
Renin-Angiotensin System in Pathogenesis of Atherosclerosis and Treatment of CVD

 ,
,  and
and 
Abstract
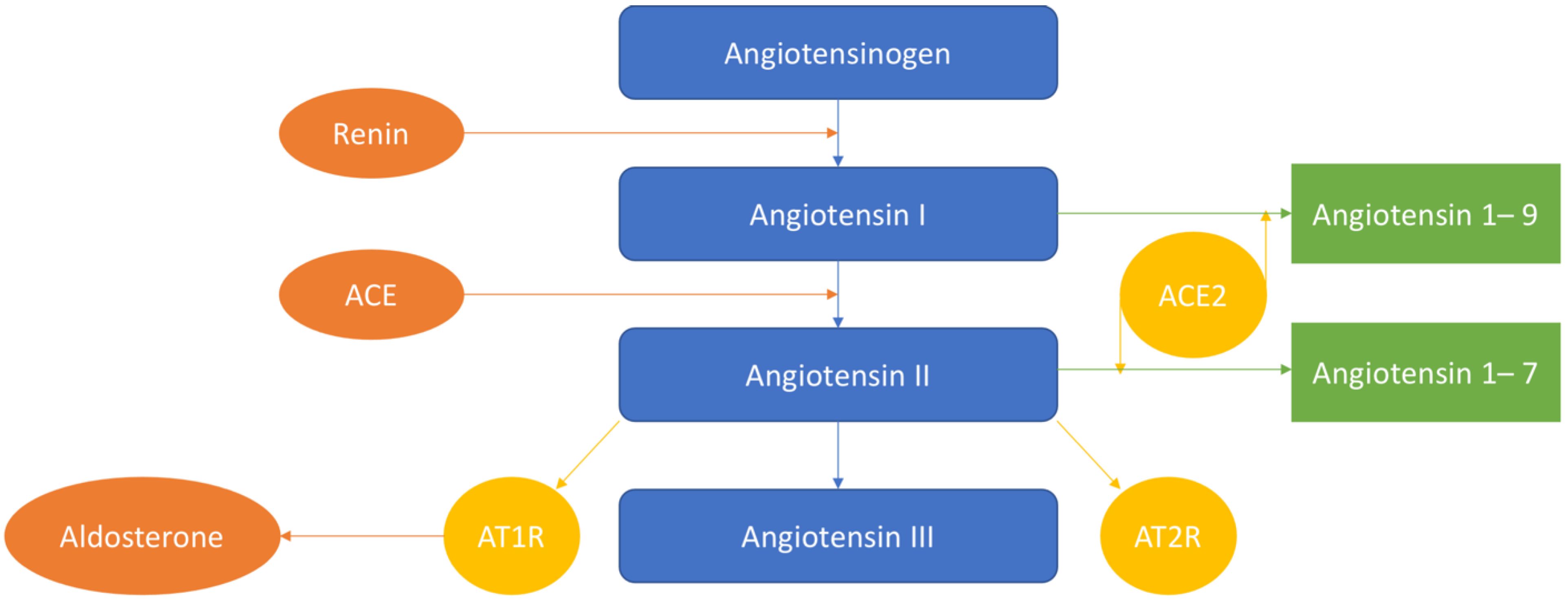
:1. Introduction
2. RAAS System: Inflammation
3. RAAS System: Oxidative Stress and ROS
4. RAAS and Endothelial Dysfunction
5. Other Actions of RAAS
6. Targeting Potential
6.1. RAAS Inhibition in Clinical Trials
6.2. Angiotensin-1–7
6.3. ACE-2
6.4. Angiotensin-II
6.5. Aldosterone
6.6. Renin
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Poznyak, A.V.; Nikiforov, N.G.; Markin, A.M.; Kashirskikh, D.A.; Myasoedova, V.A.; Gerasimova, E.V.; Orekhov, A.N. Overview of OxLDL and its impact on cardiovascular health: Focus on atherosclerosis. Front. Pharmacol. 2021, 11, 613780. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.Y.; Li, C.J.; Hou, M.F.; Chu, P.Y. new insights into the role of inflammation in the pathogenesis of atherosclerosis. Int. J. Mol. Sci. 2017, 18, 2034. [Google Scholar] [CrossRef] [PubMed]
- Kovanen, P.T. Mast cells as potential accelerators of human atherosclerosis—From early to late lesions. Int. J. Mol. Sci. 2019, 20, 4479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poznyak, A.V.; Wu, W.K.; Melnichenko, A.A.; Wetzker, R.; Sukhorukov, V.; Markin, A.M.; Khotina, V.A.; Orekhov, A.N. Signaling pathways and key genes involved in regulation of foam cell formation in atherosclerosis. Cells 2020, 9, 584. [Google Scholar] [CrossRef] [Green Version]
- Kattoor, A.J.; Pothineni, N.V.K.; Palagiri, D.; Mehta, J.L. Oxidative stress in atherosclerosis. Curr. Atheroscler. Rep. 2017, 19, 42. [Google Scholar] [CrossRef]
- Libby, P. Inflammation in atherosclerosis—No longer a theory. Clin. Chem. 2021, 67, 131–142. [Google Scholar] [CrossRef]
- Pacurari, M.; Kafoury, R.; Tchounwou, P.B.; Ndebele, K. The renin-angiotensin-aldosterone system in vascular inflammation and remodeling. Int. J. Inflamm. 2014, 2014, 689360. [Google Scholar] [CrossRef]
- Forrester, S.J.; Booz, G.W.; Sigmund, C.D.; Coffman, T.M.; Kawai, T.; Rizzo, V.; Scalia, R.; Eguchi, S. Angiotensin II signal transduction: An update on mechanisms of physiology and pathophysiology. Physiol. Rev. 2018, 98, 1627–1738. [Google Scholar] [CrossRef]
- Kolakovic, A.; Zivkovic, M.; Stankovic, A. Involvement of the renin-angiotensin system in atherosclerosis, renin-angiotensin system—Past, present and future. IntechOpen 2017. Available online: https://www.intechopen.com/books/renin-angiotensin-system-past-present-and-future/involvement-of-the-renin-angiotensin-system-in-atherosclerosis (accessed on 12 July 2017). [CrossRef] [Green Version]
- Wu, C.M.; Zheng, L.; Wang, Q.; Hu, Y.W. The emerging role of cell senescence in atherosclerosis. Clin. Chem. Lab. Med. 2020, 59, 27–38. [Google Scholar] [CrossRef]
