
Pro-Inflammatory Cytokines: Potential Links between the Endocannabinoid System and the Kynurenine Pathway in Depression

 ,
,  , and
, and 
Abstract
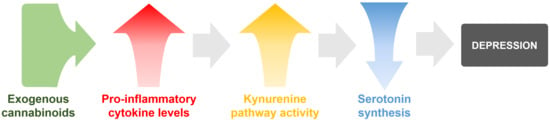
:
1. Introduction
2. Background
2.1. Pro-Inflammatory Cytokines
2.2. The Endocannabinoid System
2.3. The Kynurenine Pathway
2.4. The Serotonin and Inflammatory Hypothesis of Depression: Possible Links between ECS and KP in Depression?
3. Cannabinoids That Enhance Pro-Inflammatory Cytokine Levels
3.1. Natural Cannabinoids (Cannabis, THC, and CBD)
3.2. Semi- and Fully Synthetic Cannabinoids

{kind=link}
{kind=link}
{kind=link}
| Cannabinoid | Cytokine | Studied Sample | Ref. |
|---|---|---|---|
| THC | IFNγ | PMBC | [100,101] |
| TNFα | PMBC | [101] | |
| Adult mouse peripheral macrophage | [113] | ||
| Adult mouse hippocampus and hypothalamus | [114] | ||
| Female adol. rat microglia PFC | [115] | ||
| IL-1 | Microglia | [107] | |
| Adult mouse peripheral macrophage | [113] | ||
| Adult mouse hippocampus and hypothalamus | [114] | ||
| IL-6 | PMBC | [101] | |
| IL-8 | Eosinophilic leukemia cell line and HTLV-1 positive B cell line | [106] | |
| CBD | IFNγ | PMBC | [101] |
| IL-8 | Eosinophilic leukemia cell line | [106] | |
| Cannabis | IL-1, IL-6, IL-8 | Serum from patients with CUD | [117] |
| CP55940 | TNFα | HL-60 transfected with CB2R; mouse lung | [119,120] |
| IL-1 | Mouse lung | [119] | |
| IL-6 | Mouse lung | [119] | |
| NMSC | IL-1β, IL-6 | RAW264.7 macrophage | [123] |
| CP-47497-C8 | TNFα, IL-6 | PMBC | [122] |
| HU308 | IL-6 | Human primary leukocytes | [43] |
| Rimonabant | TNFα | rat hippocampus | [125] |
| IL-6 | rat mPFC | [125] |
4. Pro-Inflammatory Cytokines Parallelly Up-Regulated with the KP in Neuroinflammation and/or Depression
4.1. IFN-γ
4.2. IL-1
4.3. IL-6
4.4. IL-8
4.5. TNFα
| Cytokine | KP Enzyme or Metabolite | Studied Sample | Comment | Ref. |
|---|---|---|---|---|
| IFNγ | IDO mRNA ↑ | mouse hippocampus | galectin-9 synergism | [138] |
| mouse hippocampus | dexamethasone, corticosterone and aldosterone synergism | [139] | ||
| rat cortex | CMS model | [141] | ||
| KAT II mRNA ↓ | rat cortex | CMS model | [141] | |
| KYN, 3-HK, KYNA ↑ | mouse plasma | CSD model | [140] | |
| IL-1 | IDO mRNA ↑ | primary macrophage | [142] | |
| IDO activity ↑ | THP1 monocytic leukemia cell line | IFNγ pretreatment | [142] | |
| IDO, KMO mRNA ↑ | CX3CR1 K.O. mouse microglia | [143] | ||
| KMO mRNA ↑ | mouse microglia | BCG model | [144] | |
| IDO, KMO mRNA ↑ | murine microglia | LPS-induction | [145] | |
| KMO mRNA ↑ | mouse brain | nerve injury-induced depression | [146] | |
| IDO, KMO, KYNU mRNA, KYN ↑ | human hippocampal progenitor cells | [148] | ||
| IDO1 mRNA ↑ | rat hippocampus | coexisting chronic temporal lobe epilepsy and depressive behavior | [149] | |
| IDO, KMO and KYNU activity ↑ | mouse hippocampus | HMGB1 induced depressive like behavior model | [151] | |
| IL-6 | Trp ↓ | human serum | in patients with depression | [152] |
| KYN ↑ | female human serum | in early puerperium associated with anxiety and depression | [153] | |
| QUIN/KYNA ratio ↑ | human plasma | in patients with unipolar treatment-resistant depression | [154] | |
| KYN and QUIN ↑ | human plasma | did not mediate the correlation between cytokines and depressed mood | [80] | |
| QUIN ↑ | female human plasma | women with PPD | [155] | |
| KYN/Trp ratio, KYN ↑ | plasma from frailty patients | may explain high prevalence depression in frailty patients | [156] | |
| QUIN ↑ KYNA ↓ | human CSF | in suicide attempters | [159] | |
| IDO1, KMO mRNA ↑ | murine microglia | following LPS-stimulation | [145] | |
| IDO1 protein ↑ | rat hippocampus | through JAK/STAT pathway | [160] | |
| IDO1 mRNA ↑ | rat hippocampus | coexisting chronic temporal lobe epilepsy and depressive behavior | [149] | |
| IDO1 protein ↑ | rat hippocampus | ovariectomy-induced depression model | [161] | |
| IDO, KMO mRNA ↑ | rat hpc., ctx., and cultured glia cells | [162] | ||
| IL-8 | KYN/Trp quotient ↑ | human plasma | IFNα-induced depressive symptoms | [163] |
| female human serum | in early puerperium associated with anxiety and depression | [153] | ||
| TNFα | IDO ↑ | human serum | in MDD patients | [164] |
| KYN, 3-HK, KYNA ↑ | mouse plasma | CSD mouse model | [140] | |
| KYN and QUIN ↑ | human plasma | did not mediate the correlation between cytokines and depressed mood | [80] | |
| QUIN ↑ | female human plasma | women with PPD | [155] | |
| KYN, KYN/Trp ratio ↑ | plasma | associated with enhanced depression, anhedonia, and treatment nonresponse | [165] | |
| KYN, KYNA, QUIN ↑ | CSF | in unmedicated depressed patients | [165] | |
| KYN/Trp ratio, KYN ↑ | plasma from frailty patients | may explain high prevalence depression in frailty patients | [156] | |
| IDO, HAAO mRNA ↑ | mouse brain | BCG model | [166] | |
| IDO activity ↑ | mouse microglia cells | BCG model | [167] | |
| IDO, KMO mRNA ↑ | rat hpc., ctx., and cultured glia cells | [162] | ||
| IDO1, KMO mRNA ↑ | murine microglia | following LPS-stimulation | [145] | |
| IDO, KMO and KYNU activity ↑ | mouse hippocampus | HMGB1 induced depressive like behavior model | [151] |
5. Summary and Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- GBD 2017 Disease and Injury Incidence and Prevalence Collaborators. Global, regional, and national incidence, prevalence, and years lived with disability for 354 diseases and injuries for 195 countries and territories, 1990–2017: A systematic analysis for the Global Burden of Disease Study 2017. Lancet 2018, 392, 1789–1858. [Google Scholar] [CrossRef] [Green Version]
- Volkow, N.D. The reality of comorbidity: Depression and drug abuse. Biol. Psychiatry 2004, 56, 714–717. [Google Scholar] [CrossRef] [Green Version]
- Seely, K.A.; Lapoint, J.; Moran, J.H.; Fattore, L. Spice drugs are more than harmless herbal blends: A review of the pharmacology and toxicology of synthetic cannabinoids. Prog. Neuro Psychopharmacol. Biol. Psychiatry 2012, 39, 234–243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Auwärter, V.; Dresen, S.; Weinmann, W.; Müller, M.; Pütz, M.; Ferreirós, N. “Spice” and other herbal blends: Harmless incense or cannabinoid designer drugs? J. Mass Spectrom. 2009, 44, 832–837. [Google Scholar] [CrossRef] [PubMed]
