
Neutrophils in Tumorigenesis: Missing Targets for Successful Next Generation Cancer Therapies?

 , ,
, ,  ,
,
Abstract
:1. Introduction
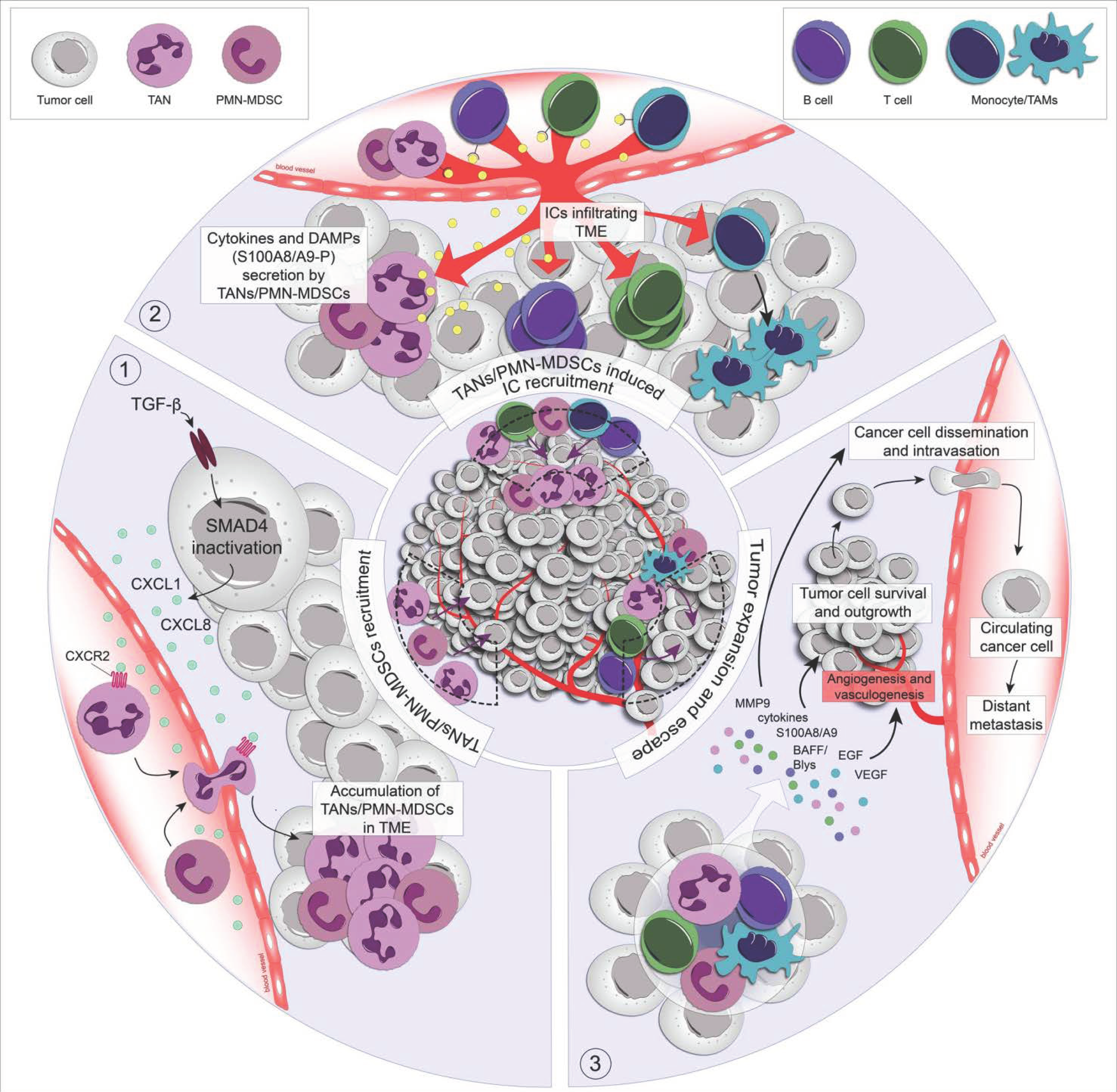
2. The Exodus of Neutrophils towards the Tumor Microenvironment
3. The Ca2+ Signaling Pathway in TANs as a Promising Target for Cancer Therapy
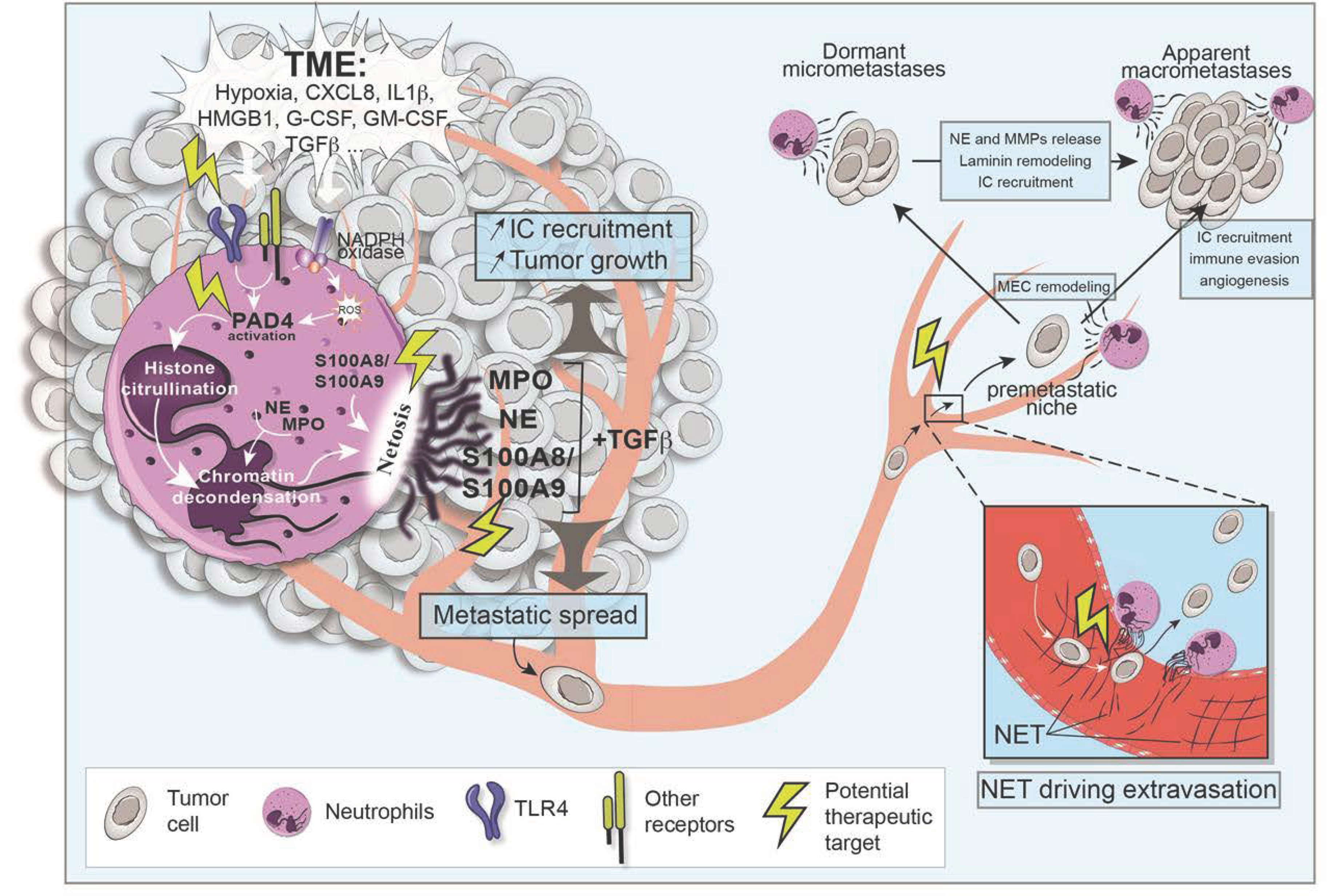
4. Targeting NETosis to Prevent Metastasis
5. Tumor Suppressive Properties of S100A8/A9 Proteins in PMN-MDSCs
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Mantovani, A.; Marchesi, F.; Malesci, A.; Laghi, L.; Allavena, P. Tumour-associated macrophages as treatment targets in oncology. Nat. Rev. Clin. Oncol. 2017, 14, 399–416. [Google Scholar] [CrossRef]
- Noy, R.; Pollard, J.W. Tumor-associated macrophages: From mechanisms to therapy. Immunity 2014, 41, 49–61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruffell, B.; Affara, N.L.; Coussens, L.M. Differential macrophage programming in the tumor microenvironment. Trends Immunol. 2012, 33, 119–126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lewis, C.E.; Pollard, J.W. Distinct role of macrophages in different tumor microenvironments. Cancer Res. 2006, 66, 605–612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pathria, P.; Louis, T.L.; Varner, J.A. Targeting tumor-associated macrophages in cancer. Trends Immunol. 2019, 40, 310–327. [Google Scholar] [CrossRef] [PubMed]
- Athens, J.W.; Haab, O.P.; Raab, S.O.; Mauer, A.M.; Ashenbrucker, H.; Cartwright, G.E.; Wintrobe, M.M. Leukokinetic studies. IV. The total blood, circulating and marginal granulocyte pools and the granulocyte turnover rate in normal subjects. J. Clin. Investig. 1961, 40, 989–995. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hidalgo, A.; Chilvers, E.R.; Summers, C.; Koenderman, L. The neutrophil life cycle. Trends Immunol. 2019, 40, 584–597. [Google Scholar] [CrossRef]
- Tecchio, C.; Micheletti, A.; Cassatella, M.A. Neutrophil-derived cytokines: Facts beyond expression. Front. Immunol. 2014, 5, 508. [Google Scholar] [CrossRef] [Green Version]
- Tamassia, N.; Bianchetto-Aguilera, F.; Arruda-Silva, F.; Gardiman, E.; Gasperini, S.; Calzetti, F.; Cassatella, M.A. Cytokine production by human neutrophils: Revisiting the “dark side of the moon”. Eur. J. Clin. Investig. 2018, 48, e12952. [Google Scholar] [CrossRef]
- Bar-Ad, V.; Palmer, J.; Li, L.; Lai, Y.; Lu, B.; Myers, R.E.; Ye, Z.; Axelrod, R.; Johnson, J.M.; Werner-Wasik, M.; et al. Neutrophil to lymphocyte ratio associated with prognosis of lung cancer. Clin. Transl. Oncol. 2017, 19, 711–717. [Google Scholar] [CrossRef] [PubMed]