- Wang, C.H.; Li, S.H.; Weisel, R.D.; Fedak, P.W.; Dumont, A.S.; Szmitko, P.; Li, R.K.; Mickle, D.A.; Verma, S. C-reactive protein upregulates angiotensin type 1 receptors in vascular smooth muscle. Circulation 2003, 107, 1783–1790. [Google Scholar] [CrossRef] [Green Version]
- Fiordelisi, A.; Iaccarino, G.; Morisco, C.; Coscioni, E.; Sorriento, D. NFkappaB is a key player in the crosstalk between inflammation and cardiovascular diseases. Int. J. Mol. Sci. 2019, 20, 1599. [Google Scholar] [CrossRef] [Green Version]
- Ismail, H.; Mitchell, R.; McFarlane, S.I.; Makaryus, A.N. Pleiotropic effects of inhibitors of the RAAS in the diabetic population: Above and beyond blood pressure lowering. Curr. Diabetes Rep. 2010, 10, 32–36. [Google Scholar] [CrossRef] [PubMed]
- Fliser, D.; Buchholz, K.; Haller, H.; European trial on Olmesartan and Pravastatin in Inflammation and Atherosclerosis (EUTOPIA) Investigators. Antiinflammatory effects of angiotensin II subtype 1 receptor blockade in hypertensive patients with microinflammation. Circulation 2004, 110, 1103–1107. [Google Scholar] [CrossRef] [Green Version]
- Van der Hoorn, J.W.; Kleemann, R.; Havekes, L.M.; Kooistra, T.; Princen, H.M.; Jukema, J.W. Olmesartan and pravastatin additively reduce development of atherosclerosis in APOE*3Leiden transgenic mice. J. Hypertens. 2007, 25, 2454–2462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sata, M.; Fukuda, D. Crucial role of renin-angiotensin system in the pathogenesis of atherosclerosis. J. Med. Investig. 2010, 57, 12–25. [Google Scholar] [CrossRef] [Green Version]
- McGraw, A.P.; Bagley, J.; Chen, W.S.; Galayda, C.; Nickerson, H.; Armani, A.; Caprio, M.; Carmeliet, P.; Jaffe, I.Z. Aldosterone increases early atherosclerosis and promotes plaque inflammation through a placental growth factor-dependent mechanism. J. Am. Heart Assoc. 2013, 2, e000018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raz-Pasteur, A.; Gamliel-Lazarovich, A.; Gantman, A.; Coleman, R.; Keidar, S. Mineralocorticoid receptor blockade inhibits accelerated atherosclerosis induced by a low sodium diet in apolipoprotein E-deficient mice. J. Renin. Angiotensin Aldosterone Syst. 2014, 15, 228–235. [Google Scholar] [CrossRef] [PubMed]
- Peng, R.; Luo, M.; Tian, R.; Lu, N. Dietary nitrate attenuated endothelial dysfunction and atherosclerosis in apolipoprotein E knockout mice fed a high-fat diet: A critical role for NADPH oxidase. Arch. Biochem. Biophys. 2020, 689, 108453. [Google Scholar] [CrossRef]
- Durante, A.; Peretto, G.; Laricchia, A.; Ancona, F.; Spartera, M.; Mangieri, A.; Cianflone, D. Role of the renin-angiotensin-aldosterone system in the pathogenesis of atherosclerosis. Curr. Pharm. Des. 2012, 18, 981–1004. [Google Scholar] [CrossRef]
- Poznyak, A.V.; Grechko, A.V.; Orekhova, V.A.; Khotina, V.; Ivanova, E.A.; Orekhov, A.N. NADPH oxidases and their role in atherosclerosis. Biomedicines 2020, 8, 206. [Google Scholar] [CrossRef]
