
Do Changes in ACE-2 Expression Affect SARS-CoV-2 Virulence and Related Complications: A Closer Look into Membrane-Bound and Soluble Forms

 ,
,  and
and 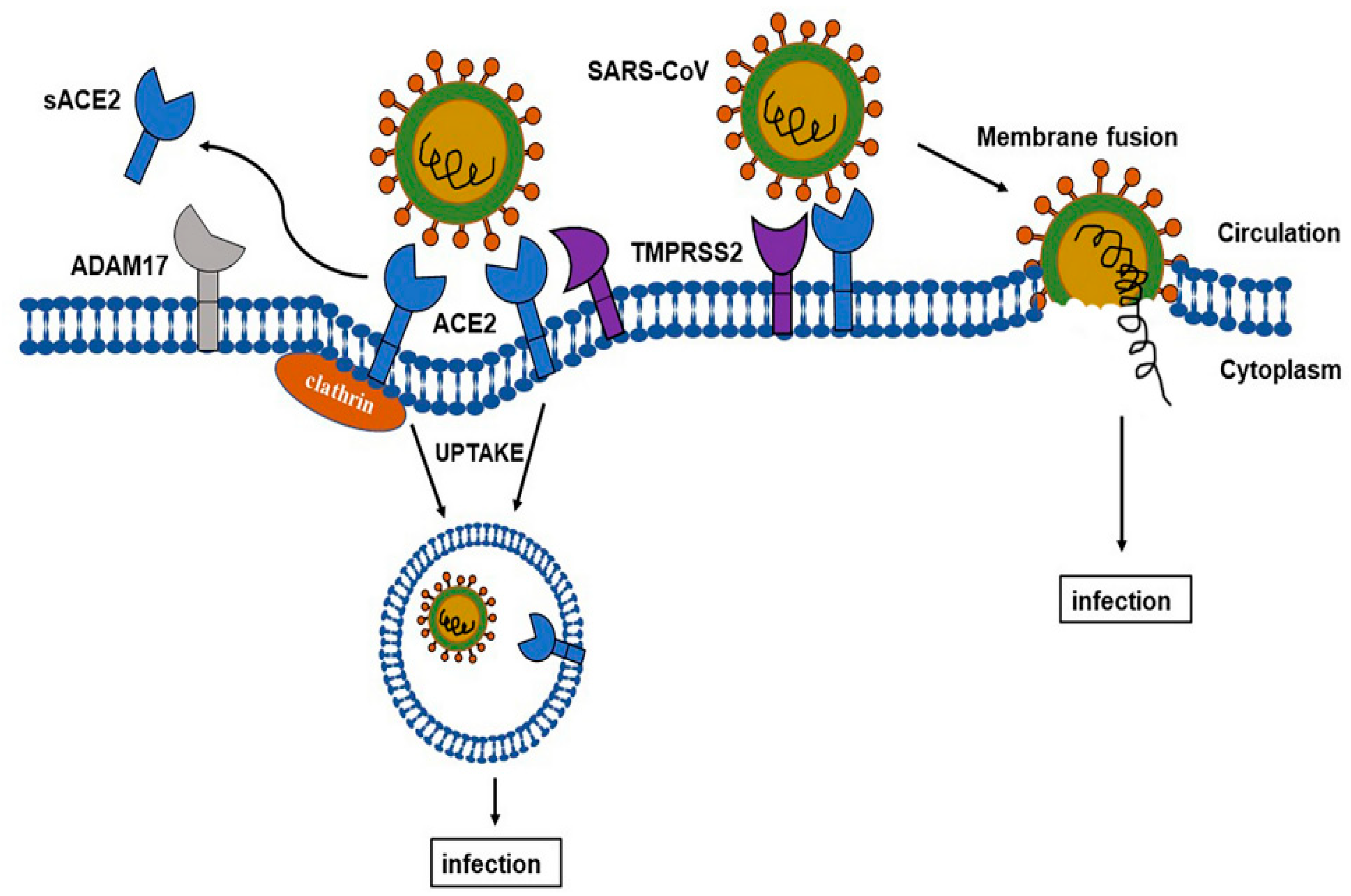{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. SARS-CoV-2 and ACE-2
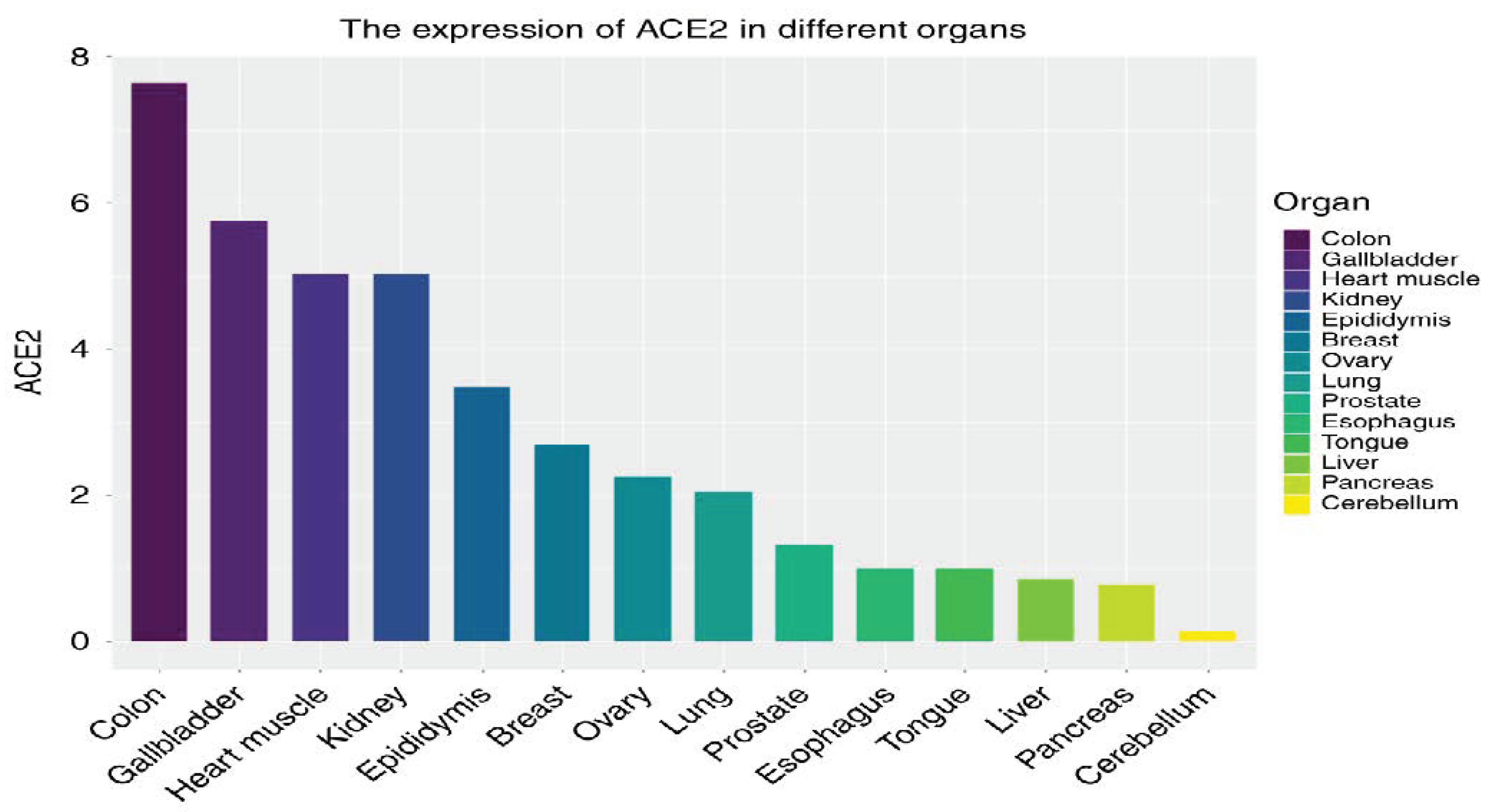
2.1. Functions and Distribution of ACE-2 in the Body
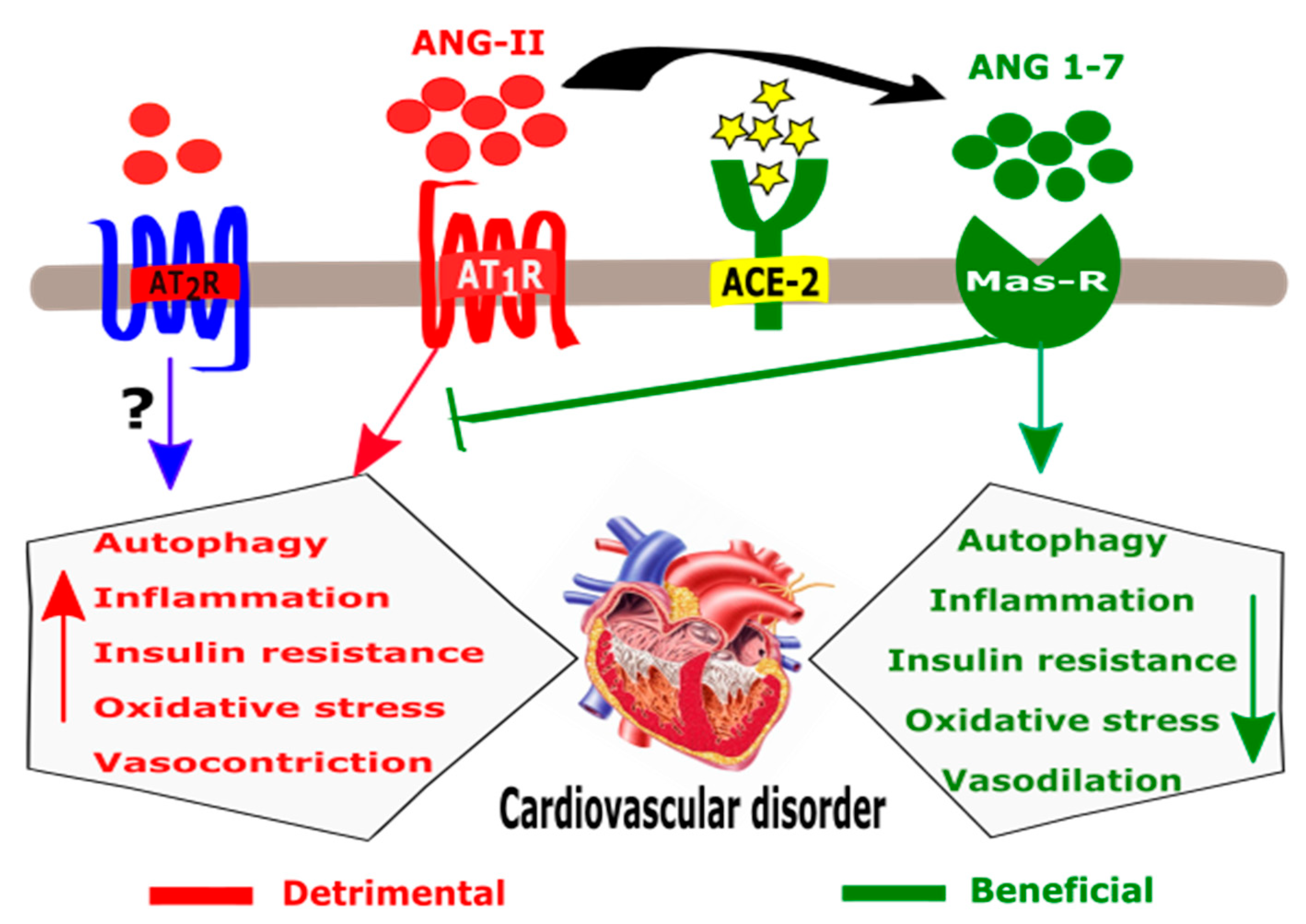
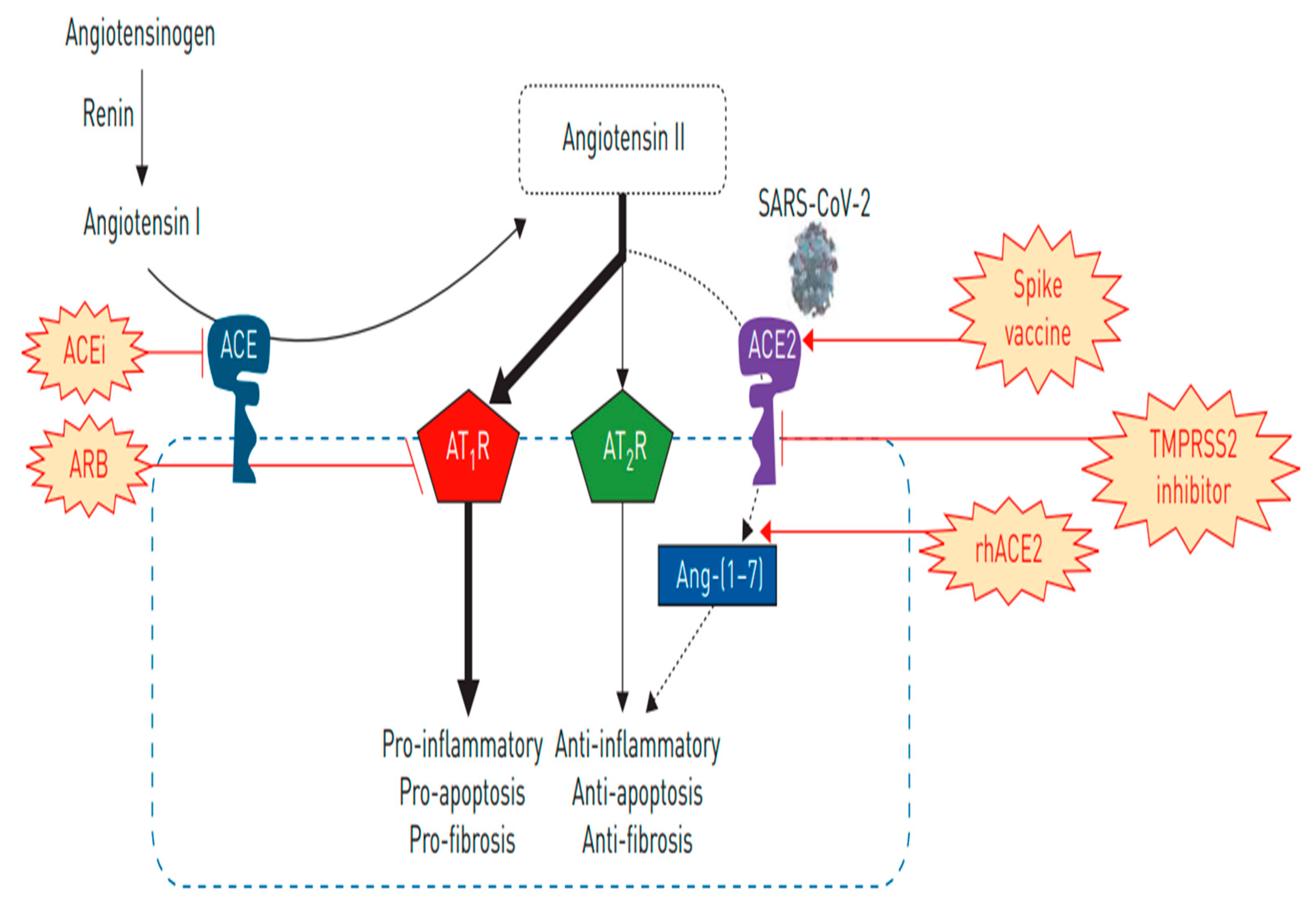
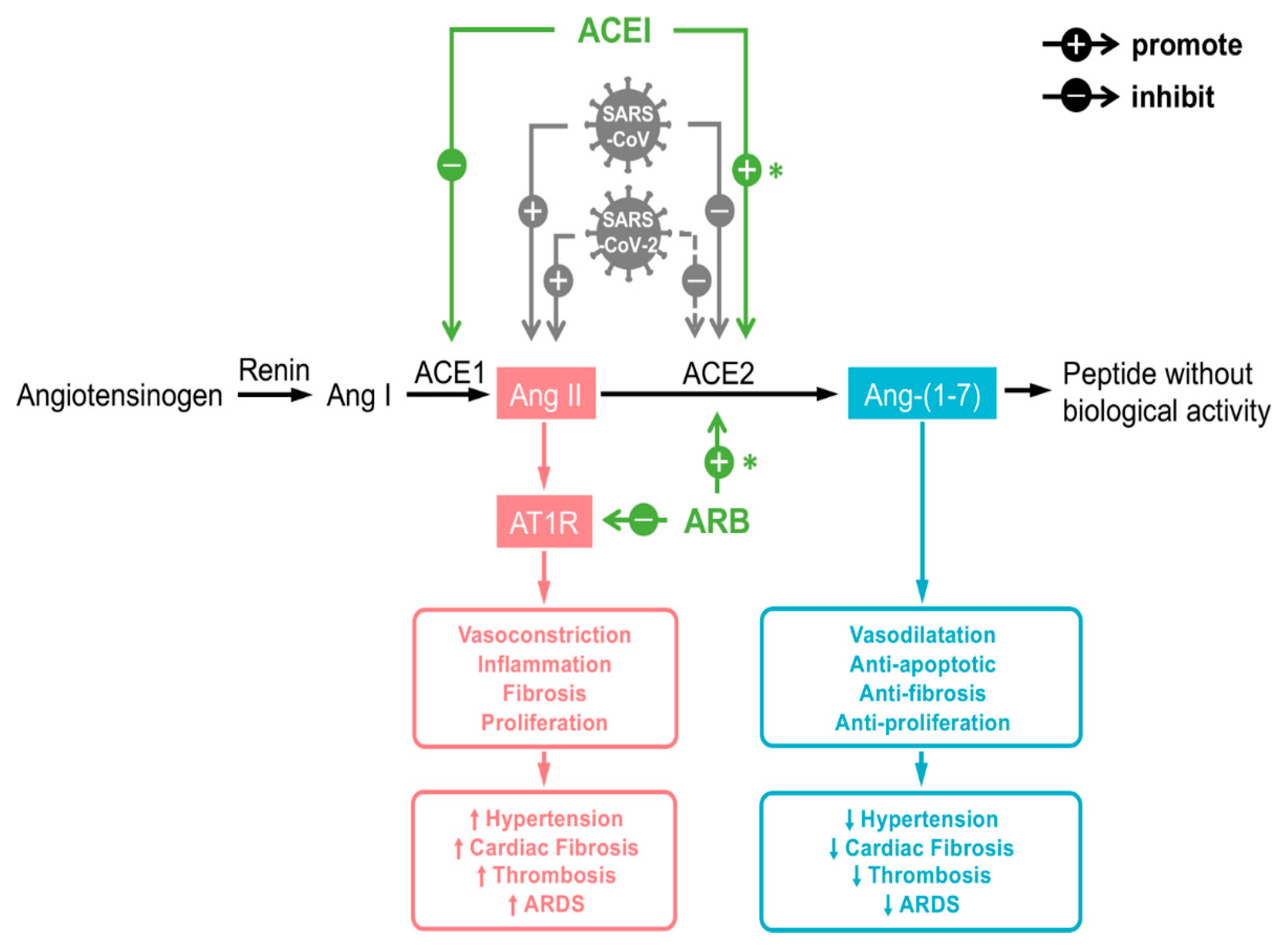
2.2. ACE-2 in the RAAS Pathway
2.3. ACE-2 in Disease
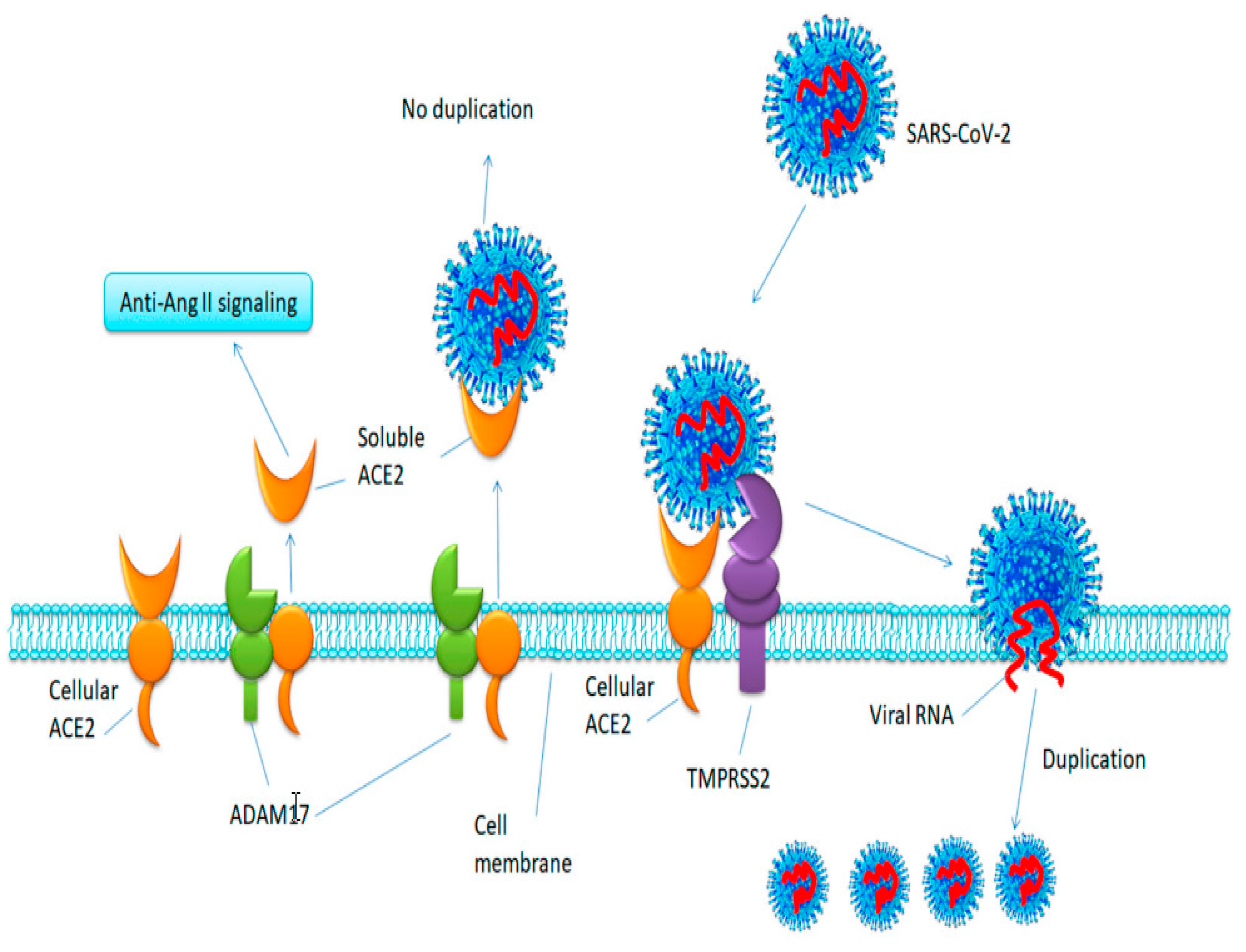
Membrane-Bound and Soluble forms of ACE-2, and Mechanism of Shedding
2.4. ACE-2 Shedding and Viral Infection
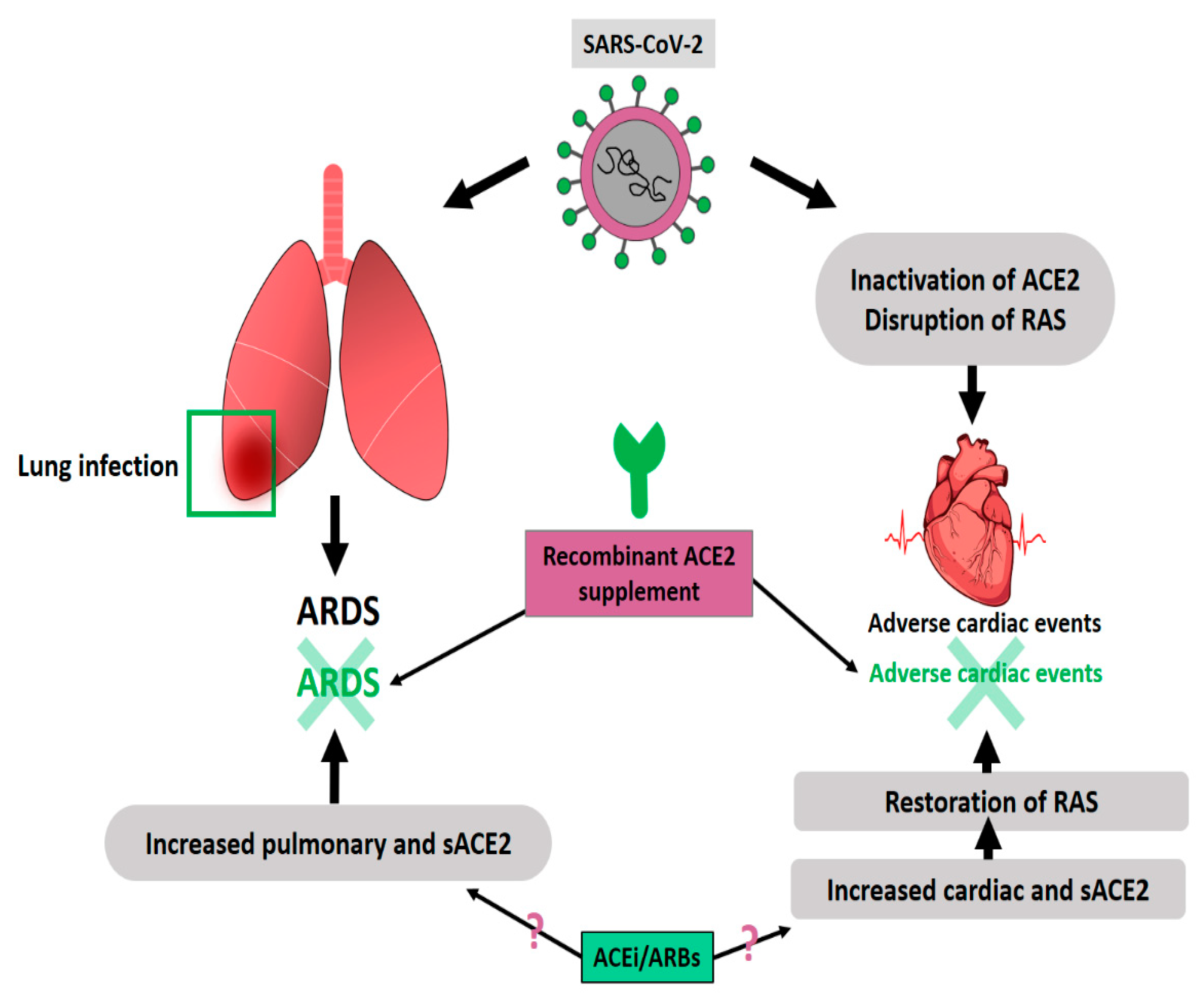
2.5. ACE-2 and ARDS
2.6. ACE-2 and CVDs
2.7. Altered ACE-2 Expression with Certain Medications: Is There Evidence of Altered Expression and SARS-CoV-2 Virulence?
3. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Matheson, N.J.; Lehner, P.J. How does SARS-CoV-2 cause COVID-19? Science 2020, 369, 510–511. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Niu, Z.; Jiang, X.; Zhang, Z.; Zheng, Y.; Wang, Z.; Zhu, Y.; Gao, L.; Wang, X.; Sun, Q. Systemic analysis of tissue cells potentially vulnerable to SARS-CoV-2 infection by the protein-proofed single-cell RNA profiling of ACE2, TMPRSS2 and Furin proteases. bioRxiv 2020. [Google Scholar] [CrossRef]
- Zhang, X.; Li, S.; Niu, S. ACE2 and COVID-19 and the resulting ARDS. Postgrad. Med. J. 2020, 96, 403–407. [Google Scholar] [CrossRef]
- Bos, J.M.; Hebl, V.B.; Oberg, A.L.; Sun, Z.; Herman, D.S.; Teekakirikul, P.; Seidman, J.G.; Seidman, C.E.; Dos Remedios, C.G.; Maleszewski, J.J.; et al. Marked Up-Regulation of ACE2 in Hearts of Patients With Obstructive Hypertrophic Cardiomyopathy: Implications for SARS-CoV-2-Mediated COVID-19. Mayo Clin. Proc. 2020, 95, 1354–1368. [Google Scholar] [CrossRef] [PubMed]
- Ferrario, C.M.; Jessup, J.; Chappell, M.C.; Averill, D.B.; Brosnihan, K.B.; Tallant, E.A.; Diz, D.I.; Gallagher, P.E. Effect of angiotensin-converting enzyme inhibition and angiotensin II receptor blockers on cardiac angiotensin-converting enzyme 2. Circulation 2005, 111, 2605–2610. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.; Deng, W.; Li, S.; Yang, X. Advances in research on ACE2 as a receptor for 2019-nCoV. Cell. Mol. Life Sci. 2020. [Google Scholar] [CrossRef]
- Tipnis, S.R.; Hooper, N.M.; Hyde, R.; Karran, E.; Christie, G.; Turner, A.J. A human homolog of angiotensin-converting enzyme. Cloning and functional expression as a captopril-insensitive carboxypeptidase. J. Biol. Chem. 2000, 275, 33238–33243. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Yang, P.; Liu, K.; Guo, F.; Zhang, Y.; Zhang, G.; Jiang, C. SARS coronavirus entry into host cells through a novel clathrin- and caveolae-independent endocytic pathway. Cell Res. 2008, 18, 290–301. [Google Scholar] [CrossRef] [Green Version]
- Heurich, A.; Hofmann-Winkler, H.; Gierer, S.; Liepold, T.; Jahn, O.; Pöhlmann, S. TMPRSS2 and ADAM17 Cleave ACE2 Differentially and Only Proteolysis by TMPRSS2 Augments Entry Driven by the Severe Acute Respiratory Syndrome Coronavirus Spike Protein. J. Virol. 2014, 88, 1293–1307. [Google Scholar] [CrossRef] [Green Version]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Krüger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.-H.; Nitsche, A.; et al. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell 2020, 181, 271–280.e8. [Google Scholar] [CrossRef]