- Capone, M.; Giannarelli, D.; Mallardo, D.; Madonna, G.; Festino, L.; Grimaldi, A.M.; Vanella, V.; Simeone, E.; Paone, M.; Palmieri, G.; et al. Baseline neutrophil-to-lymphocyte ratio (NLR) and derived NLR could predict overall survival in patients with advanced melanoma treated with nivolumab. J. Immunother. Cancer 2018, 6, 74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Howard, R.; Kanetsky, P.A.; Egan, K.M. Exploring the prognostic value of the neutrophil-to-lymphocyte ratio in cancer. Sci. Rep. 2019, 9, 19673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cupp, M.A.; Cariolou, M.; Tzoulaki, I.; Aune, D.; Evangelou, E.; ABerlanga-Taylor, A.J. Neutrophil to lymphocyte ratio and cancer prognosis: An umbrella review of systematic reviews and meta-analyses of observational studies. BMC Med. 2020, 18, 360. [Google Scholar] [CrossRef]
- Fridlender, Z.G.; Sun, J.; Kim, S.; Kapoor, V.; Noy, R.; Pollard, J.W. Polarization of tumor-associated neutrophil phenotype by TGF-beta: “N1” versus “N2” TAN. Cancer Cell 2009, 16, 183–194. [Google Scholar] [CrossRef] [Green Version]
- Mollinedo, F. Neutrophil degranulation, plasticity, and cancer metastasis. Trends Immunol. 2019, 40, 228–242. [Google Scholar] [CrossRef]
- Garley, M.; Jabłońska, E. Heterogeneity among neutrophils. Arch. Immunol. Ther. Exp. 2018, 66, 21–30. [Google Scholar] [CrossRef] [PubMed]
- Fridlender, Z.G.; Shaul, M.E. Cancer-related circulating and tumor-associated neutrophils-subtypes, sources and function. FEBS J. 2018, 285, 4316–4342. [Google Scholar]
- Masucci, T.M.; Minopoli, M.; Carriero, M.V. Tumor associated neutrophils. Their role in tumorigenesis, metastasis, prognosis and therapy. Front. Oncol. 2019, 9, 1146. [Google Scholar] [CrossRef] [Green Version]
- Silvestre-Roig, C.; Fridlender, Z.G.; Glogauer, M.; Scapini, P. Neutrophil diversity in health and disease. Trends Immunol. 2019, 40, 565–583. [Google Scholar] [CrossRef]
- Jaillon, S.; Ponzetta, A.; Di Mitri, D.; Santoni, A.; Bonecchi, R.; Mantovani, A. Neutrophil diversity and plasticity in tumour progression and therapy. Nat. Rev. Cancer. 2020, 20, 485–503. [Google Scholar] [CrossRef]
- Gershkovitz, M.; Caspi, Y.; Fainsod-Levi, T.; Katz, B.; Michaeli, J.; Khawaled, S.; Lev, S.; Polyansky, L.; Shaul, M.E.; Sionov, R.V.; et al. TRPM2 mediates neutrophil killing of disseminated tumor cells. Cancer Res. 2018, 78, 2680–2690. [Google Scholar] [CrossRef] [Green Version]
- Kennel, K.B.; Greten, F.R. Immune cell—Produced ROS and their impact on tumor growth and metastasis. Redox Biol. 2021, 42, 101891. [Google Scholar] [CrossRef]
- Shaul, M.E.; Fridlender, Z.G. Neutrophils as active regulators of the immune system in the tumor microenvironment. J. Leukoc. Biol. 2017, 102, 343–349. [Google Scholar] [CrossRef] [Green Version]
- Sun, B.; Qin, W.; Song, M.; Liu, L.; Yu, Y.; Qi, X.; Sun, H. Neutrophil suppresses tumor cell proliferation via Fas/Fas ligand pathway mediated cell cycle arrested. Int. J. Biol. Sci. 2018, 14, 2103–2113. [Google Scholar] [CrossRef]