- Montezano, A.C.; Cat, A.M.; Rios, F.J.; Touyz, R.M. Angiotensin II and vascular injury. Curr. Hypertens. Rep. 2014, 16, 431. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.; Jiang, Z.; Zhang, H.; Zhu, H.; Zhou, S.F.; Yu, X. NADPH oxidase-dependent formation of reactive oxygen species contributes to angiotensin II-induced epithelial-mesenchymal transition in rat peritoneal mesothelial cells. Int. J. Mol. Med. 2011, 28, 405–412. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, J.P.; Carmody, R.J. NF-κB and the transcriptional control of inflammation. Int. Rev. Cell Mol. Biol. 2018, 335, 41–84. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Xia, Y.; Hu, B. Infection and atherosclerosis: TLR-dependent pathways. Cell. Mol. Life Sci. 2020, 77, 2751–2769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, D.; Elner, S.G.; Bian, Z.M.; Till, G.O.; Petty, H.R.; Elner, V.M. Pro-inflammatory cytokines increase reactive oxygen species through mitochondria and NADPH oxidase in cultured RPE cells. Exp. Eye Res. 2007, 85, 462–472. [Google Scholar] [CrossRef] [Green Version]
- D’Ardes, D.; Santilli, F.; Guagnano, M.T.; Bucci, M.; Cipollone, F. From endothelium to lipids, through microRNAs and PCSK9: A fascinating travel across atherosclerosis. High Blood Press. Cardiovasc. Prev. 2020, 27, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Park, K.H.; Park, W.J. Endothelial dysfunction: Clinical implications in cardiovascular disease and therapeutic approaches. J. Korean Med. Sci. 2015, 30, 1213–1225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meza, C.A.; Favor, J.D.L.; Kim, D.H.; Hickner, R.C. Endothelial dysfunction: Is there a hyperglycemia-induced imbalance of NOX and NOS? Int. J. Mol. Sci. 2019, 20, 3775. [Google Scholar] [CrossRef] [Green Version]
- Münzel, T.; Camici, G.G.; Maack, C.; Bonetti, N.R.; Fuster, V.; Kovacic, J.C. Impact of oxidative stress on the heart and vasculature: Part 2 of a 3-part series. J. Am. Coll. Cardiol. 2017, 70, 212–229. [Google Scholar] [CrossRef]
- Bennett, M.R.; Sinha, S.; Owens, G.K. Vascular smooth muscle cells in atherosclerosis. Circ. Res. 2016, 118, 692–702. [Google Scholar] [CrossRef]
- Bauer, J.A.; Lupica, J.A.; Didonato, J.A.; Lindner, D.J. Nitric oxide inhibits NF-κB-mediated survival signaling: Possible role in overcoming TRAIL resistance. Anticancer Res. 2020, 40, 6751–6763. [Google Scholar] [CrossRef]
- Sharma, J.N.; Al-Omran, A.; Parvathy, S.S. Role of nitric oxide in inflammatory diseases. Inflammopharmacology 2007, 15, 252–259. [Google Scholar] [CrossRef]
- Rea, I.M.; Gibson, D.S.; McGilligan, V.; McNerlan, S.E.; Alexander, H.D.; Ross, O.A. Age and age-related diseases: Role of inflammation triggers and cytokines. Front. Immunol. 2018, 9, 586. [Google Scholar] [CrossRef] [PubMed]
- Takeda, Y.; Matoba, K.; Sekiguchi, K.; Nagai, Y.; Yokota, T.; Utsunomiya, K.; Nishimura, R. Endothelial dysfunction in diabetes. Biomedicines 2020, 8, 182. [Google Scholar] [CrossRef] [PubMed]