- Hou, Y.J.; Okuda, K.; Edwards, C.E.; Martinez, D.R.; Asakura, T.; Dinnon, K.H., 3rd; Kato, T.; Lee, R.E.; Yount, B.L.; Mascenik, T.M.; et al. SARS-CoV-2 reverse genetics reveals a variable infection gradient in the respiratory tract. Cell 2020, 182, 429–446.e14. [Google Scholar] [CrossRef]
- Xiao, L.; Sakagami, H.; Miwa, N. ACE2: The key Molecule for Understanding the Pathophysiology of Severe and Critical Conditions of COVID-19: Demon or Angel? Viruses 2020, 12, 491. [Google Scholar] [CrossRef]
- Xu, H.; Zhong, L.; Deng, J.; Peng, J.; Dan, H.; Zeng, X.; Li, T.; Chen, Q. High expression of ACE2 receptor of 2019-nCoV on the epithelial cells of oral mucosa. Int. J. Oral Sci. 2020, 12, 8. [Google Scholar] [CrossRef]
- Hikmet, F.; Méar, L.; Uhlén, M.; Lindskog, C. The protein expression profile of ACE2 in human tissues. bioRxiv 2020. [Google Scholar] [CrossRef] [Green Version]
- Jia, H.P.; Look, D.C.; Tan, P.; Shi, L.; Hickey, M.; Gakhar, L.; Chappell, M.C.; Wohlford-Lenane, C.; McCray, P.B., Jr. Ectodomain shedding of angiotensin converting enzyme 2 in human airway epithelia. Am. J. Physiol. Lung Cell Mol. Physiol. 2009, 297, L84–L96. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Wu, M.; Yao, J.; Guo, J.; Liao, X.; Song, S.; Li, J.; Duan, G.; Zhou, Y.; Wu, X.; et al. Caution on Kidney Dysfunctions of COVID-19 Patients. medRxiv 2020. [Google Scholar] [CrossRef]
- Donoghue, M.; Hsieh, F.; Baronas, E.; Godbout, K.; Gosselin, M.; Stagliano, N.; Donovan, M.; Woolf, B.; Robison, K.; Jeyaseelan, R.; et al. A novel angiotensin-converting enzyme-related carboxypeptidase (ACE2) converts angiotensin I to angiotensin 1-9. Circ. Res. 2000, 87, e1–e9. [Google Scholar] [CrossRef]
- Chamsi-Pasha, M.A.; Shao, Z.; Tang, W.H. Angiotensin-converting enzyme 2 as a therapeutic target for heart failure. Curr. Heart Fail. Rep. 2014, 11, 58–63. [Google Scholar] [CrossRef]
- Michikado, I.; Barry, H.G. Ectodomain Shedding of ACE and ACE2 as Regulators of Their Protein Functions. Curr. Enzym. Inhib. 2011, 7, 42–55. [Google Scholar] [CrossRef]
- Kohlstedt, K.; Brandes, R.P.; Müller-Esterl, W.; Busse, R.; Fleming, I. Angiotensin-converting enzyme is involved in outside-in signaling in endothelial cells. Circ. Res. 2004, 94, 60–67. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Moore, M.J.; Vasilieva, N.; Sui, J.; Wong, S.K.; Berne, M.A.; Somasundaran, M.; Sullivan, J.L.; Luzuriaga, K.; Greenough, T.C.; et al. Angiotensin-converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature 2003, 426, 450–454. [Google Scholar] [CrossRef] [Green Version]
- Hofmann, H.; Geier, M.; Marzi, A.; Krumbiegel, M.; Peipp, M.; Fey, G.H.; Gramberg, T.; Pöhlmann, S. Susceptibility to SARS coronavirus S protein-driven infection correlates with expression of angiotensin converting enzyme 2 and infection can be blocked by soluble receptor. Biochem. Biophys. Res. Commun. 2004, 319, 1216–1221. [Google Scholar] [CrossRef]
- Lambert, D.W.; Yarski, M.; Warner, F.J.; Thornhill, P.; Parkin, E.T.; Smith, A.I.; Hooper, N.M.; Turner, A.J. Tumor necrosis factor-alpha convertase (ADAM17) mediates regulated ectodomain shedding of the severe-acute respiratory syndrome-coronavirus (SARS-CoV) receptor, angiotensin-converting enzyme-2 (ACE2). J. Biol. Chem. 2005, 280, 30113–30119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haga, S.; Nagata, N.; Okamura, T.; Yamamoto, N.; Sata, T.; Yamamoto, N.; Sasazuki, T.; Ishizaka, Y. TACE antagonists blocking ACE2 shedding caused by the spike protein of SARS-CoV are candidate antiviral compounds. Antivir. Res. 2010, 85, 551–555. [Google Scholar] [CrossRef] [PubMed]