- Lerman, I.; Hammes, S.R. Neutrophil elastase in the tumor microenvironment. Steroids 2018, 133, 96–101. [Google Scholar] [CrossRef]
- Houghton, A.M.; Rzymkiewicz, D.M.; Ji, H.; Gregory, A.D.; Egea, E.E.; Metz, H.E.; Stolz, D.B.; Land, S.R.; Marconcini, L.A.; Kliment, C.R.; et al. Neutrophil elastase-mediated degradation of IRS-1 accelerates lung tumor growth. Nat. Med. 2010, 16, 219–223. [Google Scholar] [CrossRef] [Green Version]
- Pivetta, E.; Danussi, C.; Wassermann, B.; Modica, T.M.E.; Del Bel Belluz, L.; Canzonieri, V.; Colombatti, A.; Spessotto, P. Neutrophil elastase-dependent cleavage compromises the tumor suppressor role of EMILIN1. Matrix Biol. 2014, 34, 22–32. [Google Scholar] [CrossRef]
- Pellinen, T.; Rantala, J.K.; Arjonen, A.; Mpindi, J.-P.; Kallioniemi, O.; Ivaska, J. A functional genetic screen reveals new regulators of β1-integrin activity. J. Cell Sci. 2012, 125, 649–661. [Google Scholar] [CrossRef] [Green Version]
- Van Lint, P.; Libert, C. Chemokine and cytokine processing by matrix metalloproteinases and its effect on leukocyte migration and inflammation. J. Leukoc. Biol. 2007, 82, 1375–1381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soria-Valles, C.; Gutiérrez-Fernández, A.; Guiu, M.; Mari, B.; Fueyo, A.; Gomis, R.R.; López-Otín, C. The anti-metastatic activity of collagenase-2 in breast cancer cells is mediated by a signaling pathway involving decorin and miR-21. Oncogene 2014, 33, 3054–3063. [Google Scholar] [CrossRef] [Green Version]
- Albrengues, J.; Shields, M.A.; Park Ng, D.; Ambrico, A.; Poindexter, M.E.; Upadhyay, P.; Uyeminami, D.L.; Pommier, A.; Küttner, V.; Bružas, E.; et al. Neutrophils extracellular traps produced during inflammation awaken dormant cancer cells in mice. Science 2018, 361, eaao4227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deryugina, E.I.; Zajac, E.; Juncker-Jensen, A.; Kupriyanova, T.A.; Welter, L.; Quigley, J.P. Tissue-infiltrating neutrophils constitute the major in vivo source of angiogenesis-inducing MMP-9 in the tumor microenvironment. Neoplasia 2014, 16, 771–788. [Google Scholar] [CrossRef] [Green Version]
- Winkler, J.; Abisoye-Ogunniyan, A.; Metcalf, K.J.; Werb, Z. Concepts of extracellular matrix remodelling in tumour progression and metastasis. Nat. Commun. 2020, 11, 5120. [Google Scholar] [CrossRef] [PubMed]
- Grzywa, T.M.; Sosnowska, A.; Matryba, P.; Rydzynska, Z.; Jasinski, M.; Nowis, D.; Golab, J. Myeloid cell-derived arginase in cancer immune response. Front. Immunol. 2020, 11, 938. [Google Scholar] [CrossRef]
- Moses, K.; Brandau, S. Human neutrophils: Their role in cancer and relation to myeloid-derived suppressor cells. Semin. Immunol. 2016, 28, 187–196. [Google Scholar] [CrossRef]