- Stinson, F.S.; Ruan, W.J.; Pickering, R.; Grant, B.F. Cannabis use disorders in the USA: Prevalence, correlates and co-morbidity. Psychol. Med. 2006, 36, 1447–1460. [Google Scholar] [CrossRef] [PubMed]
- Dierker, L.; Selya, A.; Lanza, S.; Li, R.; Rose, J. Depression and marijuana use disorder symptoms among current marijuana users. Addict. Behav. 2018, 76, 161–168. [Google Scholar] [CrossRef]
- Armenian, P.; Darracq, M.; Gevorkyan, J.; Clark, S.; Kaye, B.; Brandehoff, N.P. Intoxication from the novel synthetic cannabinoids AB-PINACA and ADB-PINACA: A case series and review of the literature. Neuropharmacology 2018, 134, 82–91. [Google Scholar] [CrossRef]
- Lev-Ran, S.; Roerecke, M.; Le Foll, B.; George, T.P.; McKenzie, K.; Rehm, J. The association between cannabis use and depression: A systematic review and meta-analysis of longitudinal studies. Psychol. Med. 2014, 44, 797–810. [Google Scholar] [CrossRef]
- Richter, L.; Pugh, B.S.; Ball, S.A. Assessing the risk of marijuana use disorder among adolescents and adults who use marijuana. Am. J. Drug Alcohol Abus. 2017, 43, 247–260. [Google Scholar] [CrossRef] [PubMed]
- Mensen, V.T.; Vreeker, A.; Nordgren, J.; Atkinson, A.; de la Torre, R.; Farré, M.; Ramaekers, J.G.; Brunt, T.M. Psychopathological symptoms associated with synthetic cannabinoid use: A comparison with natural cannabis. Psychopharmacology 2019, 236, 2677–2685. [Google Scholar] [CrossRef] [Green Version]
- Evren, C.; Bozkurt, M. Synthetic Cannabinoids: Crisis of The Decade. Dusunen Adam J. Psychiatry Neurol. Sci. 2013, 26, 1. [Google Scholar]
- Nagy-Grócz, G.; Zádor, F.; Dvorácskó, S.; Bohár, Z.; Benyhe, S.; Tömböly, C.; Párdutz, Á.; Vécsei, L. Interactions between the Kynurenine and the Endocannabinoid System with Special Emphasis on Migraine. Int. J. Mol. Sci. 2017, 18, 1617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zádor, F.; Nagy-Grócz, G.; Kekesi, G.; Dvorácskó, S.; Szűcs, E.; Tömböly, C.; Horvath, G.; Benyhe, S.; Vécsei, L. Kynurenines and the Endocannabinoid System in Schizophrenia: Common Points and Potential Interactions. Molecules 2019, 24, 3709. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zádor, F.; Nagy-Grócz, G.; Dvorácskó, S.; Bohár, Z.; Cseh, E.K.; Zádori, D.; Párdutz, Á.; Szűcs, E.; Tömböly, C.; Borsodi, A.; et al. Long-term systemic administration of kynurenic acid brain region specifically elevates the abundance of functional CB1 receptors in rats. Neurochem. Int. 2020, 138, 104752. [Google Scholar] [CrossRef]
- Beggiato, S.; Borelli, A.C.; Tomasini, M.C.; Morgano, L.; Antonelli, T.; Tanganelli, S.; Cuomo, V.; Ferraro, L. Long-lasting alterations of hippocampal GABAergic neurotransmission in adult rats following perinatal Δ9-THC exposure. Neurobiol. Learn. Mem. 2017, 139, 135–143. [Google Scholar] [CrossRef] [PubMed]
- Secci, M.E.; Mascia, P.; Sagheddu, C.; Beggiato, S.; Melis, M.; Borelli, A.C.; Tomasini, M.C.; Panlilio, L.V.; Schindler, C.W.; Tanda, G.; et al. Astrocytic Mechanisms Involving Kynurenic Acid Control Δ9-Tetrahydrocannabinol-Induced Increases in Glutamate Release in Brain Reward-Processing Areas. Mol. Neurobiol. 2018, 56, 3563–3575. [Google Scholar] [CrossRef] [PubMed]
- Beggiato, S.; Ieraci, A.; Tomasini, M.C.; Schwarcz, R.; Ferraro, L. Prenatal THC exposure raises kynurenic acid levels in the prefrontal cortex of adult rats. Prog. Neuro Psychopharmacol. Biol. Psychiatry 2020, 100, 109883. [Google Scholar] [CrossRef] [PubMed]
- Colín-González, A.L.; Aguilera, G.; Santamaría, A. Cannabinoids: Glutamatergic Transmission and Kynurenines. Adv. Neurobiol. 2016, 12, 173–198. [Google Scholar]
- Dinarello, C.A. Proinflammatory Cytokines. Chest 2000, 118, 503–508. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.-M.; An, J. Cytokines, inflammation, and pain. Int. Anesthesiol. Clin. 2007, 45, 27–37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, Q.-Q.; Yu, J. Inflammation: A mechanism of depression? Neurosci. Bull. 2014, 30, 515–523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brooks, A.J.; Dehkhoda, F.; Kragelund, B.B. Cytokine Receptors; Springer: Cham, Switzerland, 2017; pp. 1–29. [Google Scholar]
- Kronfol, Z.; Remick, D.G. Cytokines and the Brain: Implications for Clinical Psychiatry. Am. J. Psychiatry 2000, 157, 683–694. [Google Scholar] [CrossRef]
- Huang, W.-J.; Chen, W.-W.; Zhang, X. Endocannabinoid system: Role in depression, reward and pain control (Review). Mol. Med. Rep. 2016, 14, 2899–2903. [Google Scholar] [CrossRef] [Green Version]
- Vinod, K.Y.; Hungund, B.L. Role of the endocannabinoid system in depression and suicide. Trends Pharmacol. Sci. 2006, 27, 539–545. [Google Scholar] [CrossRef]
- Patel, S.; Hillard, C.J. Role of endocannabinoid signaling in anxiety and depression. Curr. Top. Behav. Neurosci. 2009, 1, 347–371. [Google Scholar] [PubMed] [Green Version]
- Valverde, O.; Torrens, M. CB1 receptor-deficient mice as a model for depression. Neuroscience 2012, 204, 193–206. [Google Scholar] [CrossRef]
- Battista, N.; Di Tommaso, M.; Bari, M.; Maccarrone, M. The endocannabinoid system: An overview. Front. Behav. Neurosci. 2012, 6, 9. [Google Scholar] [CrossRef] [Green Version]
- Rodríguez de Fonseca, F.; Del Arco, I.; Bermudez-Silva, F.J.; Bilbao, A.; Cippitelli, A.; Navarro, M. The endocannabinoid system: Physiology and pharmacology. Alcohol Alcohol. 2005, 40, 2–14. [Google Scholar] [CrossRef] [PubMed]
- Bisogno, T. Endogenous cannabinoids: Structure and metabolism. J. Neuroendocr. 2008, 20, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Basavarajappa, B.S. Critical enzymes involved in endocannabinoid metabolism. Protein Pept. Lett. 2007, 14, 237–246. [Google Scholar] [CrossRef] [PubMed]
- Busquets-Garcia, A.; Bains, J.; Marsicano, G. CB1 Receptor Signaling in the Brain: Extracting Specificity from Ubiquity. Neuropsychopharmacology 2018, 43, 4–20. [Google Scholar] [CrossRef]
- Howlett, A.C.; Abood, M.E. CB1 and CB2 Receptor Pharmacology. Adv. Pharmacol. 2017, 80, 169–206. [Google Scholar]
- Pertwee, R.G. The pharmacology of cannabinoid receptors and their ligands: An overview. Int. J. Obes. 2006, 30 (Suppl. 1), S13–S18. [Google Scholar] [CrossRef] [Green Version]
- Pertwee, R.G.; Howlett, A.C.; Abood, M.E.; Alexander, S.P.H.; Di Marzo, V.; Elphick, M.R.; Greasley, P.J.; Hansen, H.S.; Kunos, G.; Mackie, K.; et al. International Union of Basic and Clinical Pharmacology. LXXIX. Cannabinoid Receptors and Their Ligands: Beyond CB1 and CB2. Pharmacol. Rev. 2010, 62, 588–631. [Google Scholar] [CrossRef] [Green Version]