- Glowacka, I.; Bertram, S.; Herzog, P.; Pfefferle, S.; Steffen, I.; Muench, M.O.; Simmons, G.; Hofmann, H.; Kuri, T.; Weber, F.; et al. Differential downregulation of ACE2 by the spike proteins of severe acute respiratory syndrome coronavirus and human coronavirus NL63. J. Virol. 2010, 84, 1198–1205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shulla, A.; Heald-Sargent, T.; Subramanya, G.; Zhao, J.; Perlman, S.; Gallagher, T. A transmembrane serine protease is linked to the severe acute respiratory syndrome coronavirus receptor and activates virus entry. J. Virol. 2011, 85, 873–882. [Google Scholar] [CrossRef] [Green Version]
- Donaldson, S.H.; Hirsh, A.; Li, D.C.; Holloway, G.; Chao, J.; Boucher, R.C.; Gabriel, S.E. Regulation of the epithelial sodium channel by serine proteases in human airways. J. Biol. Chem. 2002, 277, 8338–8345. [Google Scholar] [CrossRef] [Green Version]
- Böttcher, E.; Matrosovich, T.; Beyerle, M.; Klenk, H.D.; Garten, W.; Matrosovich, M. Proteolytic activation of influenza viruses by serine proteases TMPRSS2 and HAT from human airway epithelium. J. Virol. 2006, 80, 9896–9898. [Google Scholar] [CrossRef] [Green Version]
- Kam, Y.-W.; Okumura, Y.; Kido, H.; Ng, L.F.P.; Bruzzone, R.; Altmeyer, R. Cleavage of the SARS coronavirus spike glycoprotein by airway proteases enhances virus entry into human bronchial epithelial cells in vitro. PLoS ONE 2009, 4, e7870. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Zhao, Z.; Wang, Y.; Zhou, Y.; Ma, Y.; Zuo, W. Single-cell RNA expression profiling of ACE2, the receptor of SARS-CoV-2. bioRxiv 2020. [Google Scholar] [CrossRef]
- Patel, S.; Rauf, A.; Khan, H.; Abu-Izneid, T. Renin-angiotensin-aldosterone (RAAS): The ubiquitous system for homeostasis and pathologies. Biomed. Pharm. 2017, 94, 317–325. [Google Scholar] [CrossRef]
- Turner, A.J.; Hiscox, J.A.; Hooper, N.M. ACE2: From vasopeptidase to SARS virus receptor. Trends Pharm. Sci. 2004, 25, 291–294. [Google Scholar] [CrossRef] [PubMed]
- Papp, M.; Li, X.; Zhuang, J.; Wang, R.; Uhal, B.D. Angiotensin receptor subtype AT(1) mediates alveolar epithelial cell apoptosis in response to ANG II. Am. J. Physiol. Lung Cell Mol. Physiol. 2002, 282, L713–L718. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sriram, K.; Insel, P.A. A hypothesis for pathobiology and treatment of COVID-19: The centrality of ACE1/ACE2 imbalance. Br. J. Pharm. 2020, 177, 4825–4844. [Google Scholar] [CrossRef]
- Ismael-Badarneh, R.; Guetta, J.; Klorin, G.; Berger, G.; Abu-saleh, N.; Abassi, Z.; Azzam, Z.S. The Role of Angiotensin II and Cyclic AMP in Alveolar Active Sodium Transport. PLoS ONE 2015, 10, e0134175. [Google Scholar] [CrossRef] [Green Version]
- Imai, Y.; Kuba, K.; Rao, S.; Huan, Y.; Guo, F.; Guan, B.; Yang, P.; Sarao, R.; Wada, T.; Leong-Poi, H.; et al. Angiotensin-converting enzyme 2 protects from severe acute lung failure. Nature 2005, 436, 112–116. [Google Scholar] [CrossRef]
- Kuba, K.; Imai, Y.; Rao, S.; Gao, H.; Guo, F.; Guan, B.; Huan, Y.; Yang, P.; Zhang, Y.; Deng, W.; et al. A crucial role of angiotensin converting enzyme 2 (ACE2) in SARS coronavirus–induced lung injury. Nat. Med. 2005, 11, 875–879. [Google Scholar] [CrossRef]
- Li, Y.; Zeng, Z.; Cao, Y.; Liu, Y.; Ping, F.; Liang, M.; Xue, Y.; Xi, C.; Zhou, M.; Jiang, W. Angiotensin-converting enzyme 2 prevents lipopolysaccharide-induced rat acute lung injury via suppressing the ERK1/2 and NF-κB signaling pathways. Sci. Rep. 2016, 6, 27911. [Google Scholar] [CrossRef] [Green Version]