- Pylaeva, E.; Harati, M.D.; Spyra, I.; Bordbari, S.; Strachan, S.; Thakur, B.K.; Höing, B.; Franklin, C.; Skokowa, J.; Welte, K.; et al. NAMPT signaling is critical for the proangiogenic activity of tumor-associated neutrophils. Int. J. Cancer 2019, 144, 136–149. [Google Scholar] [CrossRef]
- Singel, K.L.; Segal, B.H. Neutrophils in the tumor microenvironment: Trying to heal the wound that cannot heal. Immunol. Rev. 2016, 273, 329–343. [Google Scholar] [CrossRef] [Green Version]
- Martins, F.; Sofiya, L.; Sykiotis, G.P.; Lamine, F.; Maillard, M.; Fraga, M.; Shabafrouz, K.; Ribi, C.; Cairoli, A.; Guex-Crosier, Y.; et al. Adverse effects of immune-checkpoint inhibitors: Epidemiology, management and surveillance. Nat. Rev. Clin. Oncol. 2019, 16, 563–580. [Google Scholar] [CrossRef]
- Ostrand-Rosenberg, S. Immune surveillance: A balance between protumor and antitumor immunity. Curr. Opin. Genet. Dev. 2008, 18, 11–18. [Google Scholar] [CrossRef] [Green Version]
- Michaeli, J.; Shaul, M.E.; Mishalian, I.; Hovav, A.-H.; Levy, L.; Zolotriov, L.; Granot, Z.; Zvi, G.; Fridlender, Z.G. Tumor-associated neutrophils induce apoptosis of non-activated CD8 T-cells in a TNFα and NO-dependent mechanism, promoting a tumor-supportive environment. Oncoimmunology 2017, 6, e1356965. [Google Scholar] [CrossRef] [PubMed]
- Powell, D.; Lou, M.; Barros Becker, F.; Huttenlocher, A. Cxcr1 mediates recruitment of neutrophils and supports proliferation of tumor-initiating astrocytes in vivo. Sci. Rep. 2018, 8, 13285. [Google Scholar] [CrossRef]
- Eash, K.J.; Greenbaum, A.M.; Gopalan, P.K.; Link, D.C. CXCR2 and CXCR4 antagonistically regulate neutrophil trafficking from murine bone marrow. J. Clin. Investig. 2010, 120, 2423–2431. [Google Scholar] [CrossRef] [Green Version]
- Ogawa, R.; Yamamoto, T.; Hirai, H.; Hanada, K.; Kiyasu, Y.; Nishikawa, G.; Mizuno, R.; Inamoto, S.; Itatani, Y.; Sakai, Y.; et al. Loss of SMAD4 promotes colorectal cancer progression by recruiting tumor-associated neutrophils via the CXCL1/8-CXCR2 axis. Clin. Cancer Res. 2019, 25, 2887–2899. [Google Scholar] [CrossRef] [Green Version]
- Jamieson, T.; Clarke, M.; Steele, C.W.; Samuel, M.S.; Neumann, J.; Jung, A.; Huels, D.; Olson, M.F.; Das, S.; Nibbs, R.J.; et al. Inhibition of CXCR2 profoundly suppresses inflammation-driven and spontaneous tumorigenesis. J. Clin. Investig. 2012, 122, 3127–3144. [Google Scholar] [CrossRef]
- Grenier, A.; Chollet-Martin, S.; Crestani, B.; Delarche, C.; El Benna, J.; Boutten, A.; Andrieu, V.; Durand, G.; Gougerot-Pocidalo, M.A.; Aubier, M.; et al. Presence of a mobilizable intracellular pool of hepatocyte growth factor in human polymorphonuclear neutrophils. Blood 2002, 99, 2997–3004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Finisguerra, V.; Di Conza, G.; Di Matteo, M.; Serneels, J.; Costa, S.; Thompson, A.A.; Wauters, E.; Walmsley, S.; Prenen, H.; Granot, Z.; et al. MET is required for the recruitment of anti-tumoural neutrophils. Nature 2015, 522, 349–353. [Google Scholar] [CrossRef] [PubMed]
