
Nanotechnology of Tyrosine Kinase Inhibitors in Cancer Therapy: A Perspective






Abstract
:1. Tyrosine Kinase Inhibitors
2. Nanoparticles for Cancer Treatment
2.1. Classification of Nanoparticles
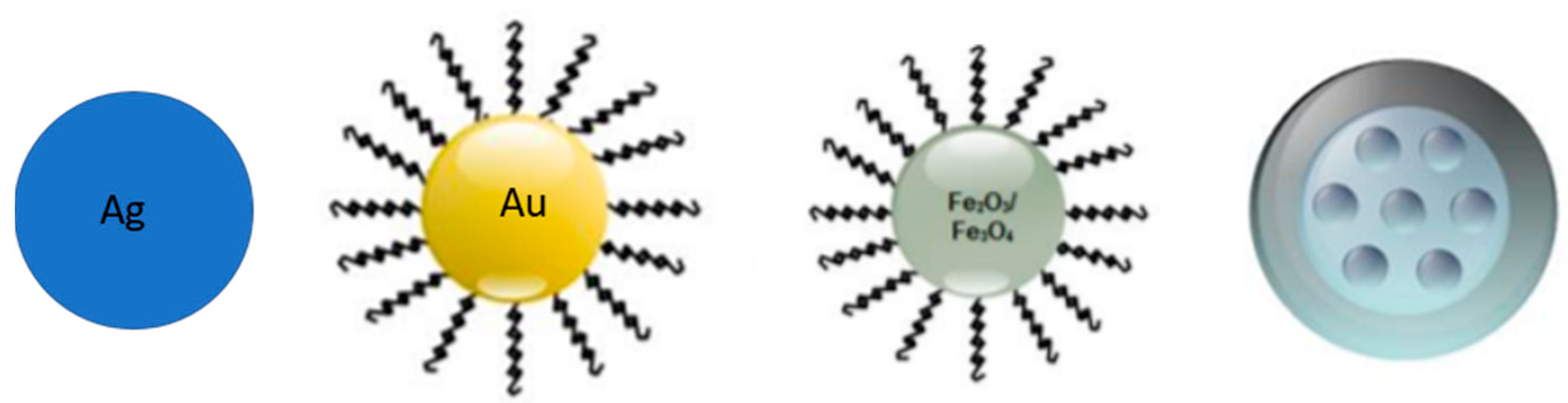
2.1.1. Inorganic Nanoparticles
Silica Nanoparticles
Silver Nanoparticles
Gold Nanoparticles
Magnetic Nanoparticles
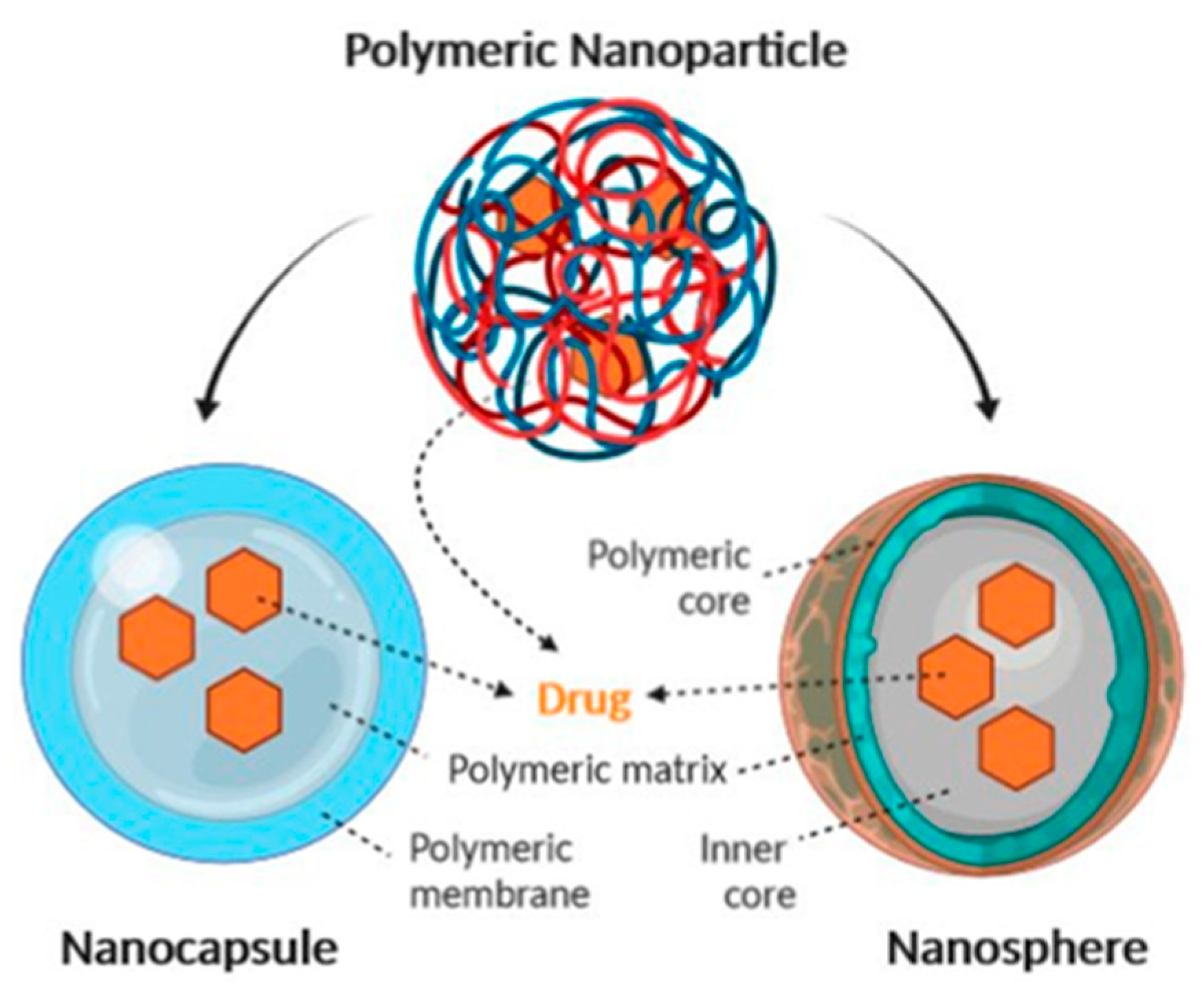
2.1.2. Organic Nanoparticles
Lipid-Based Nanoparticles (Liposomes and Solid Lipidic Nanoparticles)
Polymer-Based Nanoparticles
Albumin-Based Nanocarriers
3. Nanoparticles of Tyrosine Kinase Inhibitors
3.1. Imatinib
3.1.1. Liposome–Imatinib
3.1.2. Lipid Nanocarrier–Imatinib
3.1.3. Inorganic Nanoparticles–Imatinib
3.1.4. Polymeric Organic Nanoparticles–Imatinib
3.2. Dasatinib
3.2.1. Solid Lipid Nanoparticles (SLNs)–Dasatinib
3.2.2. Inorganic NPs–Dasatinib
3.2.3. Polymeric Organic Nanoparticles–Dasatinib
3.2.4. Targeted NPs–Dasatinib
3.3. Nilotinib
3.4. Ponatinib
3.5. Sunitinib
3.6. Sorafenib
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| Abl | Abelson kinase |
| ADR | Accumulation, distribution and retention |
| AgNPs | Silver nanoparticles |
| ALL | Acute lymphoblastic lymphoma |
| AMF | Alternative magnetic field |
| ApoA-I | Apolipoprotein A-I |
| Au-NP | Gold nanoparticles |
| BSA | Bovine serum albumin |
| Cbz | Carboxybenzyl |
| CED | Convection-enhanced delivery |
| CNTs | Carbon nanotubes |
| cRGD | Arg–Gly–Asp |
| CS | Chitosan |
| DDS | Drug delivery system |
| DexPLGA | Dextran and poly(lactide-co-glycolide) |
| DLS | Dynamic light scattering |
| DPPC | 1,2-dipalmitoyl-sn-glycero-3-phosphocholine |
| EE | Encapsulation efficiency |
| EPR | Enhanced permeability and retention |
| FDA | Food and Drug Administration |
| Fmoc | 9-fluorenylmethoxycarbonyl |
| GA | Glycolic acid |
| GVHD | Graft-versus-host disease |
| HCC | Hepatocellular carcinoma |
| HPMC | Hydroxypropyl Me cellulose |
| HSCs | Hepatic stellate cells |
| IM | Imatinib |
| JNK | C-Jun N-terminal kinase |
| LA | Lactic acid |
| LbL | Layer-by-layer |
| LNCs | Lipid nanocapsules |
| MDR | Multidrug resistant |
| MMP2 | Matrix metalloproteinase 2 |
| MNPs | Magnetic nanoparticles |
| MS | Multiple sclerosis |
| nHAp | Hydroxyapatite |
| NPs | Nanoparticles |
| NSCLC | Non-small cell lung cancer |
| PAA | Poly acrylic acid |
| PAMAM | Poly(amidoamine) |
| PBCA | Polybutylcyanoacrylate nanoparticles |
| PCL | Poly(ε-caprolactone) |
| PDGFR | Platelet-derived growth factor receptor |
| PDI | Polydispersity index |
| PDT | Photodynamic therapy |
| PEG | Polyethylene glycol |
| PEI | Polyethyleneimine |
| PET | Positron emission tomography PET |
| PGA | Poly glycolic acid |
| PK | Pharmacokinetic |
| PLA | Poly lactic acid |
| PLGA | Poly(lactide-co-glycolide) |
| PLGA-PEG-Mal | Poly(lactic-co-glycolic)-poly(ethyleneglycol)-maleimide |
| PPI | Polypropylene imine |
| PSi | Mesoporous silica |
| PVR | Proliferative vitreoretinopathy |
| RCC | Renal cell carcinoma |
| RES | Reticuloendothelial system |
| RGD | Arg–Gly–Asp peptides |
| SA | Sodium alginate |
| SCFR | Mast/stem cell growth factor receptor |
| SEM-EDS | Scanning Electron Microscopy–Energy Dispersive X-ray Spectroscopy |
| SLNPs | Solid lipid nanoparticles |
| SR-B1 | Scavenger type B1 receptor |
| STAT | Signal transducer and activator of transcription |
| TAM | Tumor-associated macrophages |
| TKIs | Tyrosine kinase inhibitors |
| Vd | Volume of distribution |
| XRPD | X-ray powder diffraction |
| ZnPc | Zinc phthalocyanine |
References
- Robertson, S.C.; Tynan, J.; Donoghue, D.J. RTK mutations and human syndromes: When good receptors turn bad. Trends Genet. 2000, 16, 265–271. [Google Scholar] [CrossRef]
- Moradpour, Z.; Barghi, L. Novel Approaches for Efficient Delivery of Tyrosine Kinase Inhibitors. J. Pharm. Pharm. Sci. 2019, 22, 37–48. [Google Scholar] [CrossRef] [Green Version]
- Yin, Y.; Yuan, X.; Gao, H.; Yang, Q. Nanoformulations of small molecule protein tyrosine kinases inhibitors potentiate targeted cancer therapy. Int. J. Pharm. 2020, 573, 118785. [Google Scholar] [CrossRef] [PubMed]
- Druker, B.J.; Lydon, N.B. Lessons learned from the development of an Abl tyrosine kinase inhibitor for chronic mye-logenous leukemia. J. Clin. Investig. 2000, 15, 3–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pisters, P.W.; Patel, S.R. Gastrointestinal stromal tumors: Current management. J. Surg. Oncol. 2010, 102, 530–538. [Google Scholar] [CrossRef]
- Cheah, C.Y.; Burbury, K.; Apperley, J.F.; Huguet, F.; Pitini, V.; Gardembas, M.; Ross, D.M.; Forrest, D.; Genet, P.; Rousselot, P.; et al. Patients with myeloid malignancies bearing PDGFRB fusion genes achieve durable long-term remissions with imatinib. Blood 2014, 123, 3574–3577. [Google Scholar] [CrossRef] [Green Version]
- Eilers, G.; Czaplinski, J.T.; Mayeda, M.; Bahri, N.; Tao, D.; Zhu, M.; Hornick, J.; Lindeman, N.I.; Sicinska, E.; Wagner, A.J.; et al. CDKN2A/p16 Loss Implicates CDK4 as a Therapeutic Target in Imatinib-Resistant Dermatofibrosarcoma Protuberans. Mol. Cancer Ther. 2015, 14, 1346–1353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jabbour, E.J.; Cortes, J.E.; Kantarjian, H.M. Resistance to Tyrosine Kinase Inhibition Therapy for Chronic Myelogenous Leukemia: A Clinical Perspective and Emerging Treatment Options. Clin. Lymphoma Myeloma Leuk. 2013, 13, 515–529. [Google Scholar] [CrossRef] [Green Version]
- Weisberg, E.; Manley, P.W.; Cowan-Jacob, S.W.; Hochhaus, A.; Griffin, J.D. Second generation inhibitors of BCR-ABL for the treatment of imatinib-resistant chronic myeloid leukaemia. Nat. Rev. Cancer 2007, 7, 345–356. [Google Scholar] [CrossRef]
- Schenone, S.; Brullo, C.; Botta, M. New opportunities to treat the T315I-Bcr-Abl mutant in chronic myeloid leukaemia: Tyrosine kinase inhibitors and molecules that act by alternative mechanisms. Curr. Med. Chem. 2010, 17, 1220–1245. [Google Scholar] [CrossRef]
