, 7, 241. [Google Scholar] [CrossRef] [Green Version]
- Moniruzzaman, M.; Weinheimer, A.R.; Martinez-Gutierrez, C.A.; Aylward, F.O. Widespread endogenization of giant viruses shapes genomes of green algae. Nature 2020, 588, 141–145. [Google Scholar] [CrossRef] [PubMed]
- Schvarcz, C.R.; Steward, G.F. A giant virus infecting green algae encodes key fermentation genes. Virology 2018, 518, 423–433. [Google Scholar] [CrossRef] [PubMed]
- Moniruzzaman, M.; Martinez-Gutierrez, C.A.; Weinheimer, A.R.; Aylward, F.O. Dynamic genome evolution and complex virocell metabolism of globally-distributed giant viruses. Nat. Commun. 2020, 11, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Filée, J. Multiple occurrences of giant virus core genes acquired by eukaryotic genomes: The visible part of the iceberg? Virology 2014, 466–467, 53–59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chelkha, N.; Hasni, I.; Louazani, A.C.; Levasseur, A.; Scola, B. La Vermamoeba vermiformis CDC-19 draft genome sequence reveals considerable gene trafficking including with candidate phyla radiation and giant viruses. Sci. Rep. 2020, 10, 5928. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balczun, C.; Scheid, P. Free-Living Amoebae as Hosts for and Vectors of Intracellular Microorganisms with Public Health Significance. Viruses 2017, 9, 65. [Google Scholar] [CrossRef]
- Maumus, F.; Quesneville, H. Ancestral repeats have shaped epigenome and genome composition for millions of years in Arabidopsis thaliana. Nat. Commun. 2014, 5, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chelkha, N.; Levasseur, A.; Pontarotti, P.; Raoult, D.; La Scola, B.; Colson, P. A Phylogenomic Study of Acanthamoeba polyphaga Draft Genome Sequences Suggests Genetic Exchanges With Giant Viruses. Front. Microbiol. 2018, 9, 2098. [Google Scholar] [CrossRef] [Green Version]
- Blanc, G.; Gallot-Lavallée, L.; Maumus, F. Provirophages in the Bigelowiella genome bear testimony to past encounters with giant viruses. Proc. Natl. Acad. Sci. USA 2015, 112, E5318–E5326. [Google Scholar] [CrossRef] [Green Version]
- Fischer, M.G.; Hackl, T. Host genome integration and giant virus-induced reactivation of the virophage mavirus. Nature 2016, 540, 288–291. [Google Scholar] [CrossRef]
- Aherfi, S.; Belhaouari, D.B.; Pinault, L.; Baudoin, J.P.; Decloquement, P.; Abrahao, J.; Colson, P.; Levasseur, A.; Lamb, D.C.; Chabriere, E.; et al. Tricarboxylic acid cycle and proton gradient in Pandoravirus massiliensis: Is it still a virus? bioRxiv 2020. [Google Scholar] [CrossRef]
- Fischer, M.G.; Allen, M.J.; Wilson, W.H.; Suttle, C.A. Giant virus with a remarkable complement of genes infects marine zooplankton. Proc. Natl. Acad. Sci. USA 2010, 107, 19508–19513. [Google Scholar] [CrossRef] [Green Version]
- Deeg, C.M.; Chow, C.E.T.; Suttle, C.A. The kinetoplastid-infecting bodo saltans virus (Bsv), a window into the most abundant giant viruses in the sea. eLife 2018, 7. [Google Scholar] [CrossRef]
- Boyer, M.; Madoui, M.-A.; Gimenez, G.; La Scola, B.; Raoult, D. Phylogenetic and Phyletic Studies of Informational Genes in Genomes Highlight Existence of a 4th Domain of Life Including Giant Viruses. PLoS ONE 2010, 5, e15530. [Google Scholar] [CrossRef]
- Fraser, C.M.; Eisen, J.A.; Salzberg, S.L. Microbial genome sequencing. Nature 2000, 406, 799–803. [Google Scholar] [CrossRef]
- Bordenstein, S.R.; Reznikoff, W.S. Mobile DNA in obligate intracellular bacteria. Nat. Rev. Microbiol. 2005, 3, 688–699. [Google Scholar] [CrossRef] [PubMed]
- Ogata, H.; La Scola, B.; Audic, S.; Renesto, P.; Blanc, G.; Robert, C.; Fournier, P.-E.; Claverie, J.-M.; Raoult, D. Genome Sequence of Rickettsia bellii Illuminates the Role of Amoebae in Gene Exchanges between Intracellular Pathogens. PLoS Genet. 2006, 2, e76. [Google Scholar] [CrossRef]