- Demuth, D.G.; Molleman, A. Cannabinoid signalling. Life Sci. 2006, 78, 549–563. [Google Scholar] [CrossRef]
- Bidaut-Russell, M.; Devane, W.A.; Howlett, A.C. Cannabinoid receptors and modulation of cyclic AMP accumulation in the rat brain. J. Neurochem. 1990, 55, 21–26. [Google Scholar] [CrossRef]
- Howlett, A.C.; Barth, F.; Bonner, T.I.; Cabral, G.; Casellas, P.; Devane, W.A.; Felder, C.C.; Herkenham, M.; Mackie, K.; Martin, B.R.; et al. International Union of Pharmacology. XXVII. Classification of cannabinoid receptors. Pharmacol. Rev. 2002, 54, 161–202. [Google Scholar] [CrossRef]
- Maccarrone, M.; Bab, I.; Bíró, T.; Cabral, G.A.; Dey, S.K.; Di Marzo, V.; Konje, J.C.; Kunos, G.; Mechoulam, R.; Pacher, P.; et al. Endocannabinoid signaling at the periphery: 50 years after THC. Trends Pharmacol. Sci. 2015, 36, 277–296. [Google Scholar] [CrossRef] [Green Version]
- Pertwee, R.G. Cannabinoid receptors and pain. Prog. Neurobiol. 2001, 63, 569–611. [Google Scholar] [CrossRef]
- Pagotto, U.; Marsicano, G.; Cota, D.; Lutz, B.; Pasquali, R. The emerging role of the endocannabinoid system in endocrine regulation and energy balance. Endocr. Rev. 2006, 27, 73–100. [Google Scholar] [CrossRef]
- Chen, D.; Gao, M.; Gao, F.; Su, Q.; Wu, J. Brain cannabinoid receptor 2: Expression, function and modulation. Acta Pharmacol. Sin. 2017, 38, 312–316. [Google Scholar] [CrossRef] [PubMed]
- Saroz, Y.; Kho, D.T.; Glass, M.; Graham, E.S.; Grimsey, N.L. Cannabinoid Receptor 2 (CB2) Signals via G-alpha-s and Induces IL-6 and IL-10 Cytokine Secretion in Human Primary Leukocytes. bioRxiv 2019, 2, 414–428. [Google Scholar] [CrossRef] [Green Version]
- Atwood, B.K.; Mackie, K. CB2: A cannabinoid receptor with an identity crisis. Br. J. Pharmacol. 2010, 160, 467–479. [Google Scholar] [CrossRef] [Green Version]
- Walsh, K.B.; Andersen, H.K. Molecular Pharmacology of Synthetic Cannabinoids: Delineating CB1 Receptor-Mediated Cell Signaling. Int. J. Mol. Sci. 2020, 21, 6115. [Google Scholar] [CrossRef] [PubMed]
- Potts, A.J.; Cano, C.; Thomas, S.H.L.; Hill, S.L. Synthetic cannabinoid receptor agonists: Classification and nomenclature. Clin. Toxicol. 2020, 58, 82–98. [Google Scholar] [CrossRef]
- Muccioli, G.G.; Lambert, D.M. Current knowledge on the antagonists and inverse agonists of cannabinoid receptors. Curr. Med. Chem. 2005, 12, 1361–1394. [Google Scholar] [CrossRef] [Green Version]
- Fattore, L. Synthetic Cannabinoids—Further Evidence Supporting the Relationship Between Cannabinoids and Psychosis. Biol. Psychiatry 2016, 79, 539–548. [Google Scholar] [CrossRef]
- Brents, L.K.; Prather, P.L. The K2/Spice Phenomenon: Emergence, identification, legislation and metabolic characterization of synthetic cannabinoids in herbal incense products. Drug Metab. Rev. 2014, 46, 72. [Google Scholar] [CrossRef] [Green Version]
- Volkow, N.D.; Baler, R.D.; Compton, W.M.; Weiss, S.R.B. Adverse Health Effects of Marijuana Use. N. Engl. J. Med. 2014, 370, 2219–2227. [Google Scholar] [CrossRef] [Green Version]
- Serap Akdeniz, G.; Can Sait, S. Cannabinoid Use and Depression: Comparison of Natural and Synthetic Cannabinoids. Clin. Med. Rev. Case Rep. 2020, 7, 7. [Google Scholar] [CrossRef]
- Castaneto, M.S.; Gorelick, D.A.; Desrosiers, N.A.; Hartman, R.L.; Pirard, S.; Huestis, M.A. Synthetic cannabinoids: Epidemiology, pharmacodynamics, and clinical implications. Drug Alcohol Depend. 2014, 144, 12–41. [Google Scholar] [CrossRef] [Green Version]
- Escelsior, A.; Belvederi Murri, M.; Pietro, C.G.; Serafini, G.; Aguglia, A.; Zampogna, D.; Cattedra, S.; Nebbia, J.; Trabucco, A.; Prestia, D.; et al. Cannabinoid use and self-injurious behaviours: A systematic review and meta-analysis. J. Affect. Disord. 2021, 278, 85–98. [Google Scholar] [CrossRef]
- Sajatovic, M.; Chen, P.; Young, R.C. Rating Scales in Bipolar Disorder. Clin. Trial Des. Chall. Mood Disord. 2015, 105–136. [Google Scholar] [CrossRef]
- Martinotti, G.; Santacroce, R.; Papanti, D.; Elgharably, Y.; Prilutskaya, M.; Corazza, O. Synthetic Cannabinoids: Psychopharmacology, Clinical Aspects, Psychotic Onset. CNS Neurol. Disord. Drug Targets 2017, 16, 567–575. [Google Scholar] [CrossRef]
- Guillemin, G.J.; Kerr, S.J.; Smythe, G.A.; Smith, D.G.; Kapoor, V.; Armati, P.J.; Croitoru, J.; Brew, B.J. Kynurenine pathway metabolism in human astrocytes: A paradox for neuronal protection. J. Neurochem. 2001, 78, 842–853. [Google Scholar] [CrossRef] [PubMed]
- Guillemin, G.J.; Smith, D.G.; Kerr, S.J.; Smythe, G.A.; Kapoor, V.; Armati, P.J.; Brew, B.J. Characterisation of kynurenine pathway metabolism in human astrocytes and implications in neuropathogenesis. Redox Rep. 2000, 5, 108–111. [Google Scholar] [CrossRef] [PubMed]
- Guillemin, G.J.; Cullen, K.M.; Lim, C.K.; Smythe, G.A.; Garner, B.; Kapoor, V.; Takikawa, O.; Brew, B.J. Characterization of the Kynurenine Pathway in Human Neurons. J. Neurosci. 2007, 27, 12884–12892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guillemin, G.J.; Smith, D.G.; Smythe, G.A.; Armati, P.J.; Brew, G.J. Expression of The Kynurenine Pathway Enzymes in Human Microglia and Macrophages. Adv. Exp. Med. Biol. 2003, 527, 105–112. [Google Scholar] [PubMed]
- Behan, W.M.H.; McDonald, M.; Darlington, L.G.; Stone, T.W. Oxidative stress as a mechanism for quinolinic acid-induced hippocampal damage: Protection by melatonin and deprenyl. Br. J. Pharmacol. 1999, 128, 1754–1760. [Google Scholar] [CrossRef] [PubMed]
- Lugo-Huitrón, R.; Ugalde Muñiz, P.; Pineda, B.; Pedraza-Chaverrí, J.; Ríos, C.; Pérez-de la Cruz, V. Quinolinic acid: An endogenous neurotoxin with multiple targets. Oxid. Med. Cell. Longev. 2013, 2013, 104024. [Google Scholar] [CrossRef] [Green Version]
- Vécsei, L.; Szalárdy, L.; Fülöp, F.; Toldi, J. Kynurenines in the CNS: Recent advances and new questions. Nat. Rev. Drug. Discov. 2013, 12, 64–82. [Google Scholar] [CrossRef] [PubMed]
- Coppen, A. The biochemistry of affective disorders. Br. J. Psychiatry 1967, 113, 1237–1264. [Google Scholar] [CrossRef] [PubMed]
- Joensuu, M.; Tolmunen, T.; Saarinen, P.I.; Tiihonen, J.; Kuikka, J.; Ahola, P.; Vanninen, R.; Lehtonen, J. Reduced midbrain serotonin transporter availability in drug-naïve patients with depression measured by SERT-specific [(123)I] nor-beta-CIT SPECT imaging. Psychiatry Res. 2007, 154, 125–131. [Google Scholar] [CrossRef] [PubMed]