- Pinto, B.G.G.; Oliveira, A.E.R.; Singh, Y.; Jimenez, L.; Gonçalves, A.N.A.; Ogava, R.L.T.; Creighton, R.; Peron, J.P.S.; Nakaya, H.I. ACE2 Expression is Increased in the Lungs of Patients with Comorbidities Associated with Severe COVID-19. Medrxiv Prepr. Serv. Health Sci. 2020. [Google Scholar] [CrossRef]
- Guy, J.L.; Lambert, D.W.; Warner, F.J.; Hooper, N.M.; Turner, A.J. Membrane-associated zinc peptidase families: Comparing ACE and ACE2. Biochim. Biophys. Acta 2005, 1751, 2–8. [Google Scholar] [CrossRef]
- Saponaro, F.; Rutigliano, G.; Sestito, S.; Bandini, L.; Storti, B.; Bizzarri, R.; Zucchi, R. ACE2 in the Era of SARS-CoV-2: Controversies and Novel Perspectives. Front. Mol. Biosci. 2020, 7. [Google Scholar] [CrossRef]
- Mortaz, E.; Tabarsi, P.; Varahram, M.; Folkerts, G.; Adcock, I.M. The Immune Response and Immunopathology of COVID-19. Front. Immunol. 2020, 11, 2037. [Google Scholar] [CrossRef]
- Rezaei, M.; Ziai, S.A.; Fakhri, S.; Pouriran, R. ACE2: Its potential role and regulation in severe acute respiratory syndrome and COVID-19. J. Cell Physiol. 2021, 236, 2430–2442. [Google Scholar] [CrossRef] [PubMed]
- Clarke, N.E.; Turner, A.J. Angiotensin-converting enzyme 2: The first decade. Int. J. Hypertens. 2012, 2012, 307315. [Google Scholar] [CrossRef] [Green Version]
- Imai, Y.; Kuba, K.; Penninger, J.M. Angiotensin-converting enzyme 2 in acute respiratory distress syndrome. Cell Mol. Life Sci. 2007, 64, 2006–2012. [Google Scholar] [CrossRef]
- Ingraham, N.E.; Barakat, A.G.; Reilkoff, R.; Bezdicek, T.; Schacker, T.; Chipman, J.G.; Tignanelli, C.J.; Puskarich, M.A. Understanding the renin-angiotensin-aldosterone-SARS-CoV axis: A comprehensive review. Eur. Respir. J. 2020, 56. [Google Scholar] [CrossRef]
- El-Hashim, A.Z.; Renno, W.M.; Raghupathy, R.; Abduo, H.T.; Akhtar, S.; Benter, I.F. Angiotensin-(1-7) inhibits allergic inflammation, via the MAS1 receptor, through suppression of ERK1/2- and NF-κB-dependent pathways. Br. J. Pharm. 2012, 166, 1964–1976. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reddy, R.; Asante, I.; Liu, S.; Parikh, P.; Liebler, J.; Borok, Z.; Rodgers, K.; Baydur, A.; Louie, S.G. Circulating angiotensin peptides levels in Acute Respiratory Distress Syndrome correlate with clinical outcomes: A pilot study. PLoS ONE 2019, 14, e0213096. [Google Scholar] [CrossRef] [PubMed]
- Ackermann, M.; Verleden, S.E.; Kuehnel, M.; Haverich, A.; Welte, T.; Laenger, F.; Vanstapel, A.; Werlein, C.; Stark, H.; Tzankov, A.; et al. Pulmonary Vascular Endothelialitis, Thrombosis, and Angiogenesis in Covid-19. N. Engl. J. Med. 2020, 383, 120–128. [Google Scholar] [CrossRef] [PubMed]
- Scialo, F.; Daniele, A.; Amato, F.; Pastore, L.; Matera, M.G.; Cazzola, M.; Castaldo, G.; Bianco, A. ACE2: The Major Cell Entry Receptor for SARS-CoV-2. Lung 2020, 198, 867–877. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, Y.; Luo, W.; Huang, L.; Xiao, J.; Li, F.; Qin, S.; Song, X.; Wu, Y.; Zeng, Q.; et al. A comprehensive investigation of the mRNA and protein level of ACE2, the putative receptor of SARS-CoV-2, in human tissues and blood cells. Int. J. Med. Sci. 2020, 17, 1522–1531. [Google Scholar] [CrossRef] [PubMed]
- Zou, X.; Chen, K.; Zou, J.; Han, P.; Hao, J.; Han, Z. Single-cell RNA-seq data analysis on the receptor ACE2 expression reveals the potential risk of different human organs vulnerable to 2019-nCoV infection. Front. Med. 2020, 14, 185–192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blume, C.; Jackson, C.L.; Spalluto, C.M.; Legebeke, J.; Nazlamova, L.; Conforti, F.; Perotin, J.-M.; Frank, M.; Butler, J.; Crispin, M.; et al. A novel ACE2 isoform is expressed in human respiratory epithelia and is upregulated in response to interferons and RNA respiratory virus infection. Nat. Genet. 2021, 53, 205–214. [Google Scholar] [CrossRef]
