
Targeting the NAD Salvage Synthesis Pathway as a Novel Therapeutic Strategy for Osteosarcomas with Low NAPRT Expression

 , , ,
, , ,  and
and
Abstract
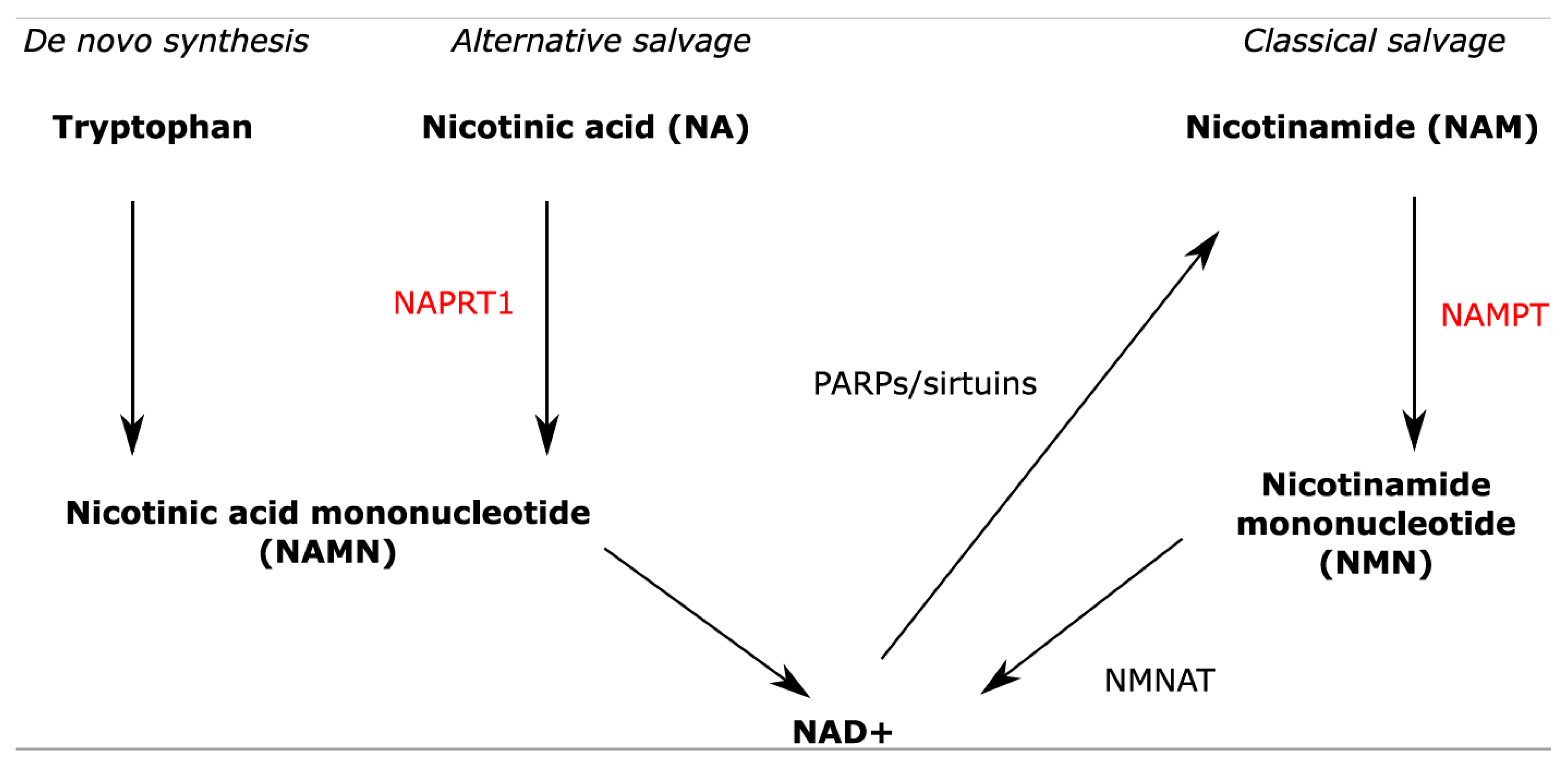
:1. Introduction
2. Results
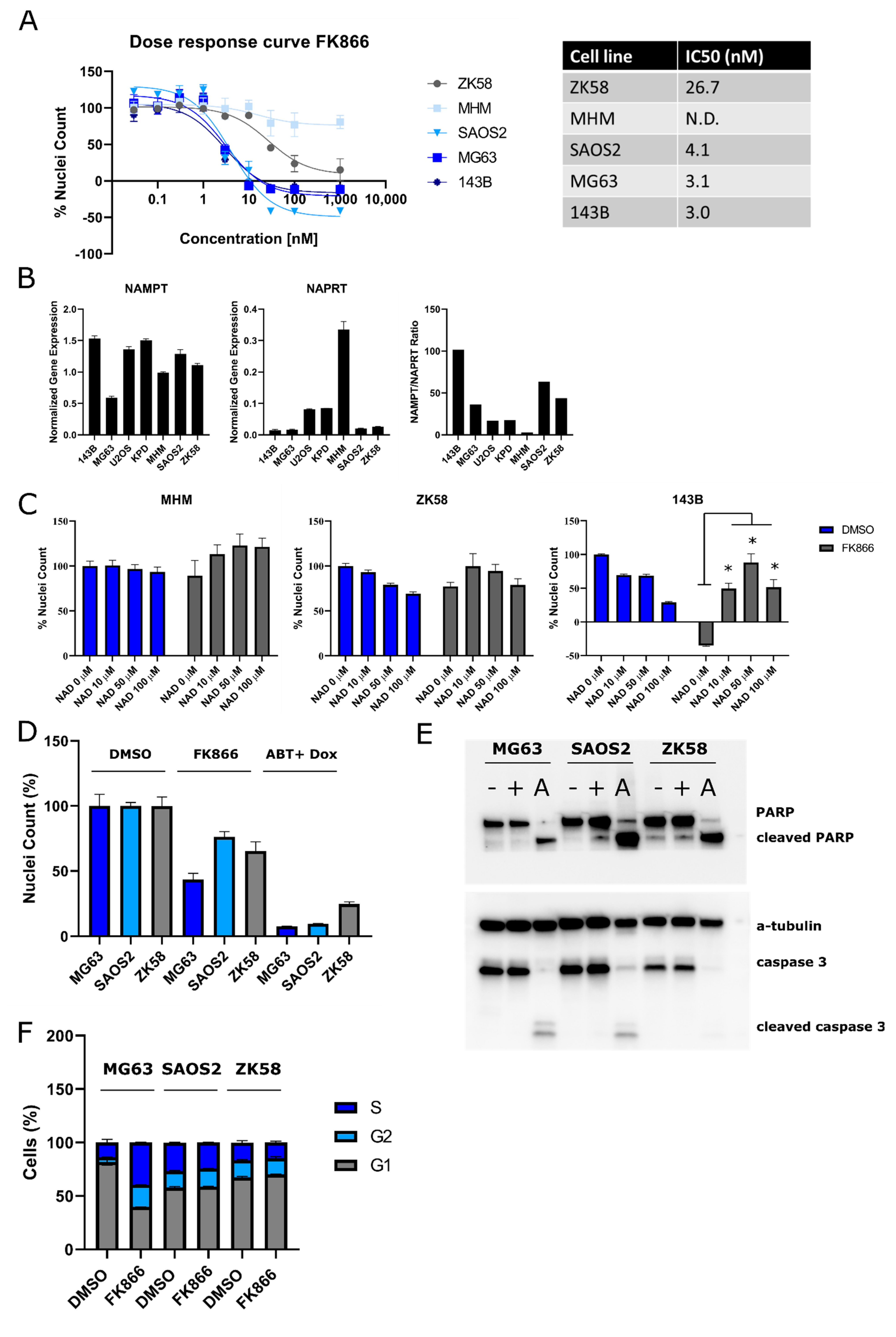
2.1. Osteosarcoma Cells Show Variable Sensitivity to NAMPT Inhibition
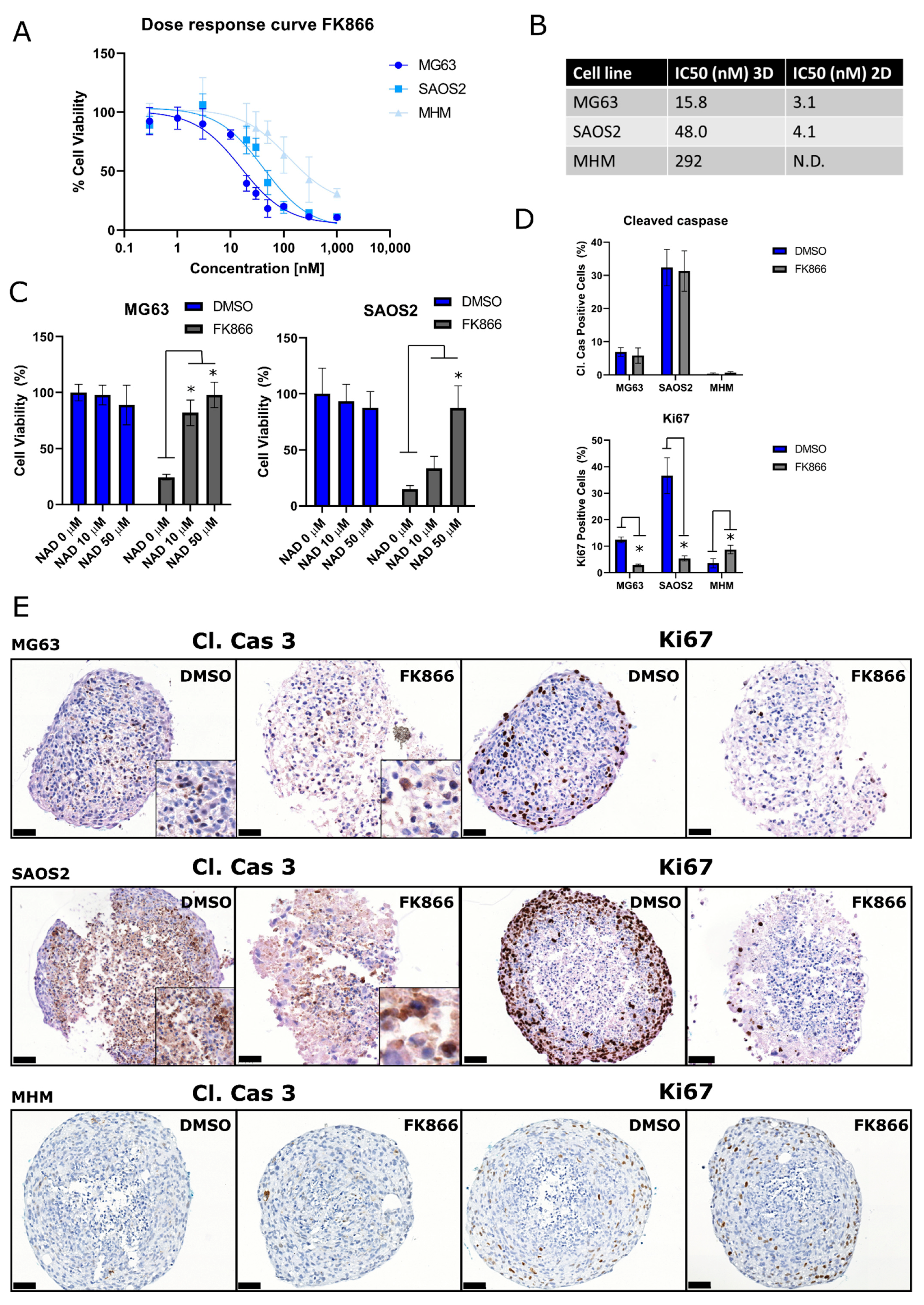
2.2. 3D Cultured Osteosarcoma Cells Are Also Sensitive to NAMPT Inhibition
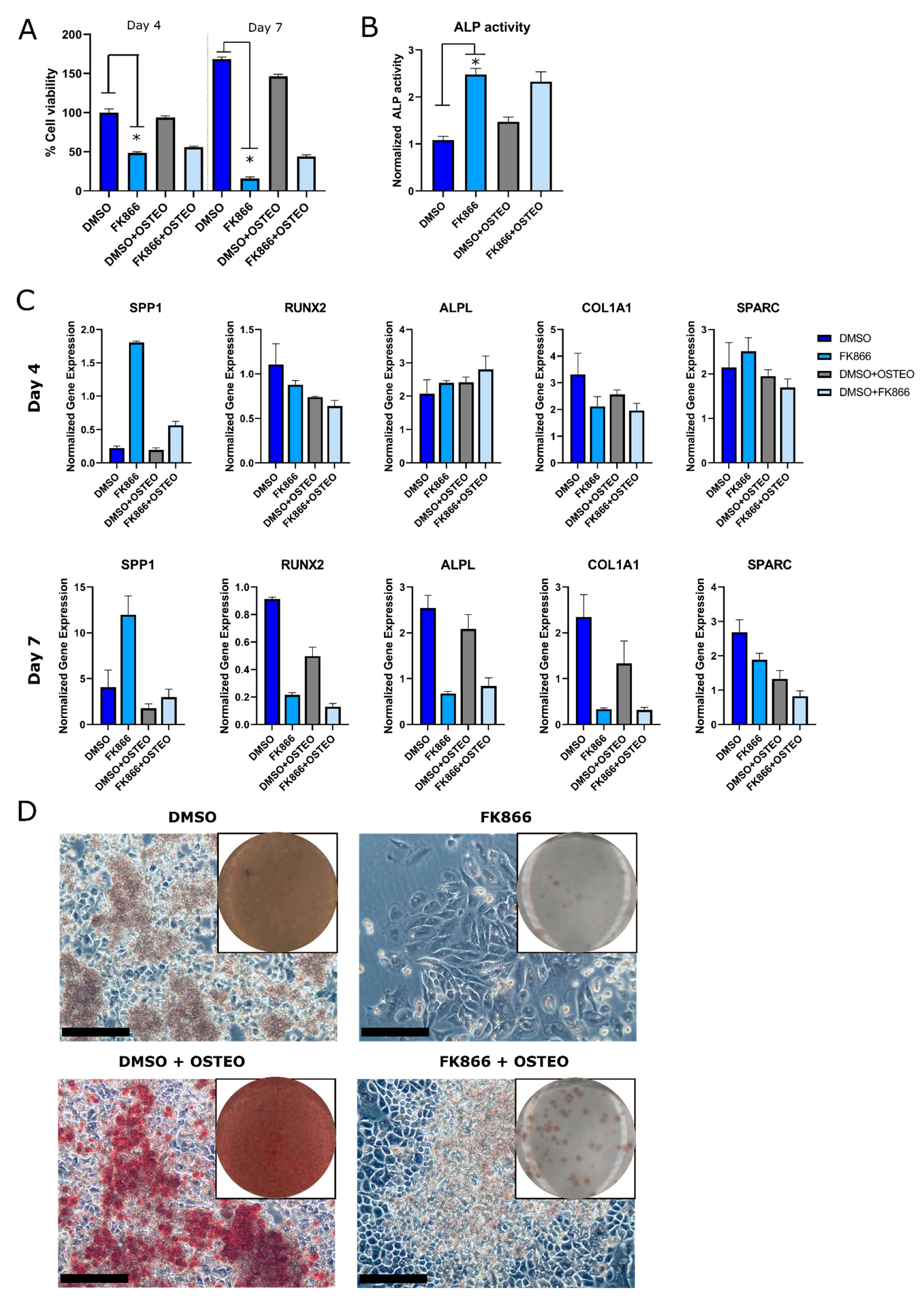
2.3. FK866 Showed a Variable and Time-Dependent Effect on Osteogenic Differentiation in the OS Cell Line ZK58
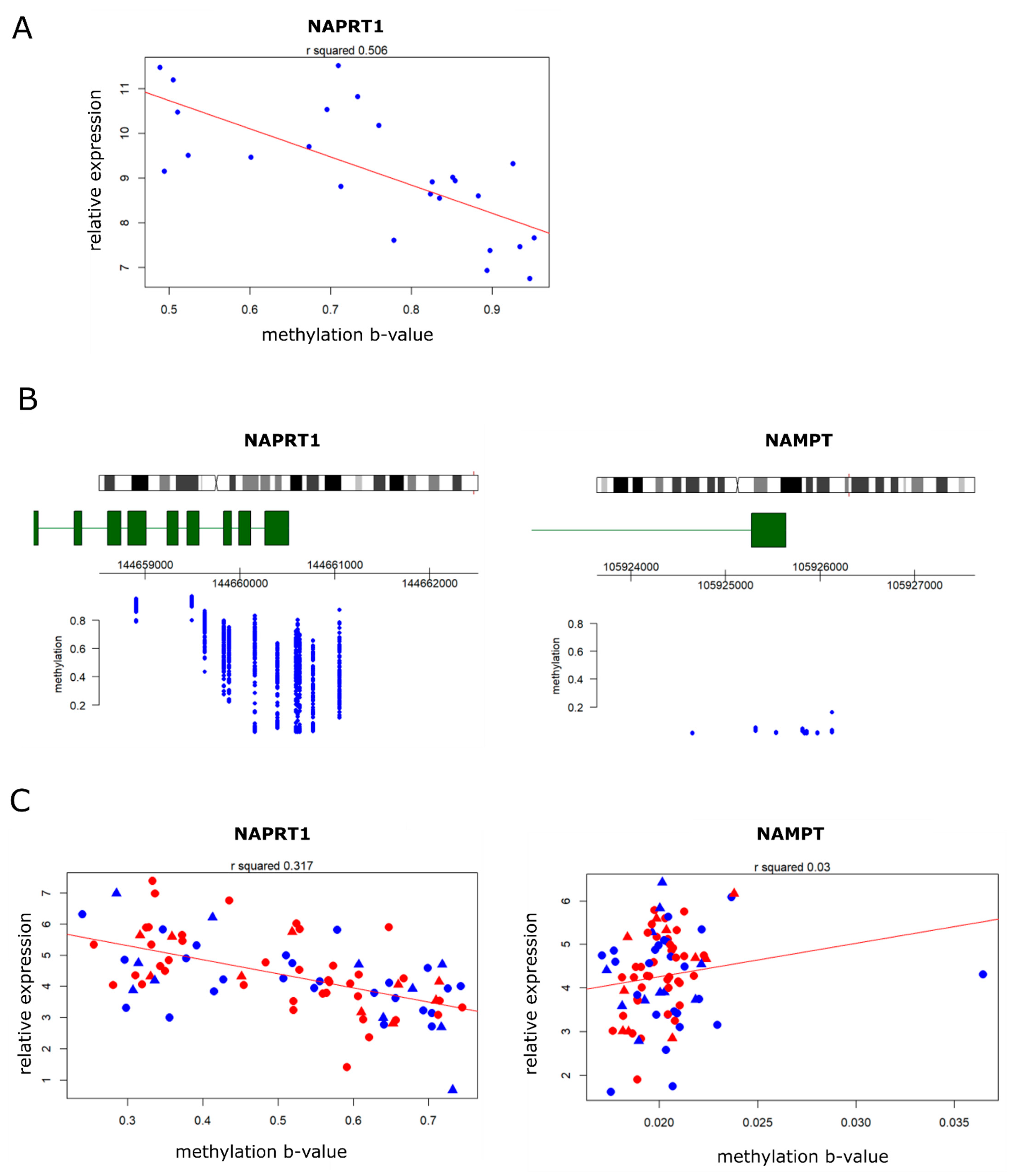
2.4. NAPRT Expression in Osteosarcomas Correlates Inversely with Methylation of the NAPRT Promotor
3. Conclusions and Discussion
4. Material and Methods
4.1. Cell Culture
4.2. Drug Treatment
4.3. Cell Cycle Analysis
4.4. Western Blotting
4.5. Immunohistochemistry
4.6. Osteogenic Differentiation
4.7. Reverse Transcriptase Quantitative PCR (RT-qPCR)
4.8. Expression and Methylation Analysis
4.9. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- WHO. Classification of Tumours of Soft Tissue and Bone, 5th ed.; WHO Classification of Tumours Editorial Board: Lyon, France, 2020; Volume 3. [Google Scholar]
- Smeland, S.; Bielack, S.S.; Whelan, J.; Bernstein, M.; Hogendoorn, P.; Krailo, M.D.; Gorlick, R.; Janeway, K.A.; Ingleby, F.C.; Anninga, J.; et al. Survival and prognosis with osteosarcoma: Outcomes in more than 2000 patients in the EURAMOS-1 (European and American Osteosarcoma Study) cohort. Eur. J. Cancer 2019, 109, 36–50. [Google Scholar] [CrossRef] [Green Version]
- Peterse, E.F.P.; van Leeuwen, T.N.; Cleton-Jansen, A.M. A researcher’s perspective on the quantity of osteosarcoma in vitro studies. J. Bone Oncol. 2017, 7, 29–31. [Google Scholar] [CrossRef] [PubMed]
- Boroughs, L.K.; DeBerardinis, R.J. Metabolic pathways promoting cancer cell survival and growth. Nat. Cell. Biol. 2015, 17, 351–359. [Google Scholar] [CrossRef] [Green Version]