- Sargent, P.A.; Kjaer, K.H.; Bench, C.J.; Rabiner, E.A.; Messa, C.; Meyer, J.; Gunn, R.N.; Grasby, P.M.; Cowen, P.J. Brain serotonin1A receptor binding measured by positron emission tomography with [11C]WAY-100635: Effects of depression and antidepressant treatment. Arch. Gen. Psychiatry 2000, 57, 174–180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drevets, W.C.; Thase, M.E.; Moses-Kolko, E.L.; Price, J.; Frank, E.; Kupfer, D.J.; Mathis, C. Serotonin-1A receptor imaging in recurrent depression: Replication and literature review. Nucl. Med. Biol. 2007, 34, 865–877. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yatham, L.N.; Liddle, P.F.; Shiah, I.S.; Scarrow, G.; Lam, R.W.; Adam, M.J.; Zis, A.P.; Ruth, T.J. Brain serotonin2 receptors in major depression: A positron emission tomography study. Arch. Gen. Psychiatry 2000, 57, 850–858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Albert, P.R.; Benkelfat, C.; Descarries, L. The neurobiology of depression--revisiting the serotonin hypothesis. I. Cellular and molecular mechanisms. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2012, 367, 2378–2381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reimold, M.; Batra, A.; Knobel, A.; Smolka, M.N.; Zimmer, A.; Mann, K.; Solbach, C.; Reischl, G.; Schwärzler, F.; Gründer, G.; et al. Anxiety is associated with reduced central serotonin transporter availability in unmedicated patients with unipolar major depression: A [11C]DASB PET study. Mol. Psychiatry 2008, 13, 606–613. [Google Scholar] [CrossRef]
- Cowen, P.J.; Browning, M. What has serotonin to do with depression? World Psychiatry 2015, 14, 158–160. [Google Scholar] [CrossRef] [Green Version]
- Lapin, I.P.; Oxenkrug, G.F. Intensification of the central serotoninergic processes as a possible determinant of the thymoleptic effect. Lancet 1969, 293, 132–136. [Google Scholar] [CrossRef]
- Salter, M.; Knowles, R.G.; Pogson, C.I. How does displacement of albumin-bound tryptophan cause sustained increases in the free tryptophan concentration in plasma and 5-hydroxytryptamine synthesis in brain? Biochem. J. 1989, 262, 365–368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Höglund, E.; Øverli, Ø.; Winberg, S. Tryptophan Metabolic Pathways and Brain Serotonergic Activity: A Comparative Review. Front. Endocrinol. 2019, 10, 158. [Google Scholar] [CrossRef] [PubMed]
- Ogyu, K.; Kubo, K.; Noda, Y.; Iwata, Y.; Tsugawa, S.; Omura, Y.; Wada, M.; Tarumi, R.; Plitman, E.; Moriguchi, S.; et al. Kynurenine pathway in depression: A systematic review and meta-analysis. Neurosci. Biobehav. Rev. 2018, 90, 16–25. [Google Scholar] [CrossRef]
- Wu, H.; Gong, J.; Liu, Y. Indoleamine 2,3-dioxygenase regulation of immune response (Review). Mol. Med. Rep. 2018, 17, 4867–4873. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Myint, A.M. Kynurenines: From the perspective of major psychiatric disorders. FEBS J. 2012, 279, 1375–1385. [Google Scholar] [CrossRef] [Green Version]
- Myint, A.-M.; Kim, Y.K.; Verkerk, R.; Scharpé, S.; Steinbusch, H.; Leonard, B. Kynurenine pathway in major depression: Evidence of impaired neuroprotection. J. Affect. Disord. 2007, 98, 143–151. [Google Scholar] [CrossRef] [PubMed]
- Myint, A.-M.; Schwarz, M.J.; Müller, N. The role of the kynurenine metabolism in major depression. J. Neural Transm. 2012, 119, 245–251. [Google Scholar] [CrossRef]
- Oxenkrug, G. Serotonin-kynurenine hypothesis of depression: Historical overview and recent developments. Curr. Drug Targets 2013, 14, 514–521. [Google Scholar] [CrossRef]
- Kruse, J.L.; Cho, J.H.-J.; Olmstead, R.; Hwang, L.; Faull, K.; Eisenberger, N.I.; Irwin, M.R. Kynurenine metabolism and inflammation-induced depressed mood: A human experimental study. Psychoneuroendocrinology 2019, 109, 104371. [Google Scholar] [CrossRef]
- Boros, F.A.; Bohár, Z.; Vécsei, L. Genetic alterations affecting the genes encoding the enzymes of the kynurenine pathway and their association with human diseases. Mutat. Res. Mutat. Res. 2018, 776, 32–45. [Google Scholar] [CrossRef] [Green Version]
- Hughes, M.M.; Carballedo, A.; McLoughlin, D.M.; Amico, F.; Harkin, A.; Frodl, T.; Connor, T.J. Tryptophan depletion in depressed patients occurs independent of kynurenine pathway activation. Brain. Behav. Immun. 2012, 26, 979–987. [Google Scholar] [CrossRef] [PubMed]
- Quak, J.; Doornbos, B.; Roest, A.M.; Duivis, H.E.; Vogelzangs, N.; Nolen, W.A.; Penninx, B.W.J.H.; Kema, I.P.; de Jonge, P. Does tryptophan degradation along the kynurenine pathway mediate the association between pro-inflammatory immune activity and depressive symptoms? Psychoneuroendocrinology 2014, 45, 202–210. [Google Scholar] [CrossRef] [PubMed]
- Dahl, J.; Andreassen, O.A.; Verkerk, R.; Malt, U.F.; Sandvik, L.; Brundin, L.; Ormstad, H. Ongoing episode of major depressive disorder is not associated with elevated plasma levels of kynurenine pathway markers. Psychoneuroendocrinology 2015, 56, 12–22. [Google Scholar] [CrossRef] [PubMed]
- Savitz, J.; Drevets, W.C.; Wurfel, B.E.; Ford, B.N.; Bellgowan, P.S.F.; Victor, T.A.; Bodurka, J.; Teague, T.K.; Dantzer, R. Reduction of kynurenic acid to quinolinic acid ratio in both the depressed and remitted phases of major depressive disorder. Brain Behav. Immun. 2015, 46, 55–59. [Google Scholar] [CrossRef] [Green Version]
- Schwarcz, R.; Bruno, J.P.; Muchowski, P.J.; Wu, H.-Q. Kynurenines in the mammalian brain: When physiology meets pathology. Nat. Rev. Neurosci. 2012, 13, 465–477. [Google Scholar] [CrossRef]
- Gururajan, A.; Clarke, G.; Dinan, T.G.; Cryan, J.F. Molecular biomarkers of depression. Neurosci. Biobehav. Rev. 2016, 64, 101–133. [Google Scholar] [CrossRef]
- Leonard, B.E. Inflammation and depression: A causal or coincidental link to the pathophysiology? Acta Neuropsychiatr. 2018, 30, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Leonard, B.E. Inflammation, depression and dementia: Are they connected? Neurochem. Res. 2007, 32, 1749–1756. [Google Scholar] [CrossRef]
- Martínez-Cengotitabengoa, M.; Carrascón, L.; O’Brien, J.; Díaz-Gutiérrez, M.-J.; Bermúdez-Ampudia, C.; Sanada, K.; Arrasate, M.; González-Pinto, A. Peripheral Inflammatory Parameters in Late-Life Depression: A Systematic Review. Int. J. Mol. Sci. 2016, 17, 2022. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strawbridge, R.; Young, A.H.; Cleare, A.J. Biomarkers for depression: Recent insights, current challenges and future prospects. Neuropsychiatr. Dis. Treat. 2017, 13, 1245–1262. [Google Scholar] [CrossRef] [Green Version]