- Muñoz-Durango, N.; Fuentes, C.A.; Castillo, A.E.; González-Gómez, L.M.; Vecchiola, A.; Fardella, C.E.; Kalergis, A.M. Role of the renin-angiotensin-aldosterone system beyond blood pressure regulation: Molecular and cellular mechanisms involved in end-organ damage during arterial hypertension. Int. J. Mol. Sci. 2016, 17, 797. [Google Scholar] [CrossRef] [Green Version]
- Maas, C. Plasminflammation–An emerging pathway to bradykinin production. Front. Immunol. 2019, 10, 2046. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poston, R.N. Atherosclerosis: Integration of its pathogenesis as a self-perpetuating propagating inflammation: A review. Cardiovasc. Endocrinol. Metab. 2019, 8, 51–61. [Google Scholar] [CrossRef]
- Nehme, A.; Zouein, F.A.; Zayeri, Z.D.; Zibara, K. An update on the tissue renin angiotensin system and its role in physiology and pathology. J. Cardiovasc. Dev. Dis. 2019, 6, 14. [Google Scholar] [CrossRef] [Green Version]
- Morato, M.; Reina-Couto, M.; Pinho, D.; Albino-Teixeira, A.; Sousa, T. Regulation of the renin-angiotensin-aldosterone system by reactive oxygen species, renin-angiotensin system—Past, present and future. IntechOpen 2020. Available online: https://www.intechopen.com/books/renin-angiotensin-system-past-present-and-future/regulation-of-the-renin-angiotensin-aldosterone-system-by-reactive-oxygen-species (accessed on 12 July 2017). [CrossRef] [Green Version]
- Paul, A.S.; Corbett, C.B.; Okune, R.; Autieri, M.V. Angiotensin II, hypercholesterolemia, and vascular smooth muscle cells: A perfect trio for vascular pathology. Int. J. Mol. Sci. 2020, 21, 4525. [Google Scholar] [CrossRef] [PubMed]
- Fukuhara, M.; Geary, R.L.; Diz, D.I.; Gallagher, P.E.; Wilson, J.A.; Glazier, S.S.; Dean, R.H.; Ferrario, C.M. Angiotensin-converting enzyme expression in human carotid artery atherosclerosis. Hypertension 2000, 35, 353–359. [Google Scholar] [CrossRef] [Green Version]
- Ferrario, C.M.; Mullick, A.E. Renin angiotensin aldosterone inhibition in the treatment of cardiovascular disease. Pharmacol. Res. 2017, 125, 57–71. [Google Scholar] [CrossRef] [PubMed]
- Jiang, F.; Yang, J.; Zhang, Y.; Dong, M.; Wang, S.; Zhang, Q.; Liu, F.F.; Zhang, K.; Zhang, C. Angiotensin-converting enzyme 2 and angiotensin 1–7: Novel therapeutic targets. Nat. Rev. Cardiol. 2014, 11, 413–426. [Google Scholar] [CrossRef]
- Yang, J.M.; Dong, M.; Meng, X.; Zhao, Y.X.; Yang, X.Y.; Liu, X.L.; Hao, P.P.; Li, J.J.; Wang, X.P.; Zhang, K.; et al. Angiotensin-(1–7) dose-dependently inhibits atherosclerotic lesion formation and enhances plaque stability by targeting vascular cells. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 1978–1985. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ocaranza, M.P.; Riquelme, J.A.; García, L.; Jalil, J.E.; Chiong, M.; Santos, R.A.S.; Lavandero, S. Counter-regulatory renin-angiotensin system in cardiovascular disease. Nat. Rev. Cardiol. 2020, 17, 116–129. [Google Scholar] [CrossRef] [Green Version]