- DeBerardinis, R.J.; Chandel, N.S. Fundamentals of cancer metabolism. Sci. Adv. 2016, 2, e1600200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ren, L.; Hong, E.S.; Mendoza, A.; Issaq, S.; Hoang, C.T.; Lizardo, M.; LeBlanc, A.; Khanna, C. Metabolomics uncovers a link between inositol metabolism and osteosarcoma metastasis. Oncotarget 2017, 8, 38541–38553. [Google Scholar] [CrossRef] [Green Version]
- Rathore, R.; Caldwell, K.E.; Schutt, C.; Brashears, C.B.; Prudner, B.C.; Ehrhardt, W.R.; Leung, C.H.; Lin, H.; Daw, N.C.; Beird, H.C.; et al. Metabolic compensation activates pro-survival mTORC1 signaling upon 3-phosphoglycerate dehydrogenase inhibition in osteosarcoma. Cell. Rep. 2021, 34, 108678. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.-W.; Wu, L.; Cao, Y.; Yang, L.; Liu, W.; E, X.-Q.; Ji, G.; Bi, Z.-G. A novel mechanism of mTORC1-mediated serine/glycine metabolism in osteosarcoma development. Cell. Signal. 2017, 29, 107–114. [Google Scholar] [CrossRef] [PubMed]
- Hu, K.; Dai, H.B.; Qiu, Z.L. mTOR signaling in osteosarcoma: Oncogenesis and therapeutic aspects (Review). Oncol. Rep. 2016, 36, 1219–1225. [Google Scholar] [CrossRef] [Green Version]
- Chiarugi, A.; Dolle, C.; Felici, R.; Ziegler, M. The NAD metabolome—A key determinant of cancer cell biology. Nat. Rev. Cancer 2012, 12, 741–752. [Google Scholar] [CrossRef]
- Canto, C.; Menzies, K.J.; Auwerx, J. NAD(+) Metabolism and the Control of Energy Homeostasis: A Balancing Act between Mitochondria and the Nucleus. Cell. Metab. 2015, 22, 31–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kennedy, B.E.; Sharif, T.; Martell, E.; Dai, C.; Kim, Y.; Lee, P.W.; Gujar, S.A. NAD(+) salvage pathway in cancer metabolism and therapy. Pharmacol. Res. 2016, 114, 274–283. [Google Scholar] [CrossRef] [PubMed]
- Sampath, D.; Zabka, T.S.; Misner, D.L.; O’Brien, T.; Dragovich, P.S. Inhibition of nicotinamide phosphoribosyltransferase (NAMPT) as a therapeutic strategy in cancer. Pharmacol. Ther. 2015, 151, 16–31. [Google Scholar] [CrossRef]