- Schenone, S.; Brullo, C.; Musumeci, F.; Botta, M. Novel dual Src/Abl inhibitors for hematologic and solid malignancies. Expert Opin. Investig. Drugs 2010, 19, 931–945. [Google Scholar] [CrossRef]
- Hantschel, O.; Rix, U.; Schmidt, U.; Bürckstümmer, T.; Kneidinger, M.; Schütze, G.; Colinge, J.; Bennett, K.L.; Ellmeier, W.; Valent, P.; et al. The Btk tyrosine kinase is a major target of the Bcr-Abl inhibitor dasatinib. Proc. Natl. Acad. Sci. USA 2007, 104, 13283–13288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilhelm, S.M.; Carter, C.; Tang, L.; Wilkie, D.; McNabola, A.; Rong, H.; Chen, C.; Zhang, X.; Vincent, P.; McHugh, M.; et al. BAY 43-9006 exhibits broad spectrum oral antitumor activity and targets the RAF/MEK/ERK pathway and receptor tyrosine kinases involved in tumor progression and angio-genesis. Cancer Res. 2004, 64, 7099–7109. [Google Scholar] [CrossRef] [Green Version]
- Jain, L.; Woo, S.; Gardner, E.R.; Dahut, W.L.; Kohn, E.C.; Kummar, S.; Mould, D.R.; Giaccone, G.; Yarchoan, R.; Venitz, J.; et al. Population pharmacokinetic analysis of sorafenib in patients with solid tumours. Br. J. Clin. Pharmacol. 2011, 72, 294–305. [Google Scholar] [CrossRef] [Green Version]
- Boudou-Rouquette, P.; Narjoz, C.; Golmard, J.L.; Thomas-Schoemann, A.; Mir, O.; Taieb, F.; Durand, J.-P.; Coriat, R.; Dauphin, A.; Vidal, M.; et al. Early Sorafenib-Induced Toxicity Is Associated with Drug Exposure and UGTIA9 Genetic Polymorphism in Patients with Solid Tumors: A Preliminary Study. PLoS ONE 2012, 7, e42875. [Google Scholar] [CrossRef] [Green Version]
- Ravandi, F.; Cortes, J.E.; Jones, D.; Faderl, S.; Garcia-Manero, G.; Konopleva, M.Y.; Susan O’Brien, S.; Zeev Estrov, Z.; Borthakur, G.; Thomas, D.; et al. Phase I/II study of combination therapy with sorafenib, idarubicin, and cytarabine in younger patients with acute myeloid leukemia. J. Clin. Oncol. 2010, 28, 1856–1862. [Google Scholar] [CrossRef]
- Duncan, J.S.; Whittle, M.C.; Nakamura, K.; Abell, A.N.; Midland, A.A.; Zawistowski, J.S.; Johnson, N.L.; Granger, D.A.; Jordan, N.V.; Darr, D.B.; et al. Dynamic Reprogramming of the Kinome in Response to Targeted MEK Inhibition in Triple-Negative Breast Cancer. Cell 2012, 149, 307–321. [Google Scholar] [CrossRef] [Green Version]
- Abdelgalil, A.A.; Alkahtani, H.M.; Al-Jenoobi, F.I. Sorafenib. In Profiles of Drug Substances, Excipients and Related Methodology; Academic Press: New York, NY, USA, 2019; Volume 44, pp. 239–266. [Google Scholar]
- Boland, P.; Wu, J. Systemic therapy for hepatocellular carcinoma: Beyond sorafenib. Chin. Clin. Oncol. 2018, 7, 50. [Google Scholar] [CrossRef]
- Tan, F.H.; Putoczki, T.L.; Stylli, S.S.; Luwor, R.B. Ponatinib: A novel multi-tyrosine kinase inhibitor against human ma-lignancies. OncoTargets Ther. 2019, 12, 635–645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wylie, A.A.; Schoepfer, J.; Jahnke, W.; Cowan-Jacob, S.W.; Loo, A.; Furet, P.; Marzinzik, A.L.; Pelle, X.; Donovan, J.; Zhu, W.; et al. The allosteric inhibitor ABL001 enables dual targeting of BCR–ABL1. Nature 2017, 543, 733–737. [Google Scholar] [CrossRef] [PubMed]
- Hughes, T.P.; Mauro, M.J.; Cortes, J.E.; Minami, H.; Rea, D.; DeAngelo, D.J.; Breccia, M.; Goh, Y.T.; Talpaz, M.; Hoch-haus, A.; et al. Asciminib in Chronic Myeloid Leukemia after ABL Kinase Inhibi-tor Failure. N. Engl. J. Med. 2019, 381, 2315–2326. [Google Scholar] [CrossRef]
- Schoepfer, J.; Jahnke, W.; Berellini, G.; Buonamici, S.; Cotesta, S.; Cowan-Jacob, S.W.; Dodd, S.; Drueckes, P.; Fabbro, D.; Gabriel, T.; et al. Discovery of Asciminib (ABL001), an Allosteric Inhibitor of the Tyrosine Kinase Activity of BCR-ABL1. J. Med. Chem. 2018, 61, 8120–8135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Li, M.; Yanli, Z.; Huanling, Z.; Jiuwei, C.; Aining, S.; Yu, H.; Jie, J.; Hao, J.; Xi, Z.; et al. Frontline flumatinib versus imatinib in patients with chronic myeloid leukemia in chronic phase: Results from the China randomized phase III study. J. Clin. Oncol. 2019, 37, 7004. [Google Scholar]
- Goldman, A.; Kulkarni, A.; Kohandel, M.; Pandey, P.; Rao, P.; Natarajan, S.K.; Sabbisetti, V.; Sengupta, S. Rationally Designed 2-in-1 Nanoparticles Can Overcome Adaptive Resistance in Cancer. ACS Nano 2016, 10, 5823–5834. [Google Scholar] [CrossRef] [PubMed]
- Golombek, S.K.; May, J.-N.; Theek, B.; Appold, L.; Drude, N.; Kiessling, F.; Lammers, T. Tumor targeting via EPR: Strategies to enhance patient responses. Adv. Drug Deliv. Rev. 2018, 130, 17–38. [Google Scholar] [CrossRef]
- Svenson, S. What nanomedicine in the clinic right now really forms nanoparticles? Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2014, 6, 125–135. [Google Scholar] [CrossRef]
- Torchilin, V. Multifunctional and stimuli-sensitive pharmaceutical nanocarriers. Eur. J. Pharm. Biopharm. 2009, 71, 431–444. [Google Scholar] [CrossRef] [Green Version]
- Matsumura, Y.; Maeda, H. A new concept for macromolecular therapeutics in cancer chemotherapy: Mechanism of tumoritropic accumulation of proteins and the antitumor agent smancs. Cancer Res. 1986, 46, 6387–6392. [Google Scholar]
- Byrne, J.D.; Betancourt, T.; Brannon-Peppas, L. Active targeting schemes for nanoparticle systems in cancer therapeutics. Adv. Drug Deliv. Rev. 2008, 60, 1615–1626. [Google Scholar] [CrossRef]
- Lee, J.Y.; Chung, S.J.; Cho, H.J.; Kim, D.D. Bile acid-conjugated chondroitin sulfate: A-based nanoparticles for tumor-targeted anticancer drug delivery. Eur. J. Pharm. Biopharm. 2015, 94, 532–541. [Google Scholar] [CrossRef]
- Danhier, F. To exploit the tumor microenvironment: Since the EPR effect fails in the clinic, what is the future of nanomedicine? J. Control. Release 2016, 244, 108–121. [Google Scholar] [CrossRef]
- Lammers, T.; Kiessling, F.; Hennink, W.E.; Storm, G. Drug targeting to tumors: Principles, pitfalls and (pre-) clinical progress. J. Control. Release 2012, 161, 175–187. [Google Scholar] [CrossRef]
- Wang, S.; Low, P.S. Folate-mediated targeting of antineoplastic drugs, imaging agents, and nucleic acids to cancer cells. J. Control. Release 1998, 53, 39–48. [Google Scholar] [CrossRef]