- Santos, S.H.; Braga, J.F.; Mario, E.G.; Pôrto, L.C.; Rodrigues-Machado, M.D.G.; Murari, A.; Botion, L.M.; Alenina, N.; Bader, M.; Santos, R.A. Improved lipid and glucose metabolism in transgenic rats with increased circulating angiotensin-(1–7). Arterioscler. Thromb. Vasc. Biol. 2010, 30, 953–961. [Google Scholar] [CrossRef] [Green Version]
- Qaradakhi, T.; Gadanec, L.K.; McSweeney, K.R.; Tacey, A.; Apostolopoulos, V.; Levinger, I.; Rimarova, K.; Egom, E.E.; Rodrigo, L.; Kruzliak, P.; et al. The potential actions of angiotensin-converting enzyme II (ACE2) activator diminazene aceturate (DIZE) in various diseases. Clin. Exp. Pharmacol. Physiol. 2020, 47, 751–758. [Google Scholar] [CrossRef]
- Macedo, S.M.D.; Guimarares, T.A.; Andrade, J.M.; Guimaraes, A.L.; Paula, A.M.B.D.; Ferreira, A.J.; Santos, S.H.S. Angiotensin converting enzyme 2 activator (DIZE) modulates metabolic profiles in mice, decreasing lipogenesis. Protein Pept. Lett. 2015, 22, 332–340. [Google Scholar] [CrossRef]
- Maria, M.L.D.; Araújo, L.D.; Fraga-Silva, R.A.; Pereira, L.A.; Ribeiro, H.J.; Menezes, G.B.; Shenoy, V.; Raizada, M.K.; Ferreira, A.J. Anti-hypertensive effects of diminazene aceturate: An angiotensin—Converting enzyme 2 activator in rats. Protein Pept. Lett. 2016, 23, 9–16. [Google Scholar] [CrossRef]
- Silva, G.M.; França-Falcão, M.S.; Calzerra, N.T.M.; Luz, M.S.; Gadelha, D.D.A.; Balarini, C.M.; Queiroz, T.M. Role of renin-angiotensin system components in atherosclerosis: Focus on Ang-II, ACE2, and Ang-1–7. Front. Physiol. 2020, 11, 1067. [Google Scholar] [CrossRef]
- Husain, K.; Hernandez, W.; Ansari, R.A.; Ferder, L. Inflammation, oxidative stress and renin angiotensin system in atherosclerosis. World J. Biol. Chem. 2015, 6, 209–217. [Google Scholar] [CrossRef]
- Elgendy, I.Y.; Mahtta, D.; Pepine, C.J. Medical therapy for heart failure caused by ischemic heart disease. Circ. Res. 2019, 124, 1520–1535. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Sun, Y.; Dong, M.; Yang, X.; Meng, X.; Niu, R.; Guan, J.; Zhang, Y.; Zhang, C. Comparison of angiotensin-(1–7), losartan and their combination on atherosclerotic plaque formation in apolipoprotein E knockout mice. Atherosclerosis 2015, 240, 544–549. [Google Scholar] [CrossRef] [PubMed]
- Jawien, J.; Toton-Zuranska, J.; Gajda, M.; Niepsuj, A.; Gebska, A.; Kus, K.; Suski, M.; Pyka-Fosciak, G.; Nowak, B.; Guzik, T.J.; et al. Angiotensin-(1–7) receptor Mas agonist ameliorates progress of atherosclerosis in apoE-knockout mice. J. Physiol. Pharmacol. 2012, 63, 77–85. [Google Scholar]