- Xiao, Y.; Elkins, K.; Durieux, J.K.; Lee, L.; Oeh, J.; Yang, L.X.; Liang, X.; DelNagro, C.; Tremayne, J.; Kwong, M.; et al. Dependence of tumor cell lines and patient-derived tumors on the NAD salvage pathway renders them sensitive to NAMPT inhibition with GNE-618. Neoplasia 2013, 15, 1151–1160. [Google Scholar] [CrossRef] [PubMed]
- Meram, A.T.; Alzubaidi, Y.; Cotelingam, J.; Ghali, G.; Lopez, L.; Coppola, D.; Shackelford, R. Nicotinamide Phosphoribosyl Transferase Is Increased in Osteosarcomas and Chondrosarcomas Compared to Benign Bone and Cartilage. Anticancer. Res. 2019, 39, 1761–1765. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; He, J.; He, X.; Li, Y.; Lindgren, U. Nampt expression increases during osteogenic differentiation of multi- and omnipotent progenitors. Biochem. Biophys. Res. Commun. 2013, 434, 117–123. [Google Scholar] [CrossRef]
- Baranski, Z.; de Jong, Y.; Ilkova, T.; Peterse, E.F.P.; Cleton-Jansen, A.M.; Van de Water, B.; Hogendoorn, P.; Bovee, J.V.M.G.; Danen, E.H.J. Pharmacological inhibition of Bcl-xL sensitizes osteosarcoma to doxorubicin. Oncotarget 2015, 6, 36113–36125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hasmann, M.; Schemainda, I. FK866, a Highly Specific Noncompetitive Inhibitor of Nicotinamide Phosphoribosyltransferase, Represents a Novel Mechanism for Induction of Tumor Cell Apoptosis. Cancer Res. 2003, 163, 7436–7442. [Google Scholar]
- Mohseny, A.B.; Machado, I.; Cai, Y.; Schaefer, K.L.; Serra, M.; Hogendoorn, P.C.; Llombart-Bosch, A.; Cleton-Jansen, A.M. Functional characterization of osteosarcoma cell lines provides representative models to study the human disease. Lab. Investig. 2011, 91, 1195–1205. [Google Scholar] [CrossRef] [PubMed]
- Peterse, E.F.P.; van den Akker, B.; Niessen, B.; Oosting, J.; Suijker, J.; de Jong, Y.; Danen, E.H.J.; Cleton-Jansen, A.M.; Bovee, J. NAD Synthesis Pathway Interference Is a Viable Therapeutic Strategy for Chondrosarcoma. Mol. Cancer Res. 2017, 15, 1714–1721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kresse, S.H.; Rydbeck, H.; Skarn, M.; Namlos, H.M.; Barragan-Polania, A.H.; Cleton-Jansen, A.M.; Serra, M.; Liestol, K.; Hogendoorn, P.C.; Hovig, E.; et