- Hacimusalar, Y.; Eşel, E. Suggested Biomarkers for Major Depressive Disorder. Noro Psikiyatr. Ars. 2018, 55, 280–290. [Google Scholar] [CrossRef] [PubMed]
- Ranieri, R.; Laezza, C.; Bifulco, M.; Marasco, D.; Malfitano, A.M. Cannabinoids and Neuro-Inflammation: Regulation of Brain Immune Response. Recent Pat. CNS Drug Discov. 2016, 10, 178–203. [Google Scholar] [CrossRef] [Green Version]
- Klein, T.; Lane, B.; Newton, C.; Friedman, H. The cannabinoid system and cytokine network. Proc. Soc. Exp. Biol. Med. 2000, 225, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Massi, P.; Vaccani, A.; Parolaro, D. Cannabinoids, immune system and cytokine network. Curr. Pharm. Des. 2006, 12, 3135–3146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jean-Gilles, L.; Braitch, M.; Latif, M.L.; Aram, J.; Fahey, A.J.; Edwards, L.J.; Robins, R.A.; Tanasescu, R.; Tighe, P.J.; Gran, B.; et al. Effects of pro-inflammatory cytokines on cannabinoid CB1 and CB2 receptors in immune cells. Acta Physiol. 2015, 214, 63. [Google Scholar] [CrossRef]
- Mandolesi, G.; Bullitta, S.; Fresegna, D.; Gentile, A.; De Vito, F.; Dolcetti, E.; Rizzo, F.R.; Strimpakos, G.; Centonze, D.; Musella, A. Interferon-γ causes mood abnormalities by altering cannabinoid CB1 receptor function in the mouse striatum. Neurobiol. Dis. 2017, 108, 45–53. [Google Scholar] [CrossRef]
- Rossi, S.; Motta, C.; Musella, A.; Centonze, D. The interplay between inflammatory cytokines and the endocannabinoid system in the regulation of synaptic transmission. Neuropharmacology 2015, 96, 105–112. [Google Scholar] [CrossRef]
- Rossi, S.; Sacchetti, L.; Napolitano, F.; De Chiara, V.; Motta, C.; Studer, V.; Musella, A.; Barbieri, F.; Bari, M.; Bernardi, G.; et al. Interleukin-1β causes anxiety by interacting with the endocannabinoid system. J. Neurosci. 2012, 32, 13896–13905. [Google Scholar] [CrossRef] [Green Version]
- Watzl, B.; Scuderi, P.; Watson, R.R. Marijuana components stimulate human peripheral blood mononuclear cell secretion of interferon-gamma and suppress interleukin-1 alpha in vitro. Int. J. Immunopharmacol. 1991, 13, 1091–1097. [Google Scholar] [CrossRef]
- Berdyshev, E.V.; Boichot, E.; Germain, N.; Allain, N.; Anger, J.-P.; Lagente, V. Influence of fatty acid ethanolamides and Δ9-tetrahydrocannabinol on cytokine and arachidonate release by mononuclear cells. Eur. J. Pharmacol. 1997, 330, 231–240. [Google Scholar] [CrossRef]
- Klein, T.W.; Friedman, H.; Specter, S. Marijuana, immunity and infection. J. Neuroimmunol. 1998, 83, 102–115. [Google Scholar] [CrossRef]
- Klein, T.W.; Newton, C.; Zhu, W.; Daaka, Y.; Friedman, H. Delta 9-Tetrahydrocannabinol, Cytokines, and Immunity to Legionella pneumophila. Exp. Biol. Med. 1995, 209, 205–212. [Google Scholar] [CrossRef]
- Suárez-Pinilla, P.; López-Gil, J.; Crespo-Facorro, B. Immune system: A possible nexus between cannabinoids and psychosis. Brain Behav. Immun. 2014, 40, 269–282. [Google Scholar] [CrossRef] [PubMed]
- Cabral, G.A.; Griffin-Thomas, L. Emerging Role of the CB2 Cannabinoid Receptor in Immune Regulation and Therapeutic Prospects. Expert Rev. Mol. Med. 2010, 11, e3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Srivastava, M.D.; Srivastava, B.I.; Brouhard, B. Δ9 Tetrahydrocannabinol and cannabidiol alter cytokine production by human immune cells. Immunopharmacology 1998, 40, 179–185. [Google Scholar] [CrossRef]
- Cutando, L.; Busquets-Garcia, A.; Puighermanal, E.; Gomis-González, M.; Delgado-García, J.M.; Gruart, A.; Maldonado, R.; Ozaita, A. Microglial activation underlies cerebellar deficits produced by repeated cannabis exposure. J. Clin. Investig. 2013, 123, 2816–2831. [Google Scholar] [CrossRef] [PubMed]
- García-Gutiérrez, M.S.; Navarrete, F.; Gasparyan, A.; Austrich-Olivares, A.; Sala, F.; Manzanares, J. Cannabidiol: A Potential New Alternative for the Treatment of Anxiety, Depression, and Psychotic Disorders. Biomolecules 2020, 10, 1575. [Google Scholar] [CrossRef] [PubMed]
- Nichols, J.M.; Kaplan, B.L.F. Immune Responses Regulated by Cannabidiol. Cannabis Cannabinoid Res. 2020, 5, 12–31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burstein, S. Cannabidiol (CBD) and its analogs: A review of their effects on inflammation. Bioorg. Med. Chem. 2015, 23, 1377–1385. [Google Scholar] [CrossRef]
- Hahn, B. The Potential of Cannabidiol Treatment for Cannabis Users With Recent-Onset Psychosis. Schizophr. Bull. 2018, 44, 46–53. [Google Scholar] [CrossRef]
- Calpe-López, C.; García-Pardo, M.P.; Aguilar, M.A. Cannabidiol Treatment Might Promote Resilience to Cocaine and Methamphetamine Use Disorders: A Review of Possible Mechanisms. Molecules 2019, 24, 2583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moretti, S.; Castelli, M.; Franchi, S.; Raggi, M.A.; Mercolini, L.; Protti, M.; Somaini, L.; Panerai, A.E.; Sacerdote, P. Δ9-Tetrahydrocannabinol-induced anti-inflammatory responses in adolescent mice switch to proinflammatory in adulthood. J. Leukoc. Biol. 2014, 96, 523–534. [Google Scholar] [CrossRef]
- Moretti, S.; Franchi, S.; Castelli, M.; Amodeo, G.; Somaini, L.; Panerai, A.; Sacerdote, P. Exposure of Adolescent Mice to Delta-9-Tetrahydrocannabinol Induces Long-Lasting Modulation of Pro- and Anti-Inflammatory Cytokines in Hypothalamus and Hippocampus Similar to that Observed for Peripheral Macrophages. J. Neuroimmune Pharmacol. 2015, 10, 371–379. [Google Scholar] [CrossRef]
- Zamberletti, E.; Gabaglio, M.; Prini, P.; Rubino, T.; Parolaro, D. Cortical neuroinflammation contributes to long-term cognitive dysfunctions following adolescent delta-9-tetrahydrocannabinol treatment in female rats. Eur. Neuropsychopharmacol. 2015, 25, 2404–2415. [Google Scholar] [CrossRef]
- American Psychiatric Association. Diagnostic and Statistical Manual of DSM-5, 5th ed.; American Psychiatric Publishing: London, UK; Washington, DC, USA, 2013; ISBN 978-0-89042-554-1. [Google Scholar]
- Bayazit, H.; Selek, S.; Karababa, I.F.; Cicek, E.; Aksoy, N. Evaluation of Oxidant/Antioxidant Status and Cytokine Levels in Patients with Cannabis Use Disorder. Clin. Psychopharmacol. Neurosci. 2017, 15, 237–242. [Google Scholar] [CrossRef] [Green Version]