- Jawien, J.; Toton-Zuranska, J.; Kus, K.; Pawlowska, M.; Olszanecki, R.; Korbut, R. The effect of AVE 0991, nebivolol and doxycycline on inflammatory mediators in an apoE-knockout mouse model of atherosclerosis. Med. Sci. Monit. 2012, 18, BR389–BR393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McKinney, C.A.; Fattah, C.; Loughrey, C.M.; Milligan, G.; Nicklin, S.A. Angiotensin-(1–7) and angiotensin-(1–9): Function in cardiac and vascular remodelling. Clin. Sci. 2014, 126, 815–827. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tesanovic, S.; Vinh, A.; Gaspari, T.A.; Casley, D.; Widdop, R.E. Vasoprotective and atheroprotective effects of angiotensin (1–7) in apolipoprotein E-deficient mice. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 1606–1613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teixeira, L.B.; Parreiras-E-Silva, L.T.; Bruder-Nascimento, T.; Duarte, D.A.; Simões, S.C.; Costa, R.M.; Rodríguez, D.Y.; Ferreira, P.A.B.; Silva, C.A.A.; Abrao, E.P.; et al. Ang-(1–7) is an endogenous β-arrestin-biased agonist of the AT1 receptor with protective action in cardiac hypertrophy. Sci. Rep. 2017, 7, 11903. [Google Scholar] [CrossRef]
- Trojanowicz, B.; Ulrich, C.; Kohler, F.; Bode, V.; Seibert, E.; Fiedler, R.; Girndt, M. Monocytic angiotensin-converting enzyme 2 relates to atherosclerosis in patients with chronic kidney disease. Nephrol. Dial. Transplant. 2017, 32, 287–298. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.H.; Hao, Q.Q.; Wang, X.Y.; Chen, X.; Wang, N.; Zhu, L.; Li, S.Y.; Yu, Q.T.; Dong, B. ACE2 activity was increased in atherosclerotic plaque by losartan: Possible relation to anti-atherosclerosis. J. Renin. Angiotensin Aldosterone Syst. 2015, 16, 292–300. [Google Scholar] [CrossRef]
- Shenoy, V.; Gjymishka, A.; Jarajapu, Y.P.; Qi, Y.; Afzal, A.; Rigatto, K.; Ferreira, A.J.; Fraga-Silva, R.A.; Kearns, P.; Douglas, J.Y.; et al. Diminazene attenuates pulmonary hypertension and improves angiogenic progenitor cell functions in experimental models. Am. J. Respir. Crit. Care Med. 2013, 187, 648–657. [Google Scholar] [CrossRef]
- Fraga-Silva, R.A.; Montecucco, F.; Costa-Fraga, F.P.; Nencioni, A.; Caffa, I.; Bragina, M.E.; Mach, F.; Raizada, M.K.; Santos, R.A.S.; Silva, R.F.D.; et al. Diminazene enhances stability of atherosclerotic plaques in ApoE-deficient mice. Vascul. Pharmacol. 2015, 74, 103–113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mirabito Colafella, K.M.; Bovée, D.M.; Danser, A.H.J. The renin-angiotensin-aldosterone system and its therapeutic targets. Exp. Eye Res. 2019, 186, 107680. [Google Scholar] [CrossRef] [PubMed]
- Tousoulis, D.; Psaltopoulou, T.; Androulakis, E.; Papageorgiou, N.; Papaioannou, S.; Oikonomou, E.; Synetos, A.; Stefanadis, C. Oxidative stress and early atherosclerosis: Novel antioxidant treatment. Cardiovasc. Drugs Ther. 2015, 29, 75–88. [Google Scholar] [CrossRef]