- Ziegler, C.G.K.; Allon, S.J.; Nyquist, S.K.; Mbano, I.M.; Miao, V.N.; Tzouanas, C.N.; Cao, Y.; Yousif, A.S.; Bals, J.; Hauser, B.M.; et al. SARS-CoV-2 Receptor ACE2 Is an Interferon-Stimulated Gene in Human Airway Epithelial Cells and Is Detected in Specific Cell Subsets across Tissues. Cell 2020, 181, 1016–1035.e9. [Google Scholar] [CrossRef]
- Chua, R.L.; Lukassen, S.; Trump, S.; Hennig, B.P.; Wendisch, D.; Pott, F.; Debnath, O.; Thürmann, L.; Kurth, F.; Völker, M.T.; et al. COVID-19 severity correlates with airway epithelium–immune cell interactions identified by single-cell analysis. Nat. Biotechnol. 2020, 38, 970–979. [Google Scholar] [CrossRef]
- Zhuang, M.-W.; Cheng, Y.; Zhang, J.; Jiang, X.-M.; Wang, L.; Deng, J.; Wang, P.-H. Increasing host cellular receptor-angiotensin-converting enzyme 2 expression by coronavirus may facilitate 2019-nCoV (or SARS-CoV-2) infection. J. Med. Virol. 2020, 92, 2693–2701. [Google Scholar] [CrossRef]
- Wösten-van Asperen, R.M.; Lutter, R.; Specht, P.A.; Moll, G.N.; van Woensel, J.B.; van der Loos, C.M.; van Goor, H.; Kamilic, J.; Florquin, S.; Bos, A.P. Acute respiratory distress syndrome leads to reduced ratio of ACE/ACE2 activities and is prevented by angiotensin-(1-7) or an angiotensin II receptor antagonist. J. Pathol. 2011, 225, 618–627. [Google Scholar] [CrossRef] [PubMed]
- Kimura, H.; Francisco, D.; Conway, M.; Martinez, F.D.; Vercelli, D.; Polverino, F.; Billheimer, D.; Kraft, M. Type 2 inflammation modulates ACE2 and TMPRSS2 in airway epithelial cells. J. Allergy Clin. Immunol. 2020, 146, 80–88.e8. [Google Scholar] [CrossRef]
- Liu, Y.; Yang, Y.; Zhang, C.; Huang, F.; Wang, F.; Yuan, J.; Wang, Z.; Li, J.; Li, J.; Feng, C.; et al. Clinical and biochemical indexes from 2019-nCoV infected patients linked to viral loads and lung injury. Sci. China Life Sci. 2020, 63, 364–374. [Google Scholar] [CrossRef] [Green Version]
- Yang, P.; Gu, H.; Zhao, Z.; Wang, W.; Cao, B.; Lai, C.; Yang, X.; Zhang, L.; Duan, Y.; Zhang, S.; et al. Angiotensin-converting enzyme 2 (ACE2) mediates influenza H7N9 virus-induced acute lung injury. Sci. Rep. 2014, 4, 7027. [Google Scholar] [CrossRef] [Green Version]
- Ciaglia, E.; Vecchione, C.; Puca, A.A. COVID-19 Infection and Circulating ACE2 Levels: Protective Role in Women and Children. Front. Pediatr 2020, 8, 206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sodhi, C.P.; Wohlford-Lenane, C.; Yamaguchi, Y.; Prindle, T.; Fulton, W.B.; Wang, S.; McCray, P.B., Jr.; Chappell, M.; Hackam, D.J.; Jia, H. Attenuation of pulmonary ACE2 activity impairs inactivation of des-Arg(9) bradykinin/BKB1R axis and facilitates LPS-induced neutrophil infiltration. Am. J. Physiol. Lung Cell Mol. Physiol. 2018, 314, L17–L31. [Google Scholar] [CrossRef] [PubMed]
- Treml, B.; Neu, N.; Kleinsasser, A.; Gritsch, C.; Finsterwalder, T.; Geiger, R.; Schuster, M.; Janzek, E.; Loibner, H.; Penninger, J.; et al. Recombinant angiotensin-converting enzyme 2 improves pulmonary blood flow and oxygenation in lipopolysaccharide-induced lung injury in piglets. Crit. Care Med. 2010, 38, 596–601. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.; Benthin, C.; Zeno, B.; Albertson, T.E.; Boyd, J.; Christie, J.D.; Hall, R.; Poirier, G.; Ronco, J.J.; Tidswell, M.; et al. A pilot clinical trial of recombinant human angiotensin-converting enzyme 2 in acute respiratory distress syndrome. Crit. Care 2017, 21, 234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Donoghue, M.; Wakimoto, H.; Maguire, C.T.; Acton, S.; Hales, P.; Stagliano, N.; Fairchild-Huntress, V.; Xu, J.; Lorenz, J.N.; Kadambi, V.; et al. Heart block, ventricular tachycardia, and sudden death in ACE2 transgenic mice with downregulated connexins. J. Mol. Cell. Cardiol. 2003, 35, 1043–1053. [Google Scholar] [CrossRef]