- Lisano, J.K.; Kisiolek, J.N.; Smoak, P.; Phillips, K.T.; Stewart, L.K. Chronic cannabis use and circulating biomarkers of neural health, stress, and inflammation in physically active individuals. Appl. Physiol. Nutr. Metab. 2020, 45, 258–263. [Google Scholar] [CrossRef]
- Zawatsky, C.N.; Abdalla, J.; Cinar, R. Synthetic cannabinoids induce acute lung inflammation via cannabinoid receptor 1 activation. ERJ Open Res. 2020, 6, 00121–02020. [Google Scholar] [CrossRef] [PubMed]
- Derocq, J.-M.; Jbilo, O.; Bouaboula, M.; Ségui, M.; Clère, C.; Casellas, P. Genomic and Functional Changes Induced by the Activation of the Peripheral Cannabinoid Receptor CB2 in the Promyelocytic Cells HL-60. J. Biol. Chem. 2000, 275, 15621–15628. [Google Scholar] [CrossRef] [Green Version]
- Nahoko, U.; Ruri, K.-H.; Nobuo, K.; Yuji, H.; Yukihiro, G. Identification of a cannabinoid analog as a new type of designer drug in a herbal product. Chem. Pharm. Bull. 2009, 57, 439–441. [Google Scholar]
- Bileck, A.; Ferk, F.; Al-Serori, H.; Koller, V.J.; Muqaku, B.; Haslberger, A.; Auwärter, V.; Gerner, C.; Knasmüller, S. Impact of a synthetic cannabinoid (CP-47,497-C8) on protein expression in human cells: Evidence for induction of inflammation and DNA damage. Arch. Toxicol. 2016, 90, 1369–1382. [Google Scholar] [CrossRef]
- Zádor, F.; Mohammadzadeh, A.; Balogh, M.; Zádori, Z.S.; Király, K.; Barsi, S.; Galambos, A.R.; László, S.B.; Hutka, B.; Váradi, A.; et al. Comparisons of In Vivo and In Vitro Opioid Effects of Newly Synthesized 14-Methoxycodeine-6-O-sulfate and Codeine-6-O-sulfate. Molecules 2020, 25, 1370. [Google Scholar] [CrossRef] [Green Version]
- Christensen, R.; Kristensen, P.K.; Bartels, E.M.; Bliddal, H.; Astrup, A. Efficacy and safety of the weight-loss drug rimonabant: A meta-analysis of randomised trials. Lancet 2007, 370, 1706–1713. [Google Scholar] [CrossRef]
- Beyer, C.E.; Dwyer, J.M.; Piesla, M.J.; Platt, B.J.; Shen, R.; Rahman, Z.; Chan, K.; Manners, M.T.; Samad, T.A.; Kennedy, J.D.; et al. Depression-like phenotype following chronic CB1 receptor antagonism. Neurobiol. Dis. 2010, 39, 148–155. [Google Scholar] [CrossRef]
- Guler, E.; Bektay, M.; Akyildiz, A.; Sisman, B.; Izzettin, F.; Kocyigit, A. Investigation of DNA damage, oxidative stress, and inflammation in synthetic cannabinoid users. Hum. Exp. Toxicol. 2020, 39, 1454–1462. [Google Scholar] [CrossRef]
- Campbell, B.M.; Charych, E.; Lee, A.W.; Möller, T. Kynurenines in CNS disease: Regulation by inflammatory cytokines. Front. Neurosci. 2014, 8, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strasser, B.; Becker, K.; Fuchs, D.; Gostner, J.M. Kynurenine pathway metabolism and immune activation: Peripheral measurements in psychiatric and co-morbid conditions. Neuropharmacology 2017, 112, 286–296. [Google Scholar] [CrossRef] [PubMed]
- Baumgartner, R.; Forteza, M.J.; Ketelhuth, D.F.J. The interplay between cytokines and the Kynurenine pathway in inflammation and atherosclerosis. Cytokine 2019, 122, 154148. [Google Scholar] [CrossRef]
- Mándi, Y.; Vécsei, L. The kynurenine system and immunoregulation. J. Neural Transm. 2012, 119, 197–209. [Google Scholar] [CrossRef]
- Clark, S.M.; Pocivavsek, A.; Nicholson, J.D.; Notarangelo, F.M.; Langenberg, P.; McMahon, R.P.; Kleinman, J.E.; Hyde, T.M.; Stiller, J.; Postolache, T.T.; et al. Reduced kynurenine pathway metabolism and cytokine expression in the prefrontal cortex of depressed individuals. J. Psychiatry Neurosci. 2016, 41, 386–394. [Google Scholar] [CrossRef] [Green Version]
- Byrne, G.I.; Lehmann, L.K.; Landry, G.J. Induction of tryptophan catabolism is the mechanism for gamma-interferon-mediated inhibition of intracellular Chlamydia psittaci replication in T24 cells. Infect. Immun. 1986, 53, 347–351. [Google Scholar] [CrossRef] [Green Version]
- Pfefferkorn, E.R.; Rebhun, S.; Eckel, M. Characterization of an indoleamine 2,3-dioxygenase induced by gamma-interferon in cultured human fibroblasts. J. Interferon Res. 1986, 6, 267–279. [Google Scholar] [CrossRef] [PubMed]
- Hissong, B.D.; Byrne, G.I.; Padilla, M.L.; Carlin, J.M. Upregulation of interferon-induced indoleamine 2,3-dioxygenase in human macrophage cultures by lipopolysaccharide, muramyl tripeptide, and interleukin-1. Cell. Immunol. 1995, 160, 264–269. [Google Scholar] [CrossRef]
- Hissong, B.D.; Carlin, J.M. Potentiation of Interferon-Induced Indoleamine 2,3-Dioxygenase mRNA in Human Mononuclear Phagocytes by Lipopolysaccharide and Interleukin-1. J. Interf. Cytokine Res. 1997, 17, 387–393. [Google Scholar] [CrossRef] [PubMed]
- Babcock, T.A.; Carlin, J.M. Transcriptional activation of indoleamine dioxygenase by interleukin 1 and tumor necrosis factor α in interferon-treated epithelial cells. Cytokine 2000, 12, 588–594. [Google Scholar] [CrossRef] [PubMed]
- Fujigaki, H.; Saito, K.; Fujigaki, S.; Takemura, M.; Sudo, K.; Ishiguro, H.; Seishima, M. The Signal Transducer and Activator of Transcription 1α and Interferon Regulatory Factor 1 Are Not Essential for the Induction of Indoleamine 2,3-Dioxygenase by Lipopolysaccharide: Involvement of p38 Mitogen-Activated Protein Kinase and Nuclear Factor-κB. J. Biochem. 2006, 139, 655–662. [Google Scholar] [CrossRef]
- Brooks, A.K.; Lawson, M.A.; Rytych, J.L.; Yu, K.C.; Janda, T.M.; Steelman, A.J.; McCusker, R.H. Immunomodulatory Factors Galectin-9 and Interferon-Gamma Synergize to Induce Expression of Rate-Limiting Enzymes of the Kynurenine Pathway in the Mouse Hippocampus. Front. Immunol. 2016, 7, 422. [Google Scholar] [CrossRef] [Green Version]
- Brooks, A.K.; Lawson, M.A.; Smith, R.A.; Janda, T.M.; Kelley, K.W.; McCusker, R.H. Interactions between inflammatory mediators and corticosteroids regulate transcription of genes within the Kynurenine Pathway in the mouse hippocampus. J. Neuroinflamm. 2016, 13, 98. [Google Scholar] [CrossRef] [Green Version]
- Fuertig, R.; Azzinnari, D.; Bergamini, G.; Cathomas, F.; Sigrist, H.; Seifritz, E.; Vavassori, S.; Luippold, A.; Hengerer, B.; Ceci, A.; et al. Mouse chronic social stress increases blood and brain kynurenine pathway activity and fear behaviour: Both effects are reversed by inhibition of indoleamine 2,3-dioxygenase. Brain Behav. Immun. 2016, 54, 59–72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duda, W.; Curzytek, K.; Kubera, M.; Connor, T.J.; Fagan, E.M.; Basta-Kaim, A.; Trojan, E.; Papp, M.; Gruca, P.; Budziszewska, B.; et al. Interaction of the immune-inflammatory and the kynurenine pathways in rats resistant to antidepressant treatment in model of depression. Int. Immunopharmacol. 2019, 73, 527–538. [Google Scholar] [CrossRef]