- Shintani, M. The behavior of mobile genetic elements (MGEs) in different environments. Biosci. Biotechnol. Biochem. 2017, 81, 854–862. [Google Scholar] [CrossRef] [Green Version]
- Ochman, H.; Lerat, E.; Daubin, V. Examining bacterial species under the specter of gene transfer and exchange. Proc. Natl. Acad. Sci. USA 2005, 102, 6595–6599. [Google Scholar] [CrossRef] [Green Version]
- Dokland, T. Molecular Piracy: Redirection of Bacteriophage Capsid Assembly by Mobile Genetic Elements. Viruses 2019, 11, 1003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bitto, N.J.; Chapman, R.; Pidot, S.; Costin, A.; Lo, C.; Choi, J.; D’Cruze, T.; Reynolds, E.C.; Dashper, S.G.; Turnbull, L.; et al. Bacterial membrane vesicles transport their DNA cargo into host cells. Sci. Rep. 2017, 7, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Anderson, M.T.; Seifert, H.S. Opportunity and means: Horizontal gene transfer from the human host to a bacterial pathogen. mBio 2011, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castelle, C.J.; Brown, C.T.; Anantharaman, K.; Probst, A.J.; Huang, R.H.; Banfield, J.F. Biosynthetic capacity, metabolic variety and unusual biology in the CPR and DPANN radiations. Nat. Rev. Microbiol. 2018, 16, 629–645. [Google Scholar] [CrossRef] [PubMed]
- Shao, R.; Kirkness, E.F.; Barker, S.C. The single mitochondrial chromosome typical of animals has evolved into 18 minichromosomes in the human body louse, Pediculus humanus. Genome Res. 2009, 19, 904–912. [Google Scholar] [CrossRef] [Green Version]
- Esser, C.; Martin, W.; Dagan, T. The origin of mitochondria in light of a fluid prokaryotic chromosome model. Biol. Lett. 2007, 3, 180–184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deusch, O.; Landan, G.; Roettger, M.; Gruenheit, N.; Kowallik, K.V.; Allen, J.F.; Martin, W.; Dagan, T. Genes of cyanobacterial origin in plant nuclear genomes point to a heterocyst-forming plastid ancestor. Mol. Biol. Evol. 2008, 25, 748–761. [Google Scholar] [CrossRef]
- Timmis, J.N.; Ayliff, M.A.; Huang, C.Y.; Martin, W. Endosymbiotic gene transfer: Organelle genomes forge eukaryotic chromosomes. Nat. Rev. Genet. 2004, 5, 123–135. [Google Scholar] [CrossRef]
- Zhaxybayeva, O.; Gogarten, J.P. Cladogenesis, coalescence and the evolution of the three domains of life. Trends Genet. 2004, 20, 182–187. [Google Scholar] [CrossRef]
- Peretó, J.; López-García, P.; Moreira, D. Ancestral lipid biosynthesis and early membrane evolution. Trends Biochem. Sci. 2004, 29, 469–477. [Google Scholar] [CrossRef]
- Viezens, J.; Arvard, M. Simultaneous presence of two different copies of the 16S rRNA gene in Bartonella henselae. Microbiology 2008, 154, 2881–2886. [Google Scholar] [CrossRef] [Green Version]
- Seemann, T. Prokka: Rapid prokaryotic genome annotation. Bioinformatics 2014, 30, 2068–2069. [Google Scholar] [CrossRef]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 2014, 12, 59–60. [Google Scholar] [CrossRef]
- Lechner, M.; Findeiß, S.; Steiner, L.; Marz, M.; Stadler, P.F.; Prohaska, S.J. Proteinortho: Detection of (Co-)orthologs in large-scale analysis. BMC Bioinform. 2011, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nasir, A.; Sun, F.-J.; Kim, K.M.; Caetano-Anollés, G. Untangling the origin of viruses and their impact on cellular evolution. Ann. N. Y. Acad. Sci. 2015, 1341, 61–74. [Google Scholar] [CrossRef] [PubMed]