- Huentelman, M.J.; Grobe, J.L.; Vazquez, J.; Stewart, J.M.; Mecca, A.P.; Katovich, M.J.; Ferrario, C.M.; Raizada, M.K. Protection from angiotensin II-induced cardiac hypertrophy and fibrosis by systemic lentiviral delivery of ACE2 in rats. Exp. Physiol. 2005, 90, 783–790. [Google Scholar] [CrossRef] [Green Version]
- Sukumaran, V.; Veeraveedu, P.T.; Gurusamy, N.; Yamaguchi, K.; Lakshmanan, A.P.; Ma, M.; Suzuki, K.; Kodama, M.; Watanabe, K. Cardioprotective effects of telmisartan against heart failure in rats induced by experimental autoimmune myocarditis through the modulation of angiotensin-converting enzyme-2/angiotensin 1-7/mas receptor axis. Int. J. Biol. Sci. 2011, 7, 1077–1092. [Google Scholar] [CrossRef] [Green Version]
- Sukumaran, V.; Veeraveedu, P.T.; Lakshmanan, A.P.; Gurusamy, N.; Yamaguchi, K.; Ma, M.; Suzuki, K.; Kodama, M.; Watanabe, K. Olmesartan medoxomil treatment potently improves cardiac myosin-induced dilated cardiomyopathy via the modulation of ACE-2 and ANG 1-7 mas receptor. Free Radic. Res. 2012, 46, 850–860. [Google Scholar] [CrossRef]
- Sukumaran, V.; Tsuchimochi, H.; Tatsumi, E.; Shirai, M.; Pearson, J.T. Azilsartan ameliorates diabetic cardiomyopathy in young db/db mice through the modulation of ACE-2/ANG 1-7/Mas receptor cascade. Biochem. Pharm. 2017, 144, 90–99. [Google Scholar] [CrossRef]
- Sukumaran, V.; Watanabe, K.; Veeraveedu, P.T.; Thandavarayan, R.A.; Gurusamy, N.; Ma, M.; Yamaguchi, K.; Suzuki, K.; Kodama, M.; Aizawa, Y. Telmisartan, an angiotensin-II receptor blocker ameliorates cardiac remodeling in rats with dilated cardiomyopathy. Hypertens. Res. 2010, 33, 695–702. [Google Scholar] [CrossRef]
- Sukumaran, V.; Veeraveedu, P.T.; Gurusamy, N.; Lakshmanan, A.P.; Yamaguchi, K.; Ma, M.; Suzuki, K.; Kodama, M.; Watanabe, K. Telmisartan acts through the modulation of ACE-2/ANG 1-7/mas receptor in rats with dilated cardiomyopathy induced by experimental autoimmune myocarditis. Life Sci. 2012, 90, 289–300. [Google Scholar] [CrossRef] [PubMed]
- Sukumaran, V.; Veeraveedu, P.T.; Gurusamy, N.; Lakshmanan, A.P.; Yamaguchi, K.; Ma, M.; Suzuki, K.; Nagata, M.; Takagi, R.; Kodama, M.; et al. Olmesartan attenuates the development of heart failure after experimental autoimmune myocarditis in rats through the modulation of ANG 1-7 mas receptor. Mol. Cell Endocrinol. 2012, 351, 208–219. [Google Scholar] [CrossRef]
- Crackower, M.A.; Sarao, R.; Oudit, G.Y.; Yagil, C.; Kozieradzki, I.; Scanga, S.E.; Oliveira-dos-Santos, A.J.; da Costa, J.; Zhang, L.; Pei, Y.; et al. Angiotensin-converting enzyme 2 is an essential regulator of heart function. Nature 2002, 417, 822–828. [Google Scholar] [CrossRef]
- Goulter, A.B.; Goddard, M.J.; Allen, J.C.; Clark, K.L. ACE2 gene expression is up-regulated in the human failing heart. BMC Med. 2004, 2, 19. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Li, X.; Chen, M.; Feng, Y.; Xiong, C. The ACE2 expression in human heart indicates new potential mechanism of heart injury among patients infected with SARS-CoV-2. Cardiovasc. Res. 2020, 116, 1097–1100. [Google Scholar] [CrossRef] [Green Version]
- Epelman, S.; Tang, W.H.W.; Chen, S.Y.; Van Lente, F.; Francis, G.S.; Sen, S. Detection of soluble angiotensin-converting enzyme 2 in heart failure: Insights into the endogenous counter-regulatory pathway of the renin-angiotensin-aldosterone system. J. Am. Coll Cardiol. 2008, 52, 750–754. [Google Scholar] [CrossRef] [Green Version]