al. Integrative analysis reveals relationships of genetic and epigenetic alterations in osteosarcoma. PLoS ONE 2012, 7, e48262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perry, J.A.; Kiezun, A.; Tonzi, P.; Van Allen, E.M.; Carter, S.L.; Baca, S.C.; Cowley, G.S.; Bhatt, A.S.; Rheinbay, E.; Pedamallu, C.S.; et al. Complementary genomic approaches highlight the PI3K/mTOR pathway as a common vulnerability in osteosarcoma. Proc. Natl. Acad. Sci. USA 2014, 111, E5564–E5573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kovac, M.; Blattmann, C.; Ribi, S.; Smida, J.; Mueller, N.S.; Engert, F.; Castro-Giner, F.; Weischenfeldt, J.; Kovacova, M.; Krieg, A.; et al. Exome sequencing of osteosarcoma reveals mutation signatures reminiscent of BRCA deficiency. Nat. Commun. 2015, 6, 8940. [Google Scholar] [CrossRef] [PubMed]
- Behjati, S.; Tarpey, P.S.; Haase, K.; Ye, H.; Young, M.D.; Alexandrov, L.B.; Farndon, S.J.; Collord, G.; Wedge, D.C.; Martincorena, I.; et al. Recurrent mutation of IGF signalling genes and distinct patterns of genomic rearrangement in osteosarcoma. Nat. Commun. 2017, 8, 15936. [Google Scholar] [CrossRef] [PubMed]
- Mutz, C.; Schwentner, R.; Aryee, D.; Bouchar, E.; Mjia, E.; Hatch, G.; Kauer, M.; Katschnig, A.; Ban, J.; Garten, A.; et al. EWS-FLI1 confers exquisite sensitivity to NAMPT inhibition in Ewing sarcoma cells. Oncotarget 2017, 8, 24679–24693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Del Nagro, C.; Xiao, Y.; Rangell, L.; Reichelt, M.; O’Brien, T. Depletion of the central metabolite NAD leads to oncosis-mediated cell death. J. Biol. Chem. 2014, 289, 35182–35192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weerasinghe, P.; Buja, L.M. Oncosis: An important non-apoptotic mode of cell death. Exp. Mol. Pathol. 2012, 93, 302–308. [Google Scholar] [CrossRef]