- Ramadan, R.; Dhawan, S.S.; Binongo, J.N.; Alkhoder, A.; Jones, D.P.; Oshinski, J.N.; Quyyumi, A.A. Effect of angiotensin II type I receptor blockade with valsartan on carotid artery atherosclerosis: A double blind randomized clinical trial comparing valsartan and placebo (EFFERVESCENT). Am. Heart. J. 2016, 174, 68–79. [Google Scholar] [CrossRef] [Green Version]
- Zwadlo, C.; Bauersachs, J. Mineralocorticoid receptor antagonists in the treatment of coronary artery disease, myocardial infarction and heart failure, aldosterone-mineralocorticoid receptor—Cell biology to translational medicine. IntechOpen 2019. Available online: https://www.intechopen.com/books/aldosterone-mineralocorticoid-receptor-cell-biology-to-translational-medicine/mineralocorticoid-receptor-antagonists-in-the-treatment-of-coronary-artery-disease-myocardial-infar (accessed on 25 September 2019). [CrossRef] [Green Version]
- Tsutsui, H.; Ito, H.; Kitakaze, M.; Komuro, I.; Murohara, T.; Izumi, T.; Sunagawa, K.; Yasumura, Y.; Yano, M.; Yamamoto, K.; et al. Double-blind, randomized, placebo-controlled trial evaluating the efficacy and safety of eplerenone in Japanese patients with chronic heart failure (J-EMPHASIS-HF). Circ. J. 2017, 82, 148–158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pitt, B.; Bakris, G.; Ruilope, L.M.; DiCarlo, L.; Mukherjee, R.; EPHESUS Investigators. Serum potassium and clinical outcomes in the eplerenone post-acute myocardial infarction heart failure efficacy and survival study (EPHESUS). Circulation 2008, 118, 1643–1650. [Google Scholar] [CrossRef] [Green Version]
- Beygui, F.; Cayla, G.; Roule, V.; Roubille, F.; Delarche, N.; Silvain, J.; Van Belle, E.; Belle, L.; Galinier, M.; Motreff, P.; et al. ALBATROSS Investigators. Early aldosterone blockade in acute myocardial infarction: The ALBATROSS randomized clinical trial. J. Am. Coll. Cardiol. 2016, 67, 1917–1927. [Google Scholar] [CrossRef]
- Hillaert, M.A.; Lentjes, E.G.; Beygui, F.; Kemperman, H.; Asselbergs, F.W.; Nathoe, H.M.; Agostoni, P.; Voskuil, M.; Ivanes, F.; Jude, B.; et al. Measuring and targeting aldosterone and renin in atherosclerosis-a review of clinical data. Am. Heart. J. 2011, 162, 585–596. [Google Scholar] [CrossRef] [PubMed]
- Ramya, K.; Suresh, R.; Kumar, H.Y.; Kumar, B.R.P.; Murthy, N.B.S. Decades-old renin inhibitors are still struggling to find a niche in antihypertensive therapy. A fleeting look at the old and the promising new molecules. Bioorg. Med. Chem. 2020, 28, 115466. [Google Scholar] [CrossRef] [PubMed]
- Musini, V.M.; Lawrence, K.A.; Fortin, P.M.; Bassett, K.; Wright, J.M. Blood pressure lowering efficacy of renin inhibitors for primary hypertension. Cochrane Database Syst. Rev. 2017, 4, CD007066. [Google Scholar] [CrossRef]
- Wu, T.C.; Lee, C.Y.; Lin, S.J.; Chen, J.W. Aliskiren inhibits neointimal matrix metalloproteinases in experimental atherosclerosis. Acta Cardiol. Sin. 2016, 32, 586–593. [Google Scholar] [CrossRef] [PubMed]