- Qian, Z.M.; Li, H.; Sun, H.; Ho, K. Targeted drug delivery via the transferrin receptor mediated endocytosis pathway. Pharmacol. Rev. 2002, 54, 561–587. [Google Scholar] [CrossRef] [PubMed]
- Seymour, L.W.; Ferry, D.R.; Anderson, D.; Hesslewood, S.; Julyan, P.J.; Poyner, R.; Doran, J.; Young, A.M.; Burtles, S.; Kerr, D.J. Cancer Research Campaign Phase I/II Clinical Trials Committee, hepatic drug targeting: Phase I evaluation of polymer-bound doxorubicin. J. Clin. Oncol. 2002, 20, 1668–1676. [Google Scholar] [CrossRef]
- Sutradhar, K.B.; Amin, M.L. Nanotechnology in cancer drug delivery and selective targeting. ISRN Nanotechnol. 2014, 939378. [Google Scholar] [CrossRef] [Green Version]
- Farokhzad, O.C.; Cheng, J.; Teply, B.A.; Sherifi, I.; Jon, S.; Kantoff, P.W.; Richie, J.P.; Langer, R. Targeted nanoparticle-aptamer bioconjugates for cancer chemotherapy in vivo. Proc. Natl. Acad. Sci. USA 2006, 103, 6315–6320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsushima, A.; Kodera, Y.; Hiroto, M.; Nishimura, H.; Inada, Y. Bioconjugates of proteins and polyethylene glycol: Potent tools in biotechnological processes. J. Mol. Catal. B Enzym. 1996, 2, 1–17. [Google Scholar] [CrossRef]
- Mishra, P.; Nayak, B.; Dey, R. PEGylation in anti-cancer therapy: An overview. Asian J. Pharm. Sci. 2016, 11, 337–348. [Google Scholar] [CrossRef] [Green Version]
- Hayata, S.M.G.; Bianconib, V.; Pirrob, M.; Sahebkar, A. Stealth functionalization of biomaterials and nanoparticles by CD47 mimicry. Int. J. Pharm. 2019, 569, 118628. [Google Scholar] [CrossRef]
- Guo, S.; Huang, L. Nanoparticles Escaping RES and Endosome: Challenges for siRNA Delivery for Cancer Therapy. J. Nanomater. 2011, 742895. [Google Scholar] [CrossRef] [Green Version]
- Vijayan, V.; Uthaman, S.; Park, I.-K. Cell Membrane-Camouflaged Nanoparticles: A Promising Biomimetic Strategy for Cancer Theragnostics. Polymers 2018, 10, 983. [Google Scholar] [CrossRef] [Green Version]
- Sun, H.; Su, J.; Meng, Q.; Yin, Q.; Chen, L.; Gu, W.; Zhang, P.; Zhang, Z.; Yu, H.; Wang, S.; et al. Cancer-cell-biomimetic nanoparticles for targeted therapy of homotypic tumors. Adv. Mater. 2016, 28, 9581–9588. [Google Scholar] [CrossRef]
- Anirudhan, T.S.; Nair, A.S. Temperature and ultrasound sensitive gatekeepers for the controlled release of chemother-apeutic drugs from mesoporous silica nanoparticles. J. Mater. Chem. B 2018, 6, 428–439. [Google Scholar] [CrossRef]
- Guo, Y.; Zhang, Y.; Ma, J.; Li, Q.; Li, Y.; Zhou, X.; Zhao, D.; Song, H.; Chen, Q.; Zhu, X. Light/magnetic hyperthermia triggered drug released from multifunctional thermo-sensitive magneto liposomes for precise cancer synergetic theranostics. J. Control. Release 2017, 272, 145–158. [Google Scholar] [CrossRef] [PubMed]
- Ojo, O.A.; Olayide, I.I.; Akalabu, M.C.; Ajiboye, B.O.; Ojo, A.B.; Oyinloye, B.E.; Ramalingam, M. Nanoparticles and their biomedical applications. Biointerface Res. Appl. Chem. 2021, 11, 8431–8445. [Google Scholar]
- Montaseri, H.; Kruger, C.; Abrahamse, H. Inorganic Nanoparticles Applied for Active Targeted Photodynamic Therapy of Breast Cancer. Pharmaceutics 2021, 13, 296. [Google Scholar] [CrossRef]
- Zhou, H.; Ge, J.; Miao, Q.; Zhu, R. Biodegradable Inorganic Nanoparticles for Cancer Theranostics: Insights into the Degradation Behavior. Bioconjugate Chem. 2020, 31, 315–331. [Google Scholar] [CrossRef]
- Meena, J.; Gupta, A.; Ahuja, R. Inorganic nanoparticles for natural product delivery: A review. Environ. Chem. Lett. 2020, 18, 2107–2118. [Google Scholar] [CrossRef]
- Zhao, X.; Abulikemu, A.; Lv, S.; Qi, Y.; Duan, J.; Zhang, J.; Chen, R.; Guo, C.; Li, Y.; Sun, Z. Oxidative stress- and mitochon-drial dysfunction-mediated cytotoxicity by silica nanoparticle in lung epithelial cells from metabolomic perspective. Chemosphere 2021, 275, 129969. [Google Scholar] [CrossRef] [PubMed]
- Gherasim, O.; Puiu, R.A.; Bîrcă, A.C.; Burdușel, A.-C.; Grumezescu, A.M. An Updated Review on Silver Nanoparticles in Biomedicine. Nanomaterials 2020, 10, 2318. [Google Scholar] [CrossRef]
- Nejati, K.; Dadashpour, M.; Gharibi, T.; Mellatyar, H.; Akbarzadeh, A. Biomedical Applications of Functionalized Gold Nanoparticles: A Review. J. Clust. Sci. 2021, 1–16. [Google Scholar] [CrossRef]
- Ali, A.; Zafar, H.; Zia, M.; Haq, I.; Phull, A.R.; Ali, J.S.; Hussain, A. Synthesis, characterization, applications, and challenges of iron oxide nanoparticles. Nanotechnol. Sci. Appl. 2016, 9, 49–67. [Google Scholar] [CrossRef] [Green Version]
- Rayegan, A.; Allafchian, A.; Sarsari, I.A.; Kameli, P. Synthesis and characterization of basil seed mucilage coated Fe3O4 magnetic nanoparticles as a drug carrier for the controlled delivery of cephalexin. Int. J. Biol. Macromol. 2018, 113, 317–328. [Google Scholar] [CrossRef] [PubMed]
- Li, R.N.; Da, X.H.; Li, X.; Lu, Y.S.; Gu, F.F.; Liu, Y. Functionalized magnetic nanoparticles for drug delivery in tumor therapy. Chin. Phys. B 2021, 30, 017502. [Google Scholar] [CrossRef]
- Bangham, A.; Horne, R. Negative staining of phospholipids and their structural modification by surface-active agents as observed in the electron microscope. J. Mol. Biol. 1964, 8, 660-IN10. [Google Scholar] [CrossRef]
- Barenholz, Y. Doxil(R)–the first FDA-approved nano-drug: Lessons learned. J. Control. Release 2012, 160, 117–134. [Google Scholar] [CrossRef]
- Umbarkar, M.G. Niosome as a Novel Pharmaceutical Drug Delivery: A Brief Review Highlighting Formulation, Types, Composition and Application. Indian J. Pharm. Educ. Res. 2021, 55, s11–s28. [Google Scholar] [CrossRef]
- Gabizon, A.; Chisin, R.; Amselem, S.; Druckmann, S.; Cohen, R. Pharmacokinetic and imaging studies in patients receiving a formulation of liposome-associated Adriamycin. Br. J. Cancer 1991, 64, 1125–1132. [Google Scholar] [CrossRef] [Green Version]
- Metselaar, J.M.; Storm, G. Liposomes in the treatment of inflammatory disorders. Expert Opin. Drug Deliv. 2005, 2, 465–476. [Google Scholar] [CrossRef]