- Billington, R.A.; Genazzani, A.A.; Travelli, C.; Condorelli, F. NAD depletion by FK866 induces autophagy. Autophagy 2008, 4, 385–387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, X.; He, J.; Shi, Y.; Pi, C.; Yang, Y.; Sun, Y.; Ma, C.; Lin, L.; Zhang, L.; Li, Y.; et al. Nicotinamide phosphoribosyltransferase (Nampt) may serve as the marker for osteoblast differentiation of bone marrow-derived mesenchymal stem cells. Exp. Cell. Res. 2017, 352, 45–52. [Google Scholar] [CrossRef] [PubMed]
- Duarte-Pereira, S.; Pereira-Castro, I.; Silva, S.; Correia, M.; Neto, C.; da Costa, L.; Amorim, A.; Silva, R. Extensive regulation of nicotinate phosphoribosyltransferase (NAPRT) expression in human tissues and tumors. Oncotarget 2015, 7, 1973–1983. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cole, J.; Guiot, M.; Gravel, M.; Bernier, C.; Shore, G.; Roulston, A. Novel NAPRT specific antibody identifies small cell lung cancer and neuronal cancers as promising clinical indication for a NAMPT inhibitor/niacin co-administration strategy. Oncotarget 2017, 8, 77846–77859. [Google Scholar] [CrossRef] [Green Version]
- Yaku, K.; Okabe, K.; Hikosaka, K.; Nakagawa, T. NAD Metabolism in Cancer Therapeutics. Front. Oncol. 2018, 8, 622. [Google Scholar] [CrossRef]
- Lee, J.; Kim, H.; Lee, J.E.; Shin, S.J.; Oh, S.; Kwon, G.; Kim, H.; Choi, Y.Y.; White, M.A.; Paik, S.; et al. Selective Cytotoxicity of the NAMPT Inhibitor FK866 Toward Gastric Cancer Cells With Markers of the Epithelial-Mesenchymal Transition, Due to Loss of NAPRT. Gastroenterology 2018, 155, 799–814.e713. [Google Scholar] [CrossRef]
- Gong, X.; Lin, C.; Cheng, J.; Su, J.; Zhao, H.; Liu, T.; Wen, X.; Zhao, P. Generation of Multicellular Tumor Spheroids with Microwell-Based Agarose Scaffolds for Drug Testing. PLoS ONE 2015, 10, e0130348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schreiber-Brynzak, E.; Klapproth, E.; Unger, C.; Lichtscheidl-Schultz, I.; Goschl, S.; Schweighofer, S.; Trondl, R.; Dolznig, H.; Jakupec, M.A.; Keppler, B.K. Three-dimensional and co-culture models for preclinical evaluation of metal-based anticancer drugs. Investig. New Drugs 2015, 33, 835–847. [Google Scholar] [CrossRef]