- Tecchio, C.; Scapini, P.; Pizzolo, G.; Cassatella, M.A. On the cytokines produced by human neutrophils in tumors. Semin. Cancer Biol. 2013, 23, 159–170. [Google Scholar] [CrossRef] [PubMed]
- Moreaux, J.; Legouffe, E.; Jourdan, E.; Quittet, P.; Rème, T.; Lugagne, C.; Moine, P.; Rossi, J.-F.; Klein, B.; Tarte, K. BAFF and APRIL protect myeloma cells from apoptosis induced by interleukin 6 deprivation and dexamethasone. Blood 2004, 103, 3148–3157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clemens, R.A.; Chong, J.; Grimes, D.; Hu, Y.; Lowell, C.A. STIM1 and STIM2 cooperatively regulate mouse neutrophil store-operated calcium entry and cytokine production. Blood 2017, 130, 1565–1577. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.F.; Hsu, K.F.; Shen, M.R. The store-operated Ca2+ entry-mediated signaling is important for cancer spread. Biochim. Biophys. Acta 2016, 1863, 1427–1435. [Google Scholar] [CrossRef]
- Cantonero, C.; Sanchez-Collado, J.; Gonzalez-Nuñez, M.A.; Salido, G.M.; Lopez, J.J.; Jardin, I.; Rosado, J.A. Store-independent Orai1-mediated Ca2+ entry and cancer. Cell Calcium 2019, 80, 1–7. [Google Scholar] [CrossRef]
- Chalmers, S.B.; Monteith, G.R. ORAI channels and cancer. Cell Calcium 2018, 74, 160–167. [Google Scholar] [CrossRef] [Green Version]
- Hogan, P.G.; Rao, A. Store-operated calcium entry: Mechanisms and modulation. Biochem. Biophys. Res. Commun. 2015, 460, 40–49. [Google Scholar] [CrossRef] [Green Version]
- Putney, J.W. Store-operated calcium entry: An historical overview. Adv. Exp. Med. Biol. 2017, 981, 205–214. [Google Scholar] [PubMed]
- Weidinger, C.; Shaw, P.J.; Feske, S. STIM1 and STIM2-mediated Ca2+ influx regulates antitumour immunity by CD8+ T cells. EMBO Mol. Med. 2013, 5, 1311–1321. [Google Scholar] [CrossRef] [PubMed]
- Hann, J.; Bueb, J.-L.; Tolle, F.; Bréchard, S. Calcium signaling and regulation of neutrophil functions: Still a long way to go. J. Leukoc. Biol. 2020, 107, 285–297. [Google Scholar] [CrossRef] [PubMed]
- Demaurex, N.; Saul, S. The role of STIM proteins in neutrophil functions. J. Physiol. 2018, 596, 2699–2708. [Google Scholar] [CrossRef] [Green Version]
- Steinckwich, N.; Myers, P.; Janardhan, K.S.; Flagler, N.D.; King, D.; Petranka, J.G.; Putney, J.W. Role of the store-operated calcium entry protein, STIM1, in neutrophil chemotaxis and infiltration into a murine model of psoriasis-inflamed skin. FASEB J. 2015, 29, 3003–3013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, X.; Friedmann, K.S.; Lyrmann, H.; Zhou, Y.; Schoppmeyer, R.; Knörck, A.; Mang, S.; Hoxha, C.; Angenendt, A.; Backes, C.S.; et al. A calcium optimum for cytotoxic T lymphocyte and natural killer cell cytotoxicity. J. Physiol. 2018, 596, 2681–2698. [Google Scholar] [CrossRef] [Green Version]