- Szebeni, J.; Moghimi, S.M. Liposome triggering of innate immune responses: A perspective on benefits and adverse reactions. J. Liposome Res. 2009, 19, 85–90. [Google Scholar] [CrossRef]
- Yatvin, M.B.; Kreutz, W.; Horwitz, B.A.; Shinitzky, M. pH-sensitive liposomes: Possible clinical implications. Science 1980, 210, 1253–1255. [Google Scholar] [CrossRef] [PubMed]
- Matsumura, Y.; Gotoh, M.; Muro, K.; Yamada, Y.; Shirao, K.; Shimada, Y.; Okuwa, M.; Matsumoto, S.; Miyata, Y.; Ohkura, H.; et al. Phase I and pharmacokinetic study of MCC-465, a doxorubicin (DXR) encapsulated in PEG immunoliposome, in patients with metastatic stomach cancer. Ann. Oncol. 2004, 15, 517–525. [Google Scholar] [CrossRef] [PubMed]
- Pirollo, K.F.; Chang, E.H. Does a targeting ligand influence nanoparticle tumor localization or uptake? Trends Biotechnol. 2008, 26, 552–558. [Google Scholar] [CrossRef]
- Martins, S.; Sarmento, B.; Ferreira, D.C.; Souto, E.B. Lipid-based colloidal carriers for peptide and protein delivery-liposomes versus lipid nanoparticles. Int. J. Nanomed. 2007, 2, 595–607. [Google Scholar]
- Geszke-Moritz, M.; Moritz, M. Solid lipid nanoparticles as attractive drug vehicles: Composition, properties and therapeutic strategies. Mater. Sci. Eng. C Mater. Biol. Appl. 2016, 68, 982–994. [Google Scholar] [CrossRef]
- Ekambaram, P.; Sathali, A.; Priyanka, K. Solid lipid nanoparticles: A review. Sci. Rev. Chem. Commun. 2012, 2, 80–102. [Google Scholar]
- Westesen, K.; Bunjes, H.; Koch, M. Physicochemical characterization of lipid nanoparticles and evaluation of their drug loading capacity and sustained release potential. J. Control. Release 1997, 48, 223–236. [Google Scholar] [CrossRef]
- Haider, M.; Abdin, S.M.; Kamal, L.; Orive, G. Nanostructured Lipid Carriers for Delivery of Chemotherapeutics: A Review. Pharmaceutics 2020, 12, 288. [Google Scholar] [CrossRef] [Green Version]
- Zielinska, A.; Carreiró, F.; Oliveira, A.M.; Neves, A.; Pires, B. Polymeric Nanoparticles: Production, Characterization, Toxicology and Ecotoxicology. Molecules 2020, 25, 3731. [Google Scholar] [CrossRef]
- Panyam, J.; Labhasetwar, V. Biodegradable nanoparticles for drug and gene delivery to cells and tissue. Adv. Drug Deliv. Rev. 2003, 55, 329–347. [Google Scholar] [CrossRef]
- Rancan, F.; Papakostas, D.; Hadam, S.; Hackbarth, S.; Delair, T.; Primard, C.; Verrier, B.; Sterry, W.; Blume-Peytavi, U.; Vogt, A. Investigation of Polylactic Acid (PLA) Nanoparticles as Drug Delivery Systems for Local Dermatotherapy. Pharm. Res. 2009, 26, 2027–2036. [Google Scholar] [CrossRef]
- Li, J.; Stayshich, R.M.; Meyer, T.Y. Exploiting Sequence To Control the Hydrolysis Behavior of Biodegradable PLGA Copolymers. J. Am. Chem. Soc. 2011, 133, 6910–6913. [Google Scholar] [CrossRef] [PubMed]
- Andima, M.; Costabile, G.; Isert, L.; Ndakala, A.J.; Derese, S.; Merkel, O.M. Evaluation of beta-Sitosterol Loaded PLGA and PEG-PLA Nanoparticles for Effective Treatment of Breast Cancer: Preparation, Physicochemical Characterization, and Antitumor Activity. Pharmaceutics 2018, 10, 232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghitman, J.; Biru, E.I.; Stan, R.; Iovu, H. Review of hybrid PLGA nanoparticles: Future of smart drug delivery and theranostics medicine. Mater. Des. 2020, 193, 108805. [Google Scholar] [CrossRef]
- Steichen, S.D.; Caldorera-Moore, M.; Peppas, N.A. A review of current nanoparticle and targeting moieties for the delivery of cancer therapeutics. Eur. J. Pharm. Sci. 2013, 48, 416–427. [Google Scholar] [CrossRef] [Green Version]
- Liechty, W.B.; Peppas, N.A. Expert opinion: Responsive polymer nanoparticles in cancer therapy. Eur. J. Pharm. Biopharm. 2012, 80, 241–246. [Google Scholar] [CrossRef] [Green Version]
- Lavasanifar, A.; Samuel, J.; Kwon, G.S. Poly(ethylene oxide)-block-poly(lamino acid) micelles for drug delivery. Adv. Drug Deliv. Rev. 2002, 54, 169–190. [Google Scholar] [CrossRef]
- Palazzolo, S.; Bayda, S.; Hadla, M.; Caligiuri, I.; Corona, G.; Toffoli, G.; Rizzolio, F. The Clinical Translation of Organic Nanomaterials for Cancer Therapy: A Focus on Polymeric Nanoparticles, Micelles, Liposomes and Exosomes. Curr. Med. Chem. 2018, 25, 4224–4268. [Google Scholar] [CrossRef]
- Rebrov, E.A.; Muzafarov, A.M.; Papkov, V.S.; Zhdanov, A.A. Three-dimensionally propagating polyorganosiloxanes. Dokl. Chem. 1989, 309, 376–380. [Google Scholar]
- Rengan, K.; Engel, R. Phosphonium cascade molecules. J. Chem. Soc. Chem. Commun. 1990, 1084–1085. [Google Scholar] [CrossRef]
- Zhou, L.L.; Roovers, J. Synthesis of novel carbosilane dendritic macromolecules. Macromolecules 1993, 26, 963–968. [Google Scholar] [CrossRef]
- Majoral, J.-P.; Caminade, A.-M. Dendrimers Containing Heteroatoms (Si, P, B, Ge, or Bi). Chem. Rev. 1999, 99, 845–880. [Google Scholar] [CrossRef]
- Shcharbin, D.; Dzmitruk, V.; Shakhbazau, A.; Goncharova, N.; Seviaryn, I.; Kosmacheva, S.; Potapnev, M.; Pedziwiatr-Werbicka, E.; Bryszewska, M.; Talabaev, M.; et al. Fourth Generation Phosphorus-Containing Dendrimers: Prospective Drug and Gene Delivery Carrier. Pharmaceutics 2011, 3, 458–473. [Google Scholar] [CrossRef] [Green Version]
- Iacobazzi, R.M.; Porcelli, L.; Lopedota, A.A.; Laquintana, V.; Lopalco, A.; Cutrignelli, A.; Altamura, E.; Di Fonte, R.; Azzariti, A.; Franco, M.; et al. Targeting human liver cancer cells with lactobionic acid-G(4)-PAMAM-FITC sorafenib loaded dendrimers. Int. J. Pharm. 2017, 528, 485–497. [Google Scholar] [CrossRef]
- Retnakumari, A.P.; Hanumanthu, P.L.; Malarvizhi, G.L.; Prabhu, R.; Sidharthan, N.; Thampi, M.V.; Menon, D.; Mony, U.; Menon, K.; Keechilat, P.; et al. Rationally Designed Aberrant Kinase-Targeted Endogenous Protein Nanomedicine against Oncogene Mutated/Amplified Refractory Chronic Myeloid Leukemia. Mol. Pharm. 2012, 9, 3062–3078. [Google Scholar] [CrossRef]
- Petros, R.A.; DeSimone, J.M. Strategies in the design of nanoparticles for therapeutic applications. Nat. Rev. Drug Discov. 2010, 9, 615–627. [Google Scholar] [CrossRef]