- Lagies, S.; Schlimpert, M.; Neumann, S.; Waldin, A.; Kammerer, B.; Borner, C.; Peintner, L. Cells grown in three-dimensional spheroids mirror in vivo metabolic response of epithelial cells. Commun. Biol. 2020, 3, 246. [Google Scholar] [CrossRef] [PubMed]
- Holen, K.; Saltz, L.B.; Hollywood, E.; Burk, K.; Hanauske, A.R. The pharmacokinetics, toxicities, and biologic effects of FK866, a nicotinamide adenine dinucleotide biosynthesis inhibitor. Investig. New Drugs 2008, 26, 45–51. [Google Scholar] [CrossRef]
- Watson, M.; Roulston, A.; Belec, L.; Billot, X.; Marcellus, R.; Bedard, D.; Bernier, C.; Branchaud, S.; Chan, H.; Dairi, K.; et al. The small molecule GMX1778 is a potent inhibitor of NAD+ biosynthesis: Strategy for enhanced therapy in nicotinic acid phosphoribosyltransferase 1-deficient tumors. Mol. Cell. Biol. 2009, 29, 5872–5888. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olesen, U.H.; Thougaard, A.V.; Jensen, P.B.; Sehested, M. A preclinical study on the rescue of normal tissue by nicotinic acid in high-dose treatment with APO866, a specific nicotinamide phosphoribosyltransferase inhibitor. Mol. Cancer Ther. 2010, 9, 1609–1617. [Google Scholar] [CrossRef] [Green Version]
- Beauparlant, P.; Bedard, D.; Bernier, C.; Chan, H.; Gilbert, K.; Goulet, D.; Gratton, M.O.; Lavoie, M.; Roulston, A.; Turcotte, E.; et al. Preclinical development of the nicotinamide phosphoribosyl transferase inhibitor prodrug GMX1777. Anticancer. Drugs 2009, 20, 346–354. [Google Scholar] [CrossRef] [PubMed]
- Shames, D.S.; Elkins, K.; Walter, K.; Holcomb, T.; Du, P.; Mohl, D.; Xiao, Y.; Pham, T.; Haverty, P.M.; Liederer, B.; et al. Loss of NAPRT1 expression by tumor-specific promoter methylation provides a novel predictive biomarker for NAMPT inhibitors. Clin. Cancer Res. 2013, 19, 6912–6923. [Google Scholar] [CrossRef] [Green Version]
- Ottaviano, L.; Schaefer, K.L.; Gajewski, M.; Huckenbeck, W.; Baldus, S.; Rogel, U.; Mackintosh, C.; de Alava, E.; Myklebost, O.; Kresse, S.H.; et al. Molecular characterization of commonly used cell lines for bone tumor research: A trans-European EuroBoNet effort. Genes Chromosomes Cancer 2010, 49, 40–51. [Google Scholar] [CrossRef]