- Méheust, R.; Burstein, D.; Castelle, C.J.; Banfield, J.F. The distinction of CPR bacteria from other bacteria based on protein family content. Nat. Commun. 2019, 10, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bokhari, R.H.; Amirjan, N.; Jeong, H.; Kim, K.M.; Caetano-Anollés, G.; Nasir, A.; Bapteste, E. Bacterial Origin and Reductive Evolution of the CPR Group. Genome Biol. Evol. 2020, 12, 103–121. [Google Scholar] [CrossRef] [Green Version]
- Tian, R.; Ning, D.; He, Z.; Zhang, P.; Spencer, S.J.; Gao, S.; Shi, W.; Wu, L.; Zhang, Y.; Yang, Y.; et al. Small and mighty: Adaptation of superphylum Patescibacteria to groundwater environment drives their genome simplicity. Microbiome 2020, 8, 51. [Google Scholar] [CrossRef] [Green Version]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [Green Version]
- Sharma, V.; Colson, P.; Pontarotti, P.; Raoult, D. Mimivirus inaugurated in the 21st century the beginning of a reclassification of viruses. Curr. Opin. Microbiol. 2016, 31, 16–24. [Google Scholar] [CrossRef]
- Fournier, G.P.; Poole, A.M. A briefly argued case that Asgard Archaea are part of the eukaryote tree. Front. Microbiol. 2018, 9, 15. [Google Scholar] [CrossRef]
- Sharma, V.; Colson, P.; Chabrol, O.; Scheid, P.; Pontarotti, P.; Raoult, D. Welcome to pandoraviruses at the “Fourth TRUC” club. Front. Microbiol. 2015, 6. [Google Scholar] [CrossRef] [Green Version]
- Sharma, V.; Colson, P.; Chabrol, O.; Pontarotti, P.; Raoult, D. Pithovirus sibericum, a new bona fide member of the “Fourth TRUC” club. Front. Microbiol. 2015, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moliner, C.; Fournier, P.-E.; Raoult, D. Genome analysis of microorganisms living in amoebae reveals a melting pot of evolution. FEMS Microbiol. Rev. 2010, 34, 281–294. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Term | Definition |
|---|---|
| Archaeon | Formerly known as archaebacteria, archaea are unicellular microorganisms measuring between 2 and 15 µm that were initially detected in extreme environments. Archaea have been classified in a separate domain from bacteria and eukaryotes. These organisms have transcription and translation machineries like those of eukaryotes. |
| Asgard | This superphylum consists of anaerobic prokaryotic microorganisms classified in the domain Archaea. The members of this superphylum are assumed to have an ancestor that is intermediate to archaea and eukaryotes. These organisms are considered to be most genetically similar to eukaryotes with a genome harboring some signature eukaryotic genes. |
| Bacterium | These unicellular, anucleate (prokaryotic) microorganisms have mainly a single chromosomal genome, different cell shapes and a physical size of between 1 and 10 µm. However, very large bacteria, attaining 750 µm in diameter, and very small bacteria (300 nm) have been described. Some of these organisms may host circular DNA fragments, known as plasmids. Bacteria do not have energetic organelles (mitochondria and chloroplasts); but they produce ATP by generating a proton gradient across their cell membranes through glycolysis. However, some bacteria (cyanobacteria) can use light to generate a proton gradient. Bacteria are divided into different groups according to their cell membrane morphology/Structure: Gram-positive bacteria (also known as monoderm bacteria) have a cell membrane primarily composed of peptidoglycan that retains crystal violet dye after Gram staining; Gram-negative bacteria (so called diderm bacteria) do not retain crystal violet dye after Gram staining because they have a thinner peptidoglycan layer surrounded by an outer membrane containing lipopolysaccharide; Gram-variable bacteria stain irregularly according to the peptidoglycan quantity during cell growth and Gram-indeterminate bacteria, such as Mycobacterium spp., which have a waxy layer on its surface preventing them to respond well to Gram staining. Finally, some bacteria lack a cell membrane, like Mycoplasma spp. |