- Sparreboom, A.; Scripture, C.D.; Trieu, V.; Williams, P.J.; De, T.; Yang, A.; Beals, B.; Figg, W.D.; Hawkins, M.; Desai, N. Comparative Preclinical and Clinical Pharmacokinetics of a Cremophor-Free, Nanoparticle Albumin-Bound Paclitaxel (ABI-007) and Paclitaxel Formulated in Cremophor (Taxol). Clin. Cancer Res. 2005, 11, 4136–4143. [Google Scholar] [CrossRef] [Green Version]
- Elzoghby, A.O.; Samy, W.M.; Elgindy, N.A. Albumin-based nanoparticles as potential controlled drug delivery systems. J. Control. Release 2012, 157, 168–182. [Google Scholar] [CrossRef]
- Onda, T.; Masuda, A.; Yamakawa, K.; Tomiyama, C.; Yoneta, Y.; Akatsu, Y.; Yamamoto, K.; Mochizuki, A.; Inventors; Nippon Kayaku Co Ltd. Block Copolymer Conjugate of Physiologically Active Substance. United States Patent US 10,357,573, 23 July 2019. [Google Scholar]
- Dravid, V.P.; Nandwana, V.; Inventors; Northwestern University Assignee. Nanoparticle-Lipid Composite Carriers and Uses Thereof. United States Patent Application US 16/615,260, 4 June 2020. [Google Scholar]
- Xu, H.; Ji, H.; Li, Z.; Qiao, W.; Wang, C.; Tang, J. In vivo Pharmacokinetics and in vitro Release of Imatinib Mesylate-Loaded Liposomes for Pulmonary Delivery. Int. J. Nanomed. 2021, 16, 1221–1229. [Google Scholar] [CrossRef] [PubMed]
- Soundararajan, R.; Wang, G.; Petkova, A.; Uchegbu, I.F.; Schätzlein, A.G. Hyaluronidase Coated Molecular Envelope Technology Nanoparticles Enhance Drug Absorption via the Subcutaneous Route. Mol. Pharm. 2020, 17, 2599–2611. [Google Scholar] [CrossRef] [PubMed]
- Amiri, M.; Gholami, T.; Amiri, O.; Pardakhti, A.; Ahmadi, M.; Akbari, A.; Amanatfard, A.; Salavati-Niasari, M. The magnetic inorganic-organic nanocomposite based on ZnFe2O4-Imatinib-liposome for biomedical applications, in vivo and in vitro study. J. Alloy. Compd. 2020, 849, 156604. [Google Scholar] [CrossRef]
- Peres, M.J.; dos Santos, A.P.; Nascimento, T.L.; de Ávila, R.I.; Saba Ferreira, F.; Campos Valadares, M.; Martins Lima, E. Antiproliferative Activity and VEGF Expression Reduction in MCF7 and PC-3 Cancer Cells by Paclitaxel and Imatinib Co-encapsulation in Folate-Targeted Liposomes. AAPS PharmSciTech 2018, 19, 201–212. [Google Scholar] [CrossRef]
- Gupta, B.; Poudel, B.K.; Tran, T.H.; Pradhan, R.; Cho, H.J.; Jeong, J.H.; Shin, B.S.; Choi, H.G.; Yong, C.S.; Kim, J.O. Modulation of Pharmacokinetic and Cytotoxicity Profile of Imatinib Base by Employing Optimized Nanostructured Lipid Carriers. Pharm. Res. 2015, 32, 2912–2927. [Google Scholar] [CrossRef]
- Molaahmadi, M.R.; Varshosaz, J.; Taymouri, S.; Akbari, V. Lipid Nanocapsules for Imatinib Delivery: Design, Optimization and Evaluation of Anticancer Activity Against Melanoma Cell Line. Iran. J. Pharm. Res. 2019, 18, 1676–1693. [Google Scholar]
- Sobierajska, P.; Serwotka-Suszczak, A.; Szymanski, D.; Marycz, K.; Wiglusz, R.J. Nanohydroxyapatite-Mediated Imatinib Delivery for Specific Anticancer Applications. Molecules 2020, 25, 4602. [Google Scholar] [CrossRef] [PubMed]
- Shandiz, S.A.S.; Ardestani, M.S.; Shahbazzadeh, D.; Assadi, A.; Cohan, R.A.; Asgary, V.; Salehi, S. Novel imatinib-loaded silver nanoparticles for enhanced apoptosis of human breast cancer MCF-7 cells. Artif. Cells Nanomed. Biotechnol. 2016, 45, 1082–1091. [Google Scholar] [CrossRef]
- Labala, S.; Jose, A.; Chawla, S.R.; Khan, M.S.; Bhatnagar, S.; Kulkarni, O.P.; Venuganti, V.V.K. Effective melanoma cancer suppression by iontophoretic co-delivery of STAT3 siRNA and imatinib using gold nanoparticles. Int. J. Pharm. 2017, 525, 407–417. [Google Scholar] [CrossRef] [PubMed]
- Hasandoost, L.; Akbarzadeh, A.; Attar, H.; Heydarinasab, A. In vitro effect of imatinib mesylate loaded on polybutyl-cyanoacrylate nanoparticles on leukemia cell line K562. Artif. Cells Nanomed. Biotechnol. 2017, 45, 665–669. [Google Scholar] [CrossRef] [Green Version]
- Bhattacharya, S. Fabrication and characterization of chitosan-based polymeric nanoparticles of Imatinib for colorectal cancer targeting application. Int. J. Biol. Macromol. 2020, 151, 104–115. [Google Scholar] [CrossRef]
- Gao, J.; Qiao, Z.; Liu, S.; Xu, J.; Wang, S.; Yang, X.; Wang, X.; Tang, R. Preparation of small molecule prodrug composed of pH-sensitive orthoester and dasatinib conjugate. Eur. J. Pharm. Biopharm. 2021, 163, 188–197. [Google Scholar] [CrossRef] [PubMed]
- Horner, G.; Rai, P.; Agrawal, S.; Parker, B. Remotely Triggered Therapy. United States Patent Application US 17/000,205, 17 December 2020. [Google Scholar]
- Tran, D.; Le, S.B. Methods for Targeted Treatment and Prediction of Patient Survival in Cancer. PCT Int. Appl. WO 2020163639 A1 20200813, 13 August 2020. [Google Scholar]
- Zhang, L.; Wang, S.; Zhang, M.; Sun, J. Nanocarriers for oral drug delivery. J. Drug Target. 2013, 21, 515–527. [Google Scholar] [CrossRef]
- Chauhan, R.; Balgemann, R.; Greb, C.; Nunn, B.M.; Ueda, S.; Noma, H.; McDonald, K.; Kaplan, H.J.; Tamiya, S.; O’Toole, M.G. Production of dasatinib encapsulated spray-dried poly (lactic-co-glycolic acid) particles. J. Drug Deliv. Sci. Technol. 2019, 53, 101204. [Google Scholar] [CrossRef]
- Li, Q.; Lai, K.L.; Chan, P.S.; Leung, S.C.; Li, H.Y.; Fang, Y.; To, K.; Choi, C.H.J.; Gao, Q.Y.; Lee, T.W. Micellar delivery of dasatinib for the inhibition of pathologic cellular processes of the retinal pigment epithelium. Colloids Surf. B Biointerfaces 2016, 140, 278–286. [Google Scholar] [CrossRef]
- Mohamed, Y.A.A.A.; Jaykar, B. Enhancement of oral bioavailability via solid lipid nanoparticles of anticancer drug dasatinib—An in vitro cytotoxicity and pharmacokinetic study. Asian J. Pharm. Clin. Res. 2019, 12, 143–145. [Google Scholar]
- Mohamed, Y.A.A.A.; Jaykar, B. Formulation, characterization and in-vitro evaluation of dasatinib loaded solid lipid nanoparticles for oral delivery. J. Glob. Pharm. Technol. 2019, 11, 12–20. [Google Scholar]
- Begum, M.Y.; Gudipati, P.R. Formulation and evaluation of dasatinib loaded solid lipid nanoparticles. Int. J. Pharm. Pharm. Sci. 2018, 10, 14–20. [Google Scholar] [CrossRef]