- Zhang, W.; Li, C.; Baguley, B.C.; Zhou, F.; Zhou, W.; Shaw, J.P.; Wang, Z.; Wu, Z.; Liu, J. Optimization of the formation of embedded multicellular spheroids of MCF-7 cells: How to reliably produce a biomimetic 3D model. Anal. Biochem. 2016, 515, 47–54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hafner, M.; Niepel, M.; Chung, M.; Sorger, P.K. Growth rate inhibition metrics correct for confounders in measuring sensitivity to cancer drugs. Nature Methods 2016, 13, 521–527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Haaften, C.; Boot, A.; Corver, W.E.; van Eendenburg, J.D.; Trimbos, B.J.; van Wezel, T. Synergistic effects of the sesquiterpene lactone, EPD, with cisplatin and paclitaxel in ovarian cancer cells. J. Exp. Clin. Cancer Res. 2015, 34, 38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franceschini, N.; Verbruggen, B.; Tryfonidou, M.A.; Kruisselbrink, A.B.; Baelde, H.; de Visser, K.E.; Szuhai, K.; Cleton-Jansen, A.M.; Bovée, J.V.M.G. Transformed murine and canine mesenchymal stem cells as a model for sarcoma with complex genomics. Cancers 2021, 13, 1126. [Google Scholar] [CrossRef] [PubMed]
- Bankhead, P.; Loughrey, M.B.; Fernandez, J.A.; Dombrowski, Y.; McArt, D.G.; Dunne, P.D.; McQuaid, S.; Gray, R.T.; Murray, L.J.; Coleman, H.G.; et al. QuPath: Open source software for digital pathology image analysis. Sci. Rep. 2017, 7, 16878. [Google Scholar] [CrossRef] [Green Version]
- Rozeman, L.B.; Hameetman, L.; Cleton-Jansen, A.M.; Taminiau, A.H.; Hogendoorn, P.C.; Bovee, J.V. Absence of IHH and retention of PTHrP signalling in enchondromas and central chondrosarcomas. J. Pathol. 2005, 205, 476–482. [Google Scholar] [CrossRef] [PubMed] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Forward Primer | Reverse Primer |
|---|---|---|
| ALPL | TCACTCTCCGAGATGGTGGT | GCCTGCTTGGCTTTTCCTTC |
| COL1A1 | AAGACGAAGACATCCCACCAAT | GTCACAGATCACGTCATCGCA |