- Epelman, S.; Shrestha, K.; Troughton, R.W.; Francis, G.S.; Sen, S.; Klein, A.L.; Tang, W.H. Soluble angiotensin-converting enzyme 2 in human heart failure: Relation with myocardial function and clinical outcomes. J. Card. Fail. 2009, 15, 565–571. [Google Scholar] [CrossRef] [Green Version]
- Soro-Paavonen, A.; Gordin, D.; Forsblom, C.; Rosengard-Barlund, M.; Waden, J.; Thorn, L.; Sandholm, N.; Thomas, M.C.; Groop, P.H. Circulating ACE2 activity is increased in patients with type 1 diabetes and vascular complications. J. Hypertens. 2012, 30, 375–383. [Google Scholar] [CrossRef]
- Walters, T.E.; Kalman, J.M.; Patel, S.K.; Mearns, M.; Velkoska, E.; Burrell, L.M. Angiotensin converting enzyme 2 activity and human atrial fibrillation: Increased plasma angiotensin converting enzyme 2 activity is associated with atrial fibrillation and more advanced left atrial structural remodelling. EP Eur. 2017, 19, 1280–1287. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Wang, Z.; Yang, X.; Hu, B.; Huang, Y.; Fan, S. Association between circulating angiotensin-converting enzyme 2 and cardiac remodeling in hypertensive patients. Peptides 2017, 90, 63–68. [Google Scholar] [CrossRef]
- South, A.M.; Diz, D.I.; Chappell, M.C. COVID-19, ACE2, and the cardiovascular consequences. Am. J. Physiol. Heart Circ. Physiol. 2020, 318, H1084–H1090. [Google Scholar] [CrossRef] [Green Version]
- Guo, J.; Huang, Z.; Lin, L.; Lv, J. Coronavirus Disease 2019 (COVID-19) and Cardiovascular Disease: A ViewpoInt. on the Potential Influence of Angiotensin-Converting Enzyme Inhibitors/Angiotensin Receptor Blockers on Onset and Severity of Severe Acute Respiratory Syndrome Coronavirus 2 Infection. J. Am. Heart Assoc. 2020, 9, e016219. [Google Scholar] [CrossRef] [PubMed]
- Oudit, G.Y.; Kassiri, Z.; Jiang, C.; Liu, P.P.; Poutanen, S.M.; Penninger, J.M.; Butany, J. SARS-coronavirus modulation of myocardial ACE2 expression and inflammation in patients with SARS. Eur. J. Clin. Investig. 2009, 39, 618–625. [Google Scholar] [CrossRef] [PubMed]
- Pirzada, A.; Mokhtar, A.T.; Moeller, A.D. COVID-19 and Myocarditis: What Do We Know So Far? CJC Open 2020, 2, 278–285. [Google Scholar] [CrossRef] [PubMed]
- Siddiqi, H.K.; Libby, P.; Ridker, P.M. COVID-19—A vascular disease. Trends Cardiovasc. Med. 2020. [Google Scholar] [CrossRef] [PubMed]
- Bellocchio, L.; Bordea, I.R.; Ballini, A.; Lorusso, F.; Hazballa, D.; Isacco, C.G.; Malcangi, G.; Inchingolo, A.D.; Dipalma, G.; Inchingolo, F.; et al. Environmental Issues and Neurological Manifestations Associated with COVID-19 Pandemic: New Aspects of the Disease? Int. J. Environ. Res. Public Health 2020, 17, 8049. [Google Scholar] [CrossRef]
- Hemnes, A.R.; Rathinasabapathy, A.; Austin, E.A.; Brittain, E.L.; Carrier, E.J.; Chen, X.; Fessel, J.P.; Fike, C.D.; Fong, P.; Fortune, N.; et al. A potential therapeutic role for angiotensin-converting enzyme 2 in human pulmonary arterial hypertension. Eur. Respir. J. 2018, 51. [Google Scholar] [CrossRef]
- Monteil, V.; Kwon, H.; Prado, P.; Hagelkrüys, A.; Wimmer, R.A.; Stahl, M.; Leopoldi, A.; Garreta, E.; Hurtado del Pozo, C.; Prosper, F.; et al. Inhibition of SARS-CoV-2 Infections in Engineered Human Tissues Using Clinical-Grade Soluble Human ACE2. Cell 2020, 181, 905–913.e7. [Google Scholar] [CrossRef]
- Akhtar, S.; Benter, I.F.; Danjuma, M.I.; Doi, S.A.R.; Hasan, S.S.; Habib, A.M. Pharmacotherapy in COVID-19 patients: A review of ACE2-raising drugs and their clinical safety. J. Drug Target. 2020, 28, 683–699. [Google Scholar] [CrossRef]