- Adena, S.K.R.; Upadhyay, M.; Vardhan, H.; Mishra, B. Gold nanoparticles for sustained antileukemia drug release: Development, optimization and evaluation by quality-by-design approach. Nanomedicine 2019, 14, 851–870. [Google Scholar] [CrossRef]
- Adena, S.K.R.; Upadhyay, M.; Vardhan, H.; Mishra, B. Development, optimization, and in vitro characterization of dasatinib-loaded PEG functionalized chitosan capped gold nanoparticles using Box-Behnken experimental design. Drug Dev. Ind. Pharm. 2018, 44, 493–501. [Google Scholar] [CrossRef]
- Li, N.-S.; Gossai, N.P.; Naumann, J.A.; Gordon, P.M.; Piccirilli, J.A. Efficient Synthetic Approach to Linear Dasatinib–DNA Conjugates by Click Chemistry. Bioconjugate Chem. 2016, 27, 2575–2579. [Google Scholar] [CrossRef]
- Prigodich, A.E.; Seferos, D.S.; Massich, M.D.; Giljohann, D.A.; Lane, B.C.; Mirkin, C.A. Nano-flares for mRNA regulation and detection. ACS Nano 2009, 3, 2147–2152. [Google Scholar] [CrossRef]
- Fröhling, S.; Döhner, H. Chromosomal Abnormalities in Cancer. N. Engl. J. Med. 2008, 359, 722–734. [Google Scholar] [CrossRef] [PubMed]
- Gossai, N.P.; Naumann, J.A.; Li, N.-S.; Zamora, E.A.; Gordon, D.J.; Piccirilli, J.A.; Gordon, P.M. Drug conjugated nanoparticles activated by cancer cell specific mRNA. Oncotarget 2016, 7, 38243–38256. [Google Scholar] [CrossRef] [Green Version]
- Vadia, N.; Rajput, S. Statistically Designed Formulation Development of Mesoporous Nanoparticulate Drug Delivery System of Dasatinib for Improved Dissolution and Drug Stability. In Advances in Spectroscopy: Molecules to Materials; Springer: The Gateway, Singapore, 2019; Volume 236, pp. 269–286. [Google Scholar]
- Sabra, S.A.; Sheweita, S.A.; Haroun, M.; Ragab, D.; Eldemellawy, M.A.; Xia, Y.; Goodale, D.; Allan, A.L.; Elzoghby, A.O.; Rohani, S. Magnetically Guided Self-Assembled Protein Micelles for Enhanced Delivery of Dasatinib to Human Triple-Negative Breast Cancer Cells. J. Pharm. Sci. 2019, 108, 1713–1725. [Google Scholar] [CrossRef] [PubMed]
- Valero, T.; Delgado-González, A.; Unciti-Broceta, J.D.; Cortes, V.C.; Pérez-López, A.M.; Unciti-Broceta, A.; Martín, R.M.S. Drug “Clicking” on Cell-Penetrating Fluorescent Nanoparticles for In Cellulo Chemical Proteomics. Bioconjugate Chem. 2018, 29, 3154–3160. [Google Scholar] [CrossRef] [Green Version]
- Niza, E.; Nieto-Jiménez, C.; Noblejas-López, M.D.M.; Bravo, I.; Castro-Osma, J.A.; De La Cruz-Martínez, F.; Buchaca, M.M.D.S.; Posadas, I.; Canales-Vázquez, J.; Lara-Sanchez, A.; et al. Poly(Cyclohexene Phthalate) Nanoparticles for Controlled Dasatinib Delivery in Breast Cancer Therapy. Nanomaterials 2019, 9, 1208. [Google Scholar] [CrossRef] [Green Version]
- Allotey-Babington, G.L.; Nettey, H.; D’Sa, S.; Gomes, K.B.; D’Souza, M.J. Cancer chemotherapy: Effect of poloxamer modified nanoparticles on cellular function. J. Drug Deliv. Sci. Technol. 2018, 47, 181–192. [Google Scholar] [CrossRef]
- Zhang, P.; Li, J.; Ghazwani, M.; Zhao, W.; Huang, Y.; Zhang, X.; Venkataramanan, R.; Li, S. Effective co-delivery of doxorubicin and dasatinib using a PEG-Fmoc nanocarrier for combination cancer chemotherapy. Biomaterials 2015, 67, 104–114. [Google Scholar] [CrossRef] [Green Version]
- Moore, T.L.; Grimes, S.W.; Lewis, R.L.; Alexis, F. Multilayered Polymer-Coated Carbon Nanotubes To Deliver Dasatinib. Mol. Pharm. 2013, 11, 276–282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Veach, D.R.; Namavari, M.; Pillarsetty, N.; Santos, E.B.; Beresten-Kochetkov, T.; Lambek, C.; Punzalan, B.J.; Antczak, C.; Smith-Jones, P.M.; Djaballah, H.; et al. Synthesis and biological evaluation of a fluorine-18 derivativeof dasatinib. J. Med. Chem. 2007, 50, 5853–5857. [Google Scholar] [CrossRef]
- Dong, C.; Li, B.; Li, Z.; Shetty, S.; Fu, J. Dasatinib-loaded albumin nanoparticles possess diminished endothelial cell barrier disruption and retain potent anti-leukemia cell activity. Oncotarget 2016, 7, 49699–49709. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, D.-J.; Yi, Y.W.; Kim, J.H. In Situ Monitoring of Bindings between Dasatinib and Its Target Protein Kinases Using Magnetic Nanoparticles in Live Cells. J. Am. Chem. Soc. 2008, 130, 16466–16467. [Google Scholar] [CrossRef] [PubMed]
- Egeblad, M.; Werb, Z. New functions for the matrix metalloproteinases in cancer progression. Nat. Rev. Cancer 2002, 2, 161–174. [Google Scholar] [CrossRef]
- Dai, Z.; Yao, Q.; Zhu, L. MMP2-Sensitive PEG–Lipid Copolymers: A New Type of Tumor-Targeted P-Glycoprotein Inhibitor. ACS Appl. Mater. Interfaces 2016, 8, 12661–12673. [Google Scholar] [CrossRef]
- Yao, Q.; Choi, J.H.; Dai, Z.; Wang, J.; Kim, D.; Tang, X.; Zhu, L. Improving Tumor Specificity and Anticancer Activity of Dasatinib by Dual-Targeted Polymeric Micelles. ACS Appl. Mater. Interfaces 2017, 9, 36642–36654. [Google Scholar] [CrossRef]
- Yao, Q.; Liu, Y.; Kou, L.; Tu, Y.; Tang, X.; Zhu, L. Tumor-targeted drug delivery and sensitization by MMP2-responsive polymeric micelles. Nanomed. Nanotechnol. Biol. Med. 2019, 19, 71–80. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Wang, J.; Zhang, J.; Marbach, S.; Xu, W.; Zhu, L. Targeting Tumor-Associated Macrophages by MMP2-Sensitive Apoptotic Body-Mimicking Nanoparticles. ACS Appl. Mater. Interfaces 2020, 12, 52402–52414. [Google Scholar] [CrossRef]
- Tosi, U.; Kommidi, H.; Bellat, V.; Marnell, C.S.; Guo, H.; Adeuyan, O.; Schweitzer, M.E.; Chen, N.; Su, T.; Zhang, G.; et al. Real-Time, In Vivo Correlation of Molecular Structure with Drug Distribution in the Brain Striatum Following Convection Enhanced Delivery. ACS Chem. Neurosci. 2019, 10, 2287–2298. [Google Scholar] [CrossRef]
- Benezra, M.; Phillips, E.; Overholtzer, M.; Zanzonico, P.B.; Tuominen, E.; Wiesner, U.; Bradbury, M.S. Ultrasmall integrin-targeted silica nanoparticles modulate signaling events and cellular processes in a concentration-dependent manner. Small 2015, 11, 1721–1732. [Google Scholar] [CrossRef] [Green Version]