| SPARC | CTGGACTACATCGGGCCTTG | CAGGACGTTCTTGAGCCAGT |
| RUNX2 | CCCTGAACTCTGCACCAAGT | GGCTCAGGTAGGAGGGGTAA |
| SPP1 | TTCGCAGACCTGACATCCAG | ACGGCTGTCCCAATCAGAAG |
| NAMPT | GGAGCATCTGCTCACTTGGT | TCATGGTCTTTCCCCCAAGC |
| NAPRT | GCTGGAGTCAGTCCTCATCG | TATAGACGCCACCCAGGGAA |
| CAPNS1 | ATGGTTTTGGCATTGACACATG | GCTTGCCTGTGGTGTCGC |
| SRPR | CATTGCTTTTGCACGTAACCAA | ATTGTCTTGCATGCGGCC |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Franceschini, N.; Oosting, J.; Tamsma, M.; Niessen, B.; Bruijn, I.B.-d.; van den Akker, B.; Kruisselbrink, A.B.; Palubeckaitė, I.; Bovée, J.V.M.G.; Cleton-Jansen, A.-M. Targeting the NAD Salvage Synthesis Pathway as a Novel Therapeutic Strategy for Osteosarcomas with Low NAPRT Expression. Int. J. Mol. Sci. 2021, 22, 6273. https://doi.org/10.3390/ijms22126273
Franceschini N, Oosting J, Tamsma M, Niessen B, Bruijn IB-d, van den Akker B, Kruisselbrink AB, Palubeckaitė I, Bovée JVMG, Cleton-Jansen A-M. Targeting the NAD Salvage Synthesis Pathway as a Novel Therapeutic Strategy for Osteosarcomas with Low NAPRT Expression. International Journal of Molecular Sciences. 2021; 22(12):6273. https://doi.org/10.3390/ijms22126273
Chicago/Turabian StyleFranceschini, Natasja, Jan Oosting, Maud Tamsma, Bertine Niessen, Inge Briaire-de Bruijn, Brendy van den Akker, Alwine B. Kruisselbrink, Ieva Palubeckaitė, Judith V. M. G. Bovée, and Anne-Marie Cleton-Jansen. 2021. "Targeting the NAD Salvage Synthesis Pathway as a Novel Therapeutic Strategy for Osteosarcomas with Low NAPRT Expression" International Journal of Molecular Sciences 22, no. 12: 6273. https://doi.org/10.3390/ijms22126273
APA StyleFranceschini, N., Oosting, J., Tamsma, M., Niessen, B., Bruijn, I. B. -d., van den Akker, B., Kruisselbrink, A. B., Palubeckaitė, I., Bovée, J. V. M. G., & Cleton-Jansen, A. -M. (2021). Targeting the NAD Salvage Synthesis Pathway as a Novel Therapeutic Strategy for Osteosarcomas with Low NAPRT Expression. International Journal of Molecular Sciences, 22(12), 6273. https://doi.org/10.3390/ijms22126273