- Bréchard, S.; Plançon, S.; Melchior, C.; Tschirhart, E.J. STIM1 but not STIM2 is an essential regulator of Ca2+ influx-mediated NADPH oxidase activity in neutrophil-like HL-60 cells. Biochem. Pharmacol. 2009, 78, 504–513. [Google Scholar] [CrossRef]
- Steinckwich, N.; Schenten, V.; Melchior, C.; Bréchard, S.; Tschirhart, E.J. An essential role of STIM1, Orai1, and S100A8-A9 proteins for Ca2+ signaling and FcγR-mediated phagosomal oxidative activity. J. Immunol. 2011, 186, 2182–2191. [Google Scholar] [CrossRef] [Green Version]
- Crivellato, E.; Nico, B.; Mallardi, F.; Beltrami, C.A.; Ribatti, D. Piecemeal degranulation as a general secretory mechanism. Anat. Rec. A Discov. Mol. Cell Evol. Biol. 2003, 274, 778–784. [Google Scholar] [CrossRef] [PubMed]
- Duitman, E.H.; Orinska, Z.; Bulfone-Paus, S. Mechanisms of cytokine secretion: A portfolio of distinct pathways allows flexbility in cytokine activity. Eur. J. Cell Biol. 2011, 90, 476–483. [Google Scholar] [CrossRef]
- Roth, S.; Agthe, M.; Eickhoff, S.; Möller, S.; Karsten, C.M.; Borregaard, N.; Solbach, W.; Laskay, T. Secondary necrotic neutrophils release interleukin-16C and macrophage migration inhibitory factor from stores in the cytosol. Cell Death Discov. 2015, 1, 15056. [Google Scholar] [CrossRef]
- Van Coillie, E.; Van Aelst, I.; Wuyts, A.; Vercauteren, R.; Devos, R.; De Wolf-Peeters, C.; Van Damme, J.; Opdenakker, G. Tumor angiogenesis induced by granulocyte chemotactic protein-2 as a countercurrent principle. Am. J. Pathol. 2001, 159, 1405–1414. [Google Scholar] [CrossRef] [Green Version]
- Van den Steen, P.E.; Proost, P.; Wuyts, A.; Van Damme, J.; Opdenakker, G. Neutrophil gelatinase B potentiates interleukin-8 tenfold by aminoterminal processing, whereas it degrades CTAP-III, PF-4, and GRO-alpha and leaves RANTES and MCP-2 intact. Blood 2000, 96, 2673–2681. [Google Scholar] [CrossRef] [PubMed]
- Brinkmann, V.; Reichard, U.; Goosmann, C.; Fauler, B.; Uhlemann, Y.; Weiss, D.S.; Weinrauch, Y.; Zychlinsky, A. Neutrophil extracellular traps kill bacteria. Science 2004, 303, 1532–1535. [Google Scholar] [CrossRef] [PubMed]
- Powell, D.R.; Huttenlocher, A. Neutrophils in the tumor microenvironment. Trends Immunol. 2016, 37, 41–52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitroulis, I.; Kambas, K.; Chrysanthopoulou, A.; Skendros, P.; Apostolidou, E.; Kourtzelis, I.; Drosos, G.I.; Boumpas, D.T.; Ritis, K. Neutrophil extracellular trap formation is associated with IL-1beta and autophagy-related signaling in gout. PLoS ONE 2011, 6, e293182011. [Google Scholar]