- Juthani, R.; Madajewski, B.; Yoo, B.; Zhang, L.; Chen, P.-M.; Chen, F.; Turker, M.Z.; Ma, K.; Overholtzer, M.; Longo, V.A.; et al. Ultrasmall Core-Shell Silica Nanoparticles for Precision Drug Delivery in a High-Grade Malignant Brain Tumor Model. Clin. Cancer Res. 2019, 26, 147–158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, L.; Zhou, H.; Liu, L.; He, H.; Liang, R. Preparation Method and Application of Nanoparticle Drug-Carrying System. Faming Zhuanli Shenqing CN 110859817, 6 March 2020. [Google Scholar]
- McKeon, F.; Duleba, M.; Zhang, Y.; Xie, J.; Xian, W.; Vincent, M. Screening Methods for Identifying Therapeutic Agents for Treating Chronic Inflammatory Injury, Metaplasia, Dysplasia and Cancers of Epithelial Tissues. PCT Int. Appl. WO 2020219963, 29 November 2020. [Google Scholar]
- Koehl, N.J.; Griffin, B.T.; Holm, R.; Holm, R.; Kuentz, M. Chase Dosing of Lipid Formulations to Enhance Oral Bioavailability of Nilotinib in Rats. Pharm. Res. 2020, 37, 124. [Google Scholar] [CrossRef]
- Jesson, G.; Brisander, M.; Andersson, P.; Demirbueker, M.; Derand, H.; Lennernaes, H.; Malmsten, M. Carbon Dioxide-Mediated Generation of Hybrid Nanoparticles for Improved Bioavailability of Protein Kinase Inhibitors. Pharm. Res. 2014, 31, 694–705. [Google Scholar] [CrossRef] [Green Version]
- Fan, Q.-Q.; Zhang, C.-L.; Qiao, J.-B.; Cui, P.-F.; Xing, L.; Oh, Y.-K.; Jiang, H.-L. Extracellular matrix-penetrating nanodrill micelles for liver fibrosis therapy. Biomaterials 2020, 230, 119616. [Google Scholar] [CrossRef]
- Cortese, B.; D’Amone, S.; Palamà, I.E. Wool-Like Hollow Polymeric Nanoparticles for CML Chemo-Combinatorial Therapy. Pharmaceutics 2018, 10, 52. [Google Scholar] [CrossRef] [Green Version]
- Robinson, W.H.; Postolova, A.; Raghu, H. Tyrosine Kinase Inhibitor Formulations for the Treatment of Mast Cell-Mediated Inflammatory Diseases and Methods of Use Thereof. United States Patent Application US 20170312282, 2 November 2017. [Google Scholar]
- Zhang, Y.; Zhang, H.; Peng, R. Preparation and in vitro release of ponatinib nanosuspensions. Guangdong Yaoxueyuan Xuebao 2014, 30, 544–548. [Google Scholar]
- Zinger, A.; Baudo, G.; Naoi, T.; Giordano, F.; Lenna, S.; Massaro, M.; Ewing, A.; Kim, H.R.; Tasciotti, E.; Yustein, J.T.; et al. Reproducible and Characterized Method for Ponatinib Encapsulation into Biomimetic Lipid Nanoparticles as a Platform for Multi-Tyrosine Kinase-Targeted Therapy. ACS Appl. Bio Mater. 2020, 3, 6737–6745. [Google Scholar] [CrossRef]
- Kallus, S.; Englinger, B.; Senkiv, J.; Laemmerer, A.; Heffeter, P.; Berger, W.; Kowol, C.R.; Keppler, B.K. Nanoformulations of anticancer FGFR inhibitors with improved therapeutic index. Nanomedicine 2018, 14, 2632–2643. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Tao, C.; Song, X.; Li, W. Preparation Method of Injectable Sunitinib Nanoparticle. Faming Zhuanli Shenqing CN 108030927, 15 May 2018. [Google Scholar]
- Otroj, M.; Taymouri, S.; Varshosaz, J.; Mirian, M. Preparation and characterization of dry powder containing sunitinib loaded PHBV nanoparticles for enhanced pulmonary deliver. J. Drug Deliv. Sci. Technol. 2020, 56, 101570. [Google Scholar] [CrossRef]
- Joseph, J.J.; Sangeetha, D.; Gomathi, T. Sunitinib loaded chitosan nanoparticles formulation and its evaluation. Int. J. Biol. Macromol. 2016, 82, 952–958. [Google Scholar] [CrossRef]
- Shi, J.; Ju, R.; Sun, M.; Li, X.; Zhao, Y.; Zeng, F.; Lu, W. Development of targeted sunitinib plus vinorelbine liposomes modified with DSPE-PEG2000-pemetrexed conjugate and the inhibitory effect to resistant breast cancer in vitro. J. Chin. Pharm. Sci. 2014, 23, 287–294. [Google Scholar] [CrossRef]
- Shi, J.; Sun, M.; Li, X.; Zhao, Y.; Ju, R.; Mu, L.; Yan, Y.; Li, X.; Zeng, F.; Lu, W. A combination of targeted sunitinib lipo-somes and targeted vinorelbine liposomes for treating invasive breast cancer. J. Biomed. Nanotechnol. 2015, 11, 1568–1582. [Google Scholar] [CrossRef] [PubMed]
- Bhatt, P.; Narvekar, P.; Lalani, R.; Chougule, M.B.; Pathak, Y.; Sutariya, V. An in vitro Assessment of Thermo-Reversible Gel Formulation Containing Sunitinib Nanoparticles for Neovascular Age-Related Macular Degeneration. AAPS PharmSciTech 2019, 20, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Zhang, F.; Yan, S. Preparation, in vitro release, and pharmacokinetics in rabbits of lyophilized injection of sorafenib solid lipid nanoparticles. Int. J. Nanomed. 2012, 7, 2901–2910. [Google Scholar] [CrossRef] [Green Version]
- Xu, X.; Tang, X.; Wu, X.; Feng, X. Biosynthesis of sorafenib coated graphene nanosheets for the treatment of gastric cancer in patients in nursing care. J. Photochem. Photobiol. B Biol. 2019, 191, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Almeida, P.V.; Shahbazi, M.; Correia, A.; Maekilae, E.; Kemell, M.; Salonen, J.; Hirvonen, J.; Santos, H. A multifunctional nanocomplex for enhanced cell uptake, endosomal escape and improved cancer therapeutic effect. Nanomedicine 2017, 12, 1401–1420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheng, X.; Huang, T.; Qin, J.; Li, Q.; Wang, W.; Deng, L.; Dong, A. Preparation, pharmacokinetics, tissue distribution and antitumor effect of sorafenib-incorporating nanoparticles in vivo. Oncol. Lett. 2017, 14, 6163–6169. [Google Scholar] [CrossRef] [Green Version]
- Park, S.Y.; Kang, Z.; Thapa, P.; Jin, Y.S.; Park, J.W.; Lim, H.J.; Lee, J.Y.; Lee, S.; Seo, M.; Kim, M.; et al. Development of sorafenib loaded nanoparticles to improve oral bioavailability using a quality by design approach. Intern. J. Pharm. 2019, 566, 229–238. [Google Scholar] [CrossRef]
- Kim, D.H.; Kim, M.-D.; Choi, C.-W.; Chung, C.-W.; Ha, S.H.; Kim, C.H.; Shim, Y.-H.; Jeong, Y.-I.; Kang, D.H. Antitumor activity of sorafenib-incorporated nanoparticles of dextran/poly(dl-lactide-co-glycolide) block copolymer. Nanoscale Res. Lett. 2012, 7, 91. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.-F.; Mäkilä, E.M.; Kaasalainen, M.H.; Liu, D.; Sarparanta, M.; Airaksinen, A.J.; Salonen, J.J.; Hirvonen, J.T.; Santos, H.A. Copper-free azide–alkyne cycloaddition of targeting peptides to porous silicon nanoparticles for intracellular drug uptake. Biomaterials 2014, 35, 1257–1266. [Google Scholar] [CrossRef] [PubMed]