- Hu, B.; Hissong, B.D.; Carlin, J.M. Interleukin-1 Enhances Indoleamine 2,3-Dioxygenase Activity by Increasing Specific mRNA Expression in Human Mononuclear Phagocytes. J. Interf. Cytokine Res. 1995, 15, 617–624. [Google Scholar] [CrossRef] [PubMed]
- Corona, A.W.; Huang, Y.; O’Connor, J.C.; Dantzer, R.; Kelley, K.W.; Popovich, P.G.; Godbout, J.P. Fractalkine receptor (CX3CR1) deficiency sensitizes mice to the behavioral changes induced by lipopolysaccharide. J. Neuroinflamm. 2010, 7, 93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonzalez-Pena, D.; Nixon, S.E.; O’Connor, J.C.; Southey, B.R.; Lawson, M.A.; McCusker, R.H.; Borras, T.; Machuca, D.; Hernandez, A.G.; Dantzer, R.; et al. Microglia Transcriptome Changes in a Model of Depressive Behavior after Immune Challenge. PLoS ONE 2016, 11, e150858. [Google Scholar] [CrossRef] [Green Version]
- Garrison, A.M.; Parrott, J.M.; Tuñon, A.; Delgado, J.; Redus, L.; O’connor, J.C. Kynurenine pathway metabolic balance influences microglia activity: Targeting kynurenine monooxygenase to dampen neuroinflammation HHS Public Access. Psychoneuroendocrinology 2018, 94, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Laumet, G.; Zhou, W.; Dantzer, R.; Edralin, J.D.; Huo, X.; Budac, D.P.; O’connor, J.C.; Lee, A.W.; Heijnen, C.J.; Kavelaars, A. Upregulation of neuronal kynurenine 3-monooxygenase mediates depression-like behavior in a mouse model of neuropathic pain HHS Public Access. Brain Behav. Immun. 2017, 66, 94–102. [Google Scholar] [CrossRef]
- Von Bubnoff, D.; Scheler, M.; Wilms, H.; Fimmers, R.; Bieber, T. Identification of IDO-positive and IDO-negative human dendritic cells after activation by various proinflammatory stimuli. J. Immunol. 2011, 186, 6701–6709. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zunszain, P.A.; Anacker, C.; Cattaneo, A.; Choudhury, S.; Musaelyan, K.; Myint, A.M.; Thuret, S.; Price, J.; Pariante, C.M. Interleukin-1β: A new regulator of the kynurenine pathway affecting human hippocampal neurogenesis. Neuropsychopharmacology 2012, 37, 939–949. [Google Scholar] [CrossRef] [Green Version]
- Xie, W.; Cai, L.; Yu, Y.; Gao, L.; Xiao, L.; He, Q.; Ren, Z.; Liu, Y. Activation of brain indoleamine 2,3-dioxygenase contributes to epilepsy-associated depressive-like behavior in rats with chronic temporal lobe epilepsy. J. Neuroinflamm. 2014, 11, 41. [Google Scholar] [CrossRef] [Green Version]
- Lian, Y.-J.; Gong, H.; Wu, T.-Y.; Su, W.-J.; Zhang, Y.; Yang, Y.-Y.; Peng, W.; Zhang, T.; Zhou, J.-R.; Jiang, C.-L.; et al. Ds-HMGB1 and fr-HMGB induce depressive behavior through neuroinflammation in contrast to nonoxid-HMGB1. Brain. Behav. Immun. 2017, 59, 322–332. [Google Scholar] [CrossRef]
- Wang, B.; Lian, Y.; Su, W.; Liu, L.; Li, J.; Jiang, C.; Wang, Y. Fr-HMGB1 and ds-HMGB1 activate the kynurenine pathway via different mechanisms in association with depressive-like behavior. Mol. Med. Rep. 2019, 20, 359–367. [Google Scholar] [CrossRef] [Green Version]
- Maes, M.; Meltzer, H.Y.; Scharpè, S.; Bosmans, E.; Suy, E.; De Meester, I.; Calabrese, J.; Cosyns, P. Relationships between lower plasma L-tryptophan levels and immune-inflammatory variables in depression. Psychiatry Res. 1993, 49, 151–165. [Google Scholar] [CrossRef]
- Maes, M.; Verkerk, R.; Bonaccorso, S.; Ombelet, W.; Bosmans, E.; Scharpé, S. Depressive and anxiety symptoms in the early puerperium are related to increased degradation of tryptophan into kynurenine, a phenomenon which is related to immune activation. Life Sci. 2002, 71, 1837–1848. [Google Scholar] [CrossRef]
- Schwieler, L.; Samuelsson, M.; Frye, M.A.; Bhat, M.; Schuppe-Koistinen, I.; Jungholm, O.; Johansson, A.G.; Landén, M.; Sellgren, C.M.; Erhardt, S. Electroconvulsive therapy suppresses the neurotoxic branch of the kynurenine pathway in treatment-resistant depressed patients. J. Neuroinflamm. 2016, 13, 51. [Google Scholar] [CrossRef] [Green Version]
- Achtyes, E.; Keaton, S.A.; Smart, L.; Burmeister, A.R.; Heilman, P.L.; Krzyzanowski, S.; Nagalla, M.; Guillemin, G.J.; Escobar Galvis, M.L.; Lim, C.K.; et al. Inflammation and kynurenine pathway dysregulation in post-partum women with severe and suicidal depression. Brain Behav. Immun. 2020, 83, 239–247. [Google Scholar] [CrossRef] [PubMed]
- Westbrook, R.; Chung, T.; Lovett, J.; Ward, C.; Joca, H.; Yang, H.; Khadeer, M.; Tian, J.; Xue, Q.-L.; Le, A.; et al. Kynurenines link chronic inflammation to functional decline and physical frailty. JCI Insight 2020, 5. [Google Scholar] [CrossRef]
- Anderson, G.; Kubera, M.; Duda, W.; Lasoń, W.; Berk, M.; Maes, M. Increased IL-6 trans-signaling in depression: Focus on the tryptophan catabolite pathway, melatonin and neuroprogression. Pharmacol. Rep. 2013, 65, 1647–1654. [Google Scholar] [CrossRef]
- Maes, M. Evidence for an immune response in major depression: A review and hypothesis. Prog. Neuro Psychopharmacol. Biol. Psychiatry 1995, 19, 11–38. [Google Scholar] [CrossRef]
- Bay-Richter, C.; Linderholm, K.R.; Lim, C.K.; Samuelsson, M.; Träskman-Bendz, L.; Guillemin, G.J.; Erhardt, S.; Brundin, L. A role for inflammatory metabolites as modulators of the glutamate N-methyl-d-aspartate receptor in depression and suicidality. Brain Behav. Immun. 2015, 43, 110–117. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Chen, L.; Lim, G.; Sung, B.; Wang, S.; McCabe, M.F.; Rusanescu, G.; Yang, L.; Tian, Y.; Mao, J. Brain indoleamine 2,3-dioxygenase contributes to the comorbidity of pain and depression. J. Clin. Investig. 2012, 122, 2940–2954. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, Y.; Sheng, H.; Tang, Z.; Lu, J.; Ni, X. Inflammation and increased IDO in hippocampus contribute to depression-like behavior induced by estrogen deficiency. Behav. Brain Res. 2015, 288, 71–78. [Google Scholar] [CrossRef] [PubMed]
- Connor, T.J.; Starr, N.; O’Sullivan, J.B.; Harkin, A. Induction of indolamine 2,3-dioxygenase and kynurenine 3-monooxygenase in rat brain following a systemic inflammatory challenge: A role for IFN-γ? Neurosci. Lett. 2008, 441, 29–34. [Google Scholar] [CrossRef] [PubMed]