- Gupta, A.K.; Joshi, M.B.; Philippova, M.; Erne, P.; Hasler, P.; Hahn, S.; Resink, T.J. Activated endothelial cells induce neutrophil extracellular traps and are susceptible to NETosis-mediated cell death. FEBS Lett. 2010, 584, 3193–3197. [Google Scholar] [CrossRef] [Green Version]
- Demers, M.; Krause, D.S.; Schatzberg, D.; Martinod, K.; Voorhees, J.R.; Fuchs, T.A.; Scadden, D.T.; Wagner, D.D. Cancers predispose neutrophils to release extracellular DNA traps that contribute to cancer-associated thrombosis. Proc. Natl. Acad. Sci. USA 2012, 109, 13076–13081. [Google Scholar] [CrossRef] [Green Version]
- Tadie, J.M.; Bae, H.B.; Jiang, S.; Park, D.W.; Bell, C.P.; Yang, H.; Pittet, J.F.; Tracey, K.; Thannickal, V.J.; Abraham, E.; et al. HMGB1 promotes neutrophil extracellular trap formation through interactions with Toll-like receptor 4. Am. J. Physiol. Lung Cell Mol. Physiol. 2013, 304, L342–L349. [Google Scholar] [CrossRef] [Green Version]
- Krotova, K.; Khodayari, N.; Oshins, R.; Aslanidi, G.; Brantly, M.L. Neutrophil elastase promotes macrophage cell adhesion and cytokine production through the integrin-Src kinases pathway. Sci. Rep. 2020, 10, 15874. [Google Scholar] [CrossRef] [PubMed]
- De Bont, C.M.; Boelens, W.C.; Pruijn, G.J.M. NETosis, complement, and coagulation: A triangular relationship. Cell. Mol. Immunol. 2019, 16, 19–27. [Google Scholar] [CrossRef]
- Guglietta, S.; Chiavelli, A.; Zagato, E.; Krieg, C.; Gandini, S.; Ravenda, P.S.; Bazolli, B.; Lu, B.; Penna, G.; Rescigno, M. Coagulation induced by C3aR-dependent NETosis drives protumorigenic neutrophils during small intestinal tumorigenesis. Nat. Commun. 2016, 7, 1103. [Google Scholar] [CrossRef]
- Cools-Lartigue, J.; Spicer, J.; McDonald, B.; Gowing, S.; Chow, S.; Giannias, B.; Bourdeau, F.; Kubes, P.; Ferri, L. Neutrophil extracellular traps sequester circulating tumor cells and promote metastasis. J. Clin. Investig. 2013, 123, 3446–3458. [Google Scholar] [CrossRef]
- Zhang, P.; Ozdemir, T.; Chung, C.Y.; Robertson, G.P.; Dong, C. Sequential binding of alphaVbeta3 and ICAM-1 determines fibrin-mediated melanoma capture and stable adhesion to CD11b/CD18 on neutrophils. J. Immunol. 2011, 186, 242–254. [Google Scholar] [CrossRef] [Green Version]
- Spicer, J.D.; McDonald, B.; Cools-Lartigue, J.J.; Chow, S.C.; Giannias, B.; Kubes, P.; Lorenzo, E.; Ferri, L.E. Neutrophils promote liver metastasis via Mac-1-mediated interactions with circulating tumor cells. Cancer Res. 2012, 72, 3919–3927. [Google Scholar] [CrossRef] [Green Version]
- Kolaczkowska, E.; Jenne, C.N.; Surewaard, B.G.; Thanabalasuriar, A.; Lee, W.Y.; Sanz, M.J.; Mowen, K.; Opdenakker, G.; Kubes, P. Molecular mechanisms of NET formation and degradation revealed by intravital imaging in the liver vasculature. Nat. Commun. 2015, 6, 6673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nie, M.; Yang, L.; Bi, X.; Wang, Y.; Sun, P.; Yang, H.; Liu, P.; Li, Z.; Xia, Y.; Jiang, W. Neutrophil Extracellular Traps Induced by IL8 Promote Diffuse Large B-cell Lymphoma Progression via the TLR9 Signaling. Clin. Cancer Res. 2019, 25, 1867–1879. [Google Scholar] [CrossRef] [PubMed]