| Candidate (Candidatus) | The term “candidate” refers to an undescribed species or to a single isolate of unknown species for which there is insufficient information for it to be identified as a new species according to the International Code of Bacterial Nomenclature. This term can be assigned also to uncultured prokaryotic organisms obtained by metagenomic analyses. |
| Candidate Phyla Radiation (CPR) | These ultra-small microbes have a physical size between 100 and 300 nm and a small genome (<1 megabase pair). CPR members are unable to multiply on their own and are dependent on an exo-symbiotic or parasitic interaction with bacteria. These organisms were first detected by metagenomics analyses. CPR members have a unique, characteristic 16S ribosomal RNA gene. |
| Commensalism | Eating at the same table: This term implies a biological interaction between two organisms without any advantage or disadvantage. |
| Eukaryotes | These unicellular or pluri-cellular (micro) organism have a nucleus surrounded by a membrane. The size of individual eukaryotic cells is between 10 and 50 µm. These organisms are characterized by their genomes, which are composed of several chromosomes, and some eukaryotes possess mitochondria and/or chloroplasts (plant cells) and other organelles (e.g., endoplasmic reticulum and Golgi apparatus). Eukaryote is a major branch in the tree of life alongside bacteria and archaea. |
| Genome mosaicism | This term refers to the presence in a genome of sequences with different evolutionary histories, including some that may be currently unknown. |
| Giant virus | These viruses, formerly known as nucleocytoplasmic large DNA viruses, belong to the phylum Nucleocytoviricota and the order Megavirales. The viruses have a larger physical size (>200 nm, non-filterable using Chamberland filter, visible using a light microscope) and genome size (>300,000 base pairs) in comparison to classical viruses; their size is comparable to those of microbes. The giant-virus virion contains both DNA and RNA, which includes messenger RNAs and transfer RNA, and the genome often harbors genes encoding translation components and a specific mobilome, an energy production system in some cases, which are absent in classical viruses’ genomes. Giant viruses comprise a monophyletic group with an ancient origin that may predate that of eukaryotes, and these viruses have a broad host range. |
| Intracellular bacteria | These bacteria (also known as obligate intracellular parasites) are unable to multiply independently and require host eukaryotic cells to develop and reproduce, like Rickettsia spp. These microbes are considered facultative intracellular bacteria, if they have the ability to grow inside and outside eukaryotic cells independently like Bartonella henselae and Listeria monocytogenes. |
| Lateral sequence transfer | This term refers to the transfer of genetic information (regardless of whether sequences include full-length ORFs or partial ORFs) between the genomes of a donor species and a recipient species, regardless of their evolutionary relationship. |
| LUCA (Last Universal Common Ancestor) | This organism is considered to be the starting point of life on earth. A virtual and controversial entity that would represent the common ancestor of all (micro) organisms. |
| Microbe—Microorganism | These organisms are visible by microscopy (due to a size >200 nm). |
| Mitochondria | These eukaryotic organelles possess the characteristics of prokaryotic cells, ranging in size from 0.5 to 1 µm. Mitochondria are present in most eukaryotic cells and produce ATP, providing energy for the cell. Genetically, mitochondria are of alpha-proteobacterial origin. |