- Maes, M.; Bonaccorso, S.; Marino, V.; Puzella, A.; Pasquini, M.; Biondi, M.; Artini, M.; Almerighi, C.; Meltzer, H. Treatment with interferon-alpha (IFNα) of hepatitis C patients induces lower serum dipeptidyl peptidase IV activity, which is related to IFNα-induced depressive and anxiety symptoms and immune activation. Mol. Psychiatry 2001, 6, 475–480. [Google Scholar] [CrossRef] [Green Version]
- Zoga, M.; Oulis, P.; Chatzipanagiotou, S.; Masdrakis, V.G.; Pliatsika, P.; Boufidou, F.; Foteli, S.; Soldatos, C.R.; Nikolaou, C.; Papageorgiou, C. Indoleamine 2,3-dioxygenase and immune changes under antidepressive treatment in major depression in females. In Vivo 2014, 28, 633–638. [Google Scholar]
- Haroon, E.; Welle, J.R.; Woolwine, B.J.; Goldsmith, D.R.; Baer, W.; Patel, T.; Felger, J.C.; Miller, A.H. Associations among peripheral and central kynurenine pathway metabolites and inflammation in depression. Neuropsychopharmacology 2020, 45, 998–1007. [Google Scholar] [CrossRef]
- O’Connor, J.C.; Lawson, M.A.; André, C.; Briley, E.M.; Szegedi, S.S.; Lestage, J.; Castanon, N.; Herkenham, M.; Dantzer, R.; Kelley, K.W. Induction of IDO by bacille Calmette-Guérin is responsible for development of murine depressive-like behavior. J. Immunol. 2009, 182, 3202–3212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Connor, J.C.; André, C.; Wang, Y.; Lawson, M.A.; Szegedi, S.S.; Lestage, J.; Castanon, N.; Kelley, K.W.; Dantzer, R. Interferon-gamma and tumor necrosis factor-alpha mediate the upregulation of indoleamine 2,3-dioxygenase and the induction of depressive-like behavior in mice in response to bacillus Calmette-Guerin. J. Neurosci. 2009, 29, 4200–4209. [Google Scholar] [CrossRef]
- Currier, A.R.; Ziegler, M.H.; Riley, M.M.; Babcock, T.A.; Telbis, V.P.; Carlin, J.M. Tumor Necrosis Factor-alpha and Lipopolysaccharide Enhance Interferon-Induced Antichlamydial Indoleamine Dioxygenase Activity Independently. J. Interferon Cytokine Res. 2000, 20, 369–376. [Google Scholar] [CrossRef]
- Robinson, C.M.; Hale, P.T.; Carlin, J.M. The role of IFN-gamma and TNF-alpha-responsive regulatory elements in the synergistic induction of indoleamine dioxygenase. J. Interferon Cytokine Res. 2005, 25, 20–30. [Google Scholar] [CrossRef]
- Pacher, P.; Kunos, G. Modulating the endocannabinoid system in human health and disease—Successes and failures. FEBS J. 2013, 280, 1918–1943. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pacher, P.; Bátkai, S.; Kunos, G. The endocannabinoid system as an emerging target of pharmacotherapy. Pharmacol. Rev. 2006, 58, 389–462. [Google Scholar] [CrossRef] [Green Version]
- Zimmer, A.; Zimmer, A.M.; Hohmann, A.G.; Herkenham, M.; Bonner, T.I. Increased mortality, hypoactivity, and hypoalgesia in cannabinoid CB1 receptor knockout mice. Proc. Natl. Acad. Sci. USA 1999, 96, 5780–5785. [Google Scholar] [CrossRef] [Green Version]
- Buckley, N.E.; McCoy, K.L.; Mezey, É.; Bonner, T.; Zimmer, A.; Felder, C.C.; Glass, M.; Zimmer, A. Immunomodulation by cannabinoids is absent in mice deficient for the cannabinoid CB2 receptor. Eur. J. Pharmacol. 2000, 396, 141–149. [Google Scholar] [CrossRef]
- Giorgini, F.; Huang, S.-Y.; Sathyasaikumar, K.V.; Notarangelo, F.M.; Thomas, M.A.R.; Tararina, M.; Wu, H.-Q.; Schwarcz, R.; Muchowski, P.J. Targeted deletion of kynurenine 3-monooxygenase in mice: A new tool for studying kynurenine pathway metabolism in periphery and brain. J. Biol. Chem. 2013, 288, 36554–36566. [Google Scholar] [CrossRef] [Green Version]
- Erhardt, S.; Pocivavsek, A.; Repici, M.; Liu, X.-C.; Imbeault, S.; Maddison, D.C.; Thomas, M.A.R.; Smalley, J.L.; Larsson, M.K.; Muchowski, P.J.; et al. Adaptive and Behavioral Changes in Kynurenine 3-Monooxygenase Knockout Mice: Relevance to Psychotic Disorders. Biol. Psychiatry 2017, 82, 756–765. [Google Scholar] [CrossRef] [Green Version]
- Járai, Z.; Wagner, J.A.; Varga, K.; Lake, K.D.; Compton, D.R.; Martin, B.R.; Zimmer, A.M.; Bonner, T.I.; Buckley, N.E.; Mezey, E.; et al. Cannabinoid-induced mesenteric vasodilation through an endothelial site distinct from CB1 or CB2 receptors. Proc. Natl. Acad. Sci. USA 1999, 96, 14136–14141. [Google Scholar] [CrossRef] [Green Version]
- Hirata, N.; Hattori, S.; Shoji, H.; Funakoshi, H.; Miyakawa, T. Comprehensive behavioral analysis of indoleamine 2,3-dioxygenase knockout mice. Neuropsychopharmacol. Rep. 2018, 38, 133–144. [Google Scholar] [CrossRef] [Green Version]
- Yu, P.; Di Prospero, N.A.; Sapko, M.T.; Cai, T.; Chen, A.; Melendez-Ferro, M.; Du, F.; Whetsell, W.O.; Guidetti, P.; Schwarcz, R.; et al. Biochemical and phenotypic abnormalities in kynurenine aminotransferase II-deficient mice. Mol. Cell. Biol. 2004, 24, 6919–6930. [Google Scholar] [CrossRef] [Green Version]
- Bondulich, M.K.; Fan, Y.; Song, Y.; Giorgini, F.; Bates, G.P. Ablation of kynurenine 3-monooxygenase rescues plasma inflammatory cytokine levels in the R6/2 mouse model of Huntington’s disease. Sci. Rep. 2021, 11, 5484. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zádor, F.; Joca, S.; Nagy-Grócz, G.; Dvorácskó, S.; Szűcs, E.; Tömböly, C.; Benyhe, S.; Vécsei, L. Pro-Inflammatory Cytokines: Potential Links between the Endocannabinoid System and the Kynurenine Pathway in Depression. Int. J. Mol. Sci. 2021, 22, 5903. https://doi.org/10.3390/ijms22115903
Zádor F, Joca S, Nagy-Grócz G, Dvorácskó S, Szűcs E, Tömböly C, Benyhe S, Vécsei L. Pro-Inflammatory Cytokines: Potential Links between the Endocannabinoid System and the Kynurenine Pathway in Depression. International Journal of Molecular Sciences. 2021; 22(11):5903. https://doi.org/10.3390/ijms22115903
Chicago/Turabian StyleZádor, Ferenc, Sâmia Joca, Gábor Nagy-Grócz, Szabolcs Dvorácskó, Edina Szűcs, Csaba Tömböly, Sándor Benyhe, and László Vécsei. 2021. "Pro-Inflammatory Cytokines: Potential Links between the Endocannabinoid System and the Kynurenine Pathway in Depression" International Journal of Molecular Sciences 22, no. 11: 5903. https://doi.org/10.3390/ijms22115903
APA StyleZádor, F., Joca, S., Nagy-Grócz, G., Dvorácskó, S., Szűcs, E., Tömböly, C., Benyhe, S., & Vécsei, L. (2021). Pro-Inflammatory Cytokines: Potential Links between the Endocannabinoid System and the Kynurenine Pathway in Depression. International Journal of Molecular Sciences, 22(11), 5903. https://doi.org/10.3390/ijms22115903