- Podaza, E.; Sabbione, F.; Risnik, D.; Borge, M.; Almejún, M.B.; Colado, A.; Fernández-Grecco, H.; Cabrejo, M.; Bezares, R.F.; Trevani, A.; et al. Neutrophils from chronic lymphocytic leukemia patients exhibit an increased capacity to release extracellular traps (NETs). Cancer Immunol. Immunother. 2017, 66, 77–89. [Google Scholar] [CrossRef]
- Hirz, T.; Matera, E.-L.; Chettab, K.; Jordheim, L.P.; Mathé, D.; Evesque, A.; Esmenjaud, J.; Salles, G.; Dumontet, C. Neutrophils protect lymphoma cells against cytotoxic and targeted therapies through CD11b/ICAM-1 binding. Oncotarget 2017, 8, 72818–72834. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Romano, A.; Parrinello, N.L.; Simeon, V.; Puglisi, F.; La Cava, P.; Bellofiore, C.; Giallongo, C.; Camiolo, G.; D’Auria, F.; Grieco, V.; et al. High-density neutrophils in MGUS and multiple myeloma are dysfunctional and immune-suppressive due to increased STAT3 downstream signaling. Sci. Rep. 2020, 10, 1983. [Google Scholar] [CrossRef]
- Yipp, B.G.; Kubes, P. NETosis: How vital is it? Blood 2013, 122, 2784–2794. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Biermann, M.H.; Brauner, J.M.; Liu, Y.; Zhao, Y.; Herrmann, M. New insights into neutrophil extracellular traps: Mechanisms of formation and role in inflammation. Front. Immunol. 2016, 7, 302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yousefi, S.; Stojkov, D.; Germic, N.; Simon, D.; Wang, X.; Benarafa, C.; Simon, H.-U. Untangling “NETosis” from NETs. Eur. J. Immunol. 2019, 49, 221–227. [Google Scholar] [CrossRef] [Green Version]
- Van der Weyden, L.; Speak, A.O.; Swiatkowska, A.; Clare, S.; Schejtman, A.; Santilli, G.; Arends, M.J.; Adams, D.J. Pulmonary metastatic colonisation and granulomas in NOX2-deficient mice. J. Pathol. 2018, 246, 300–310. [Google Scholar] [CrossRef]
- Tay, R.E.; Richardson, E.K.; Toh, H.C. Revisiting the role of CD4 + T cells in cancer immunotherapy-new insights into old paradigms. Cancer Gene Ther. 2020, 28, 5–17. [Google Scholar] [CrossRef]
- Wu, S.Y.; Fu, T.; Jiang, Y.Z.; Shao, Z.M. Natural killer cells in cancer biology and therapy. Mol. Cancer 2020, 19, 120. [Google Scholar] [CrossRef]
- Yang, F.; Kemp, C.J.; Henikoff, S. Anthracyclines induce double-strand DNA breaks at active gene promoters. Mutat. Res. 2015, 773, 9–15. [Google Scholar] [CrossRef] [Green Version]
- Pommier, Y.; Sun, Y.; Huang, S.N.; Nitiss, J.L. Roles of eukaryotic topoisomerases in transcription, replication and genomic stability. Nat. Rev. Mol. Cell Biol. 2016, 17, 703–721. [Google Scholar] [CrossRef]
- Khan, M.A.; D’Ovidio