- Meng, J.; Xiao, G.; Zhang, J.; He, X.; Ou, M.; Bi, J.; Yang, R.; Di, W.; Wang, Z.; Li, Z.; et al. Renin-angiotensin system inhibitors improve the clinical outcomes of COVID-19 patients with hypertension. Emerg. Microbes Infect. 2020, 9, 757–760. [Google Scholar] [CrossRef]
- Guo, X.; Zhu, Y.; Hong, Y. Decreased Mortality of COVID-19 With Renin-Angiotensin-Aldosterone System Inhibitors Therapy in Patients With Hypertension: A Meta-Analysis. Hypertension 2020, 76, e13–e14. [Google Scholar] [CrossRef]
- Bean, D.; Kraljevic, Z.; Searle, T.; Bendayan, R.; Pickles, A.; Folarin, A.; Roguski, L.; Noor, K.; Shek, A.; o’gallagher, K.; et al. Treatment with ACE-inhibitors is associated with less severe disease with SARS-Covid-19 infection in a multi-site UK acute Hospital Trust. medRxiv 2020. [Google Scholar] [CrossRef] [Green Version]
- Salah, H.M.; Calcaterra, G.; Mehta, J.L. Renin-Angiotensin System Blockade and Mortality in Patients With Hypertension and COVID-19 Infection. J. Cardiovasc. Pharm. 2020. [Google Scholar] [CrossRef]
- Gao, C.; Cai, Y.; Zhang, K.; Zhou, L.; Zhang, Y.; Zhang, X.; Li, Q.; Li, W.; Yang, S.; Zhao, X.; et al. Association of hypertension and antihypertensive treatment with COVID-19 mortality: A retrospective observational study. Eur. Heart J. 2020, 41, 2058–2066. [Google Scholar] [CrossRef]
- Mehra, M.R.; Desai, S.S.; Kuy, S.; Henry, T.D.; Patel, A.N. Cardiovascular Disease, Drug Therapy, and Mortality in Covid-19. N. Engl. J. Med. 2020, 382, e102. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Zhu, L.; Cai, J.; Lei, F.; Qin, J.J.; Xie, J.; Liu, Y.M.; Zhao, Y.C.; Huang, X.; Lin, L.; et al. Association of Inpatient Use of Angiotensin Converting Enzyme Inhibitors and Angiotensin II Receptor Blockers with Mortality Among Patients With Hypertension Hospitalized With COVID-19. Circ. Res. 2020. [Google Scholar] [CrossRef]
- Inchingolo, A.D.; Inchingolo, A.M.; Bordea, I.R.; Malcangi, G.; Xhajanka, E.; Scarano, A.; Lorusso, F.; Farronato, M.; Tartaglia, G.M.; Isacco, C.G.; et al. SARS-CoV-2 Disease Adjuvant Therapies and Supplements Breakthrough for the Infection Prevention. Microorganisms 2021, 9, 525. [Google Scholar] [CrossRef] [PubMed]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yalcin, H.C.; Sukumaran, V.; Al-Ruweidi, M.K.A.A.; Shurbaji, S. Do Changes in ACE-2 Expression Affect SARS-CoV-2 Virulence and Related Complications: A Closer Look into Membrane-Bound and Soluble Forms. Int. J. Mol. Sci. 2021, 22, 6703. https://doi.org/10.3390/ijms22136703
Yalcin HC, Sukumaran V, Al-Ruweidi MKAA, Shurbaji S. Do Changes in ACE-2 Expression Affect SARS-CoV-2 Virulence and Related Complications: A Closer Look into Membrane-Bound and Soluble Forms. International Journal of Molecular Sciences. 2021; 22(13):6703. https://doi.org/10.3390/ijms22136703
Chicago/Turabian StyleYalcin, Huseyin C., Vijayakumar Sukumaran, Mahmoud Khatib A. A. Al-Ruweidi, and Samar Shurbaji. 2021. "Do Changes in ACE-2 Expression Affect SARS-CoV-2 Virulence and Related Complications: A Closer Look into Membrane-Bound and Soluble Forms" International Journal of Molecular Sciences 22, no. 13: 6703. https://doi.org/10.3390/ijms22136703
APA StyleYalcin, H. C., Sukumaran, V., Al-Ruweidi, M. K. A. A., & Shurbaji, S. (2021). Do Changes in ACE-2 Expression Affect SARS-CoV-2 Virulence and Related Complications: A Closer Look into Membrane-Bound and Soluble Forms. International Journal of Molecular Sciences, 22(13), 6703. https://doi.org/10.3390/ijms22136703