| Chloroplast | Eukaryotic organelle of plant and algal cells with a size ranging from 1 to 2 µm conducts photosynthesis. Genetically, Chloroplast are of Cyanobacteria origin. |
| Mutualism | This term implies a lasting association between two dependent organisms with mutual benefit and no negative effect on either organism. Both organisms can reproduce, feed and multiply. |
| Nanoarchaeon | Very small archaea (measuring between 150 and 500 nm) are characterized by their reduced genome and limited biosynthetic and metabolic capacities, and this organism depends on exo-symbiotic or parasitic interactions with other archaea. Nano-archaea were detected for the first time by metagenomics. |
| ORFans | These genes have unknown evolutionary identification and origin unique to a given species. The genes may be newly created, generated by the fusion of several sequences, or produced by sequence degradation. |
| Parasitism | This function is carried out by an organism that is dependent on a host to develop and exploits the host to multiply, feed and reproduce. Parasitism exerts a negative effect on the host. |
| Rhizome | Rhizomes are a representation of the evolutionary history of a genome based on the totality of its sequence; in contrast, phylogenetic trees are based on single genes. Rhizomes consider sequences resulting from such phenomena as transfers, recombination, fusions, degradation, and de novo creation. Rhizomes consider ORFans, as well. |
| Symbiont | These organisms need other organisms to live and multiply. |
| Symbiosis | In this interaction, two organisms live together, and both benefit from the association. |
| TRUC | An abbreviation of Things Resisting Uncompleted Classification: A new term (which is French for stuff) coined in 2013 to designate major groups of microbes (bacteria, archaea, eukaryota and giant viruses) other than based on ribosomal genes (which define domains). TRUC was first used to designate giant viruses. |
| Tree of Life | This phylogenetic representation groups together currently living cells and resembles a tree with branches and nodes. This model is currently a matter of debate. |
| Virus (classical) | Viruses are compulsory parasites that are unable to multiply independently, ultra-filterable using filters with 0.2 µm pore sizes, and not visible by light microscopy. The viral genome is composed of either DNA or RNA, encodes capsid protein(s) but lacks genes encoding translation machinery and an energy production system, as well as ribosomal genes. Viruses reproduce from their nucleic acid inside the host cell only. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ibrahim, A.; Colson, P.; Merhej, V.; Zgheib, R.; Maatouk, M.; Naud, S.; Bittar, F.; Raoult, D. Rhizomal Reclassification of Living Organisms. Int. J. Mol. Sci. 2021, 22, 5643. https://doi.org/10.3390/ijms22115643
Ibrahim A, Colson P, Merhej V, Zgheib R, Maatouk M, Naud S, Bittar F, Raoult D. Rhizomal Reclassification of Living Organisms. International Journal of Molecular Sciences. 2021; 22(11):5643. https://doi.org/10.3390/ijms22115643
Chicago/Turabian StyleIbrahim, Ahmad, Philippe Colson, Vicky Merhej, Rita Zgheib, Mohamad Maatouk, Sabrina Naud, Fadi Bittar, and Didier Raoult. 2021. "Rhizomal Reclassification of Living Organisms" International Journal of Molecular Sciences 22, no. 11: 5643. https://doi.org/10.3390/ijms22115643
APA StyleIbrahim, A., Colson, P., Merhej, V., Zgheib, R., Maatouk, M., Naud, S., Bittar, F., & Raoult, D. (2021). Rhizomal Reclassification of Living Organisms. International Journal of Molecular Sciences, 22(11), 5643. https://doi.org/10.3390/ijms22115643