- Hong, F.; Tuyama, A.; Lee, T.F.; Loke, J.; Agarwal, R.; Cheng, X.; Garg, A.; Fiel, M.I.; Schwartz, M.; Walewski, J.; et al. Hepatic stellate cells express functional CXCR4: Role in stromal cell-derived factor-1α-mediated stellate cell activation. Hepatology 2009, 49, 2055–2067. [Google Scholar] [CrossRef] [Green Version]
- Sung, Y.-C.; Liu, Y.-C.; Chao, P.-H.; Chang, C.-C.; Jin, P.-R.; Lin, T.-T.; Lin, J.-A.; Cheng, H.-T.; Wang, J.; Lai, C.P.; et al. Combined delivery of sorafenib and a MEK inhibitor using CXCR4-targeted nanoparticles reduces hepatic fibrosis and prevents tumor development. Theranostics 2018, 8, 894–905. [Google Scholar] [CrossRef]
- Chen, Y.; Liu, Y.C.; Sung, Y.C.; Ramjiawan, R.R.; Lin, T.T.; Chang, C.C.; Jeng, K.S.; Chang, C.F.; Liu, C.H.; Gao, D.Y.; et al. Overcoming sorafenib evasion in hepatocellular carcinoma using CXCR4-targeted nanoparticles to co-deliver MEK-inhibitors. Sci. Rep. 2017, 7, 44123. [Google Scholar] [CrossRef]
- Yu, X.-N.; Deng, Y.; Zhang, G.-C.; Liu, J.; Liu, T.-T.; Dong, L.; Zhu, C.-F.; Shen, X.-Z.; Li, Y.-H.; Zhu, J.-M. Sorafenib-Conjugated Zinc Phthalocyanine Based Nanocapsule for Trimodal Therapy in an Orthotopic Hepatocellular Carcinoma Xenograft Mouse Model. ACS Appl. Mater. Interfaces 2020, 12, 17193–17206. [Google Scholar] [CrossRef]
- Li, Z.; Ye, L.; Liu, J.; Lian, D.; Li, X. Sorafenib-loaded nanoparticles based on biodegradable dendritic polymers for enhanced therapy of hepatocellular carcinoma. Intern. J. Nanomed. 2020, 15, 1469–1480. [Google Scholar] [CrossRef] [Green Version]
- Tang, X.; Chen, L.; Li, A.; Cai, S.; Zhang, Y.; Liu, X.; Jiang, Z.; Liu, X.; Liang, Y.; Ma, D. Anti-GPC3 antibody-modified sorafenib-loaded nanoparticles significantly inhibited HepG2 hepatocellular carcinoma. Drug Deliv. 2018, 25, 1484–1494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, H.; Wang, Y.; He, X.; Zhang, Z.; Yin, Q.; Chen, Y.; Yu, H.; Huang, Y.; Chen, L.; Xu, M.; et al. Codelivery of Sorafenib and Curcumin by Directed Self-Assembled Nanoparticles Enhances Therapeutic Effect on Hepatocellular Carcinoma. Mol. Pharm. 2015, 12, 922–931. [Google Scholar] [CrossRef]
- Zhang, J.; He, B.; Qu, W.; Cui, Z.; Wang, Y.; Zhang, H.; Wang, J.; Zhang, Q. Preparation of the albumin nanoparticle system loaded with both paclitaxel and sorafenib and its evaluation in vitro and in vivo. J. Microencapsul. 2011, 28, 528–536. [Google Scholar] [CrossRef]
- Zan, Y.; Dai, Z.; Liang, L.; Deng, Y.; Dong, L. Co-delivery of plantamajoside and sorafenib by a multi-functional nano-particle to combat the drug resistance of hepatocellular carcinoma through reprograming the tumor hypoxic microenvironment. Drug Deliv. 2019, 26, 1080–1091. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Z.; Rana, T.M. Therapeutic targeting of microRNAs: Current status and future challenges. Nat. Rev. Drug Discov. 2014, 13, 622–638. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Su, Y.; Zhang, F.; Chen, K.; Xu, X.; Xu, L.; Zhou, J.; Wang, W. A dual-targeting reconstituted high density lipoprotein leveraging the synergy of sorafenib and antimiRNA21 for enhanced hepatocellular carcinoma therapy. Acta Biomater. 2018, 75, 413–426. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Boonkaew, B.; Arora, J.; Mandava, S.H.; Maddox, M.M.; Chava, S.; Callaghan, C.; He, J.; Dash, S.; John, V.T.; et al. Comparison of Sorafenib-Loaded Poly (Lactic/Glycolic) Acid and DPPC Liposome Nanoparticles in the in Vitro Treatment of Renal Cell Carcinoma. J. Pharm. Sci. 2015, 104, 1187–1196. [Google Scholar] [CrossRef] [PubMed]
- Poojari, R.; Kini, S.; Srivastava, R.; Panda, D. Intracellular interactions of electrostatically mediated layer-by-layer assembled polyelectrolytes based sorafenib nanoparticles in oral cancer cells. Colloids Surf. B Biointerfaces 2016, 143, 131–138. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Target | Number of Clinical Trials | Diseases | FDA Approval |
|---|---|---|---|---|
| Imatinib | Abl, PDGFR, Kit | 754 | CML, GIST, GVHD, many hematological and solid tumors | 2001 |
| Dasatinib | Abl, PDGFR, Kit, Src | 320 | CML, ALL, lymphoma, NSCLC and others solid tumors | 2006 |
| Nilotinib | Abl, PDGFR, c-Kit, LCK, EPHA3, EPHA8, DDR1, DDR2, MAPK11, ZAK | 219 | CML, ALL, GIST | 2007 |
| Ponatinib | Abl, Src, FGFR, PDGFR, VEGFR, | 67 | CML, ALL | 2012 |
| Asciminib | Abl | 13 | CML | // |
| Flumatinib | Abl, PDGFR, c-Kit, CSFR | 5 | CML | // |
| Sunitinib | PDGFR, Kit, FLT3, VEGFR, CSF1R | 610 | RCC, GIST | 2006 |
| Sorafenib | PDGFR, c-Kit, FLT3, VEGFR, B-Raf | 870 | RCC, liver and thyroid cancers | 2007 |
| Axitinib | Abl, PDGFR, VEGRF, c-Kit | 161 | RCC | 2012 |
| Ruxolitinib | JAK1, JAK2 | 258 | Myelofibrosis, polycythemia vera, GVHD, many other different diseases | 2011 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Russo, E.; Spallarossa, A.; Tasso, B.; Villa, C.; Brullo, C. Nanotechnology of Tyrosine Kinase Inhibitors in Cancer Therapy: A Perspective. Int. J. Mol. Sci. 2021, 22, 6538. https://doi.org/10.3390/ijms22126538
Russo E, Spallarossa A, Tasso B, Villa C, Brullo C. Nanotechnology of Tyrosine Kinase Inhibitors in Cancer Therapy: A Perspective. International Journal of Molecular Sciences. 2021; 22(12):6538. https://doi.org/10.3390/ijms22126538
Chicago/Turabian StyleRusso, Eleonora, Andrea Spallarossa, Bruno Tasso, Carla Villa, and Chiara Brullo. 2021. "Nanotechnology of Tyrosine Kinase Inhibitors in Cancer Therapy: A Perspective" International Journal of Molecular Sciences 22, no. 12: 6538. https://doi.org/10.3390/ijms22126538
APA StyleRusso, E., Spallarossa, A., Tasso, B., Villa, C., & Brullo, C. (2021). Nanotechnology of Tyrosine Kinase Inhibitors in Cancer Therapy: A Perspective. International Journal of Molecular Sciences, 22(12), 6538. https://doi.org/10.3390/ijms22126538