- Su, J.B. Vascular endothelial dysfunction and pharmacological treatment. World J. Cardiol. 2015, 7, 719–741. [Google Scholar] [CrossRef] [PubMed]
- Mihai, G.; Varghese, J.; Kampfrath, T.; Gushchina, L.; Hafer, L.; Deiuliis, J.; Maiseyeu, A.; Simonetti, O.P.; Lu, B.; Rajagopalan, S. Aliskiren effect on plaque progression in established atherosclerosis using high resolution 3D MRI (ALPINE): A double-blind placebo-controlled trial. J. Am. Heart Assoc. 2013, 2, e004879. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
| Drug | Target | Effect | Study | Reference |
|---|---|---|---|---|
| Angiotensin-1–7 | RAAS | decrease the macrophage infiltration, oxidative stress | ApoE-KO mice | [37] |
| Losartan | ACE2 | gene expression of cellular adhesion molecules enhancement | high-cholesterol fed rabbits | [45] |
| DIZE | ACE2 | the stability of atherosclerotic plaques increase | ApoE-KO mice | [46,47] |
| Enalapril | Angiotensin-II | regulate the antioxidant defense system, lower inflammatory mediators, and inhibit NADPH oxidase activity | ApoE-deficient mice | [48] |
| Olmesartan | Angiotensin-II | decrease vascular inflammation | Hypertensive patients | [49] |
| valsartan | Angiotensin-II | atherosclerosis regression | Individuals with thickening of the carotid wall | [50] |
| Elprenone | Aldosterone (MR) | 15% decrease of risk of all-cause mortality and 13% decrease of risk of the composite end point of CV mortality/CV hospitalization | Post-myocardial infarction associated with left ventricular dysfunction and congestive heart failure | [51] |
| spironolactone | Aldosterone (MR) | no beneficial effect | Acute myocardial infarction | [52] |
| aliskiren | Renin | Approved drug for blood pressure lowering | Hypertension | [53] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Poznyak, A.V.; Bharadwaj, D.; Prasad, G.; Grechko, A.V.; Sazonova, M.A.; Orekhov, A.N. Renin-Angiotensin System in Pathogenesis of Atherosclerosis and Treatment of CVD. Int. J. Mol. Sci. 2021, 22, 6702. https://doi.org/10.3390/ijms22136702
Poznyak AV, Bharadwaj D, Prasad G, Grechko AV, Sazonova MA, Orekhov AN. Renin-Angiotensin System in Pathogenesis of Atherosclerosis and Treatment of CVD. International Journal of Molecular Sciences. 2021; 22(13):6702. https://doi.org/10.3390/ijms22136702
Chicago/Turabian StylePoznyak, Anastasia V., Dwaipayan Bharadwaj, Gauri Prasad, Andrey V. Grechko, Margarita A. Sazonova, and Alexander N. Orekhov. 2021. "Renin-Angiotensin System in Pathogenesis of Atherosclerosis and Treatment of CVD" International Journal of Molecular Sciences 22, no. 13: 6702. https://doi.org/10.3390/ijms22136702
APA StylePoznyak, A. V., Bharadwaj, D., Prasad, G., Grechko, A. V., Sazonova, M. A., & Orekhov, A. N. (2021). Renin-Angiotensin System in Pathogenesis of Atherosclerosis and Treatment of CVD. International Journal of Molecular Sciences, 22(13), 6702. https://doi.org/10.3390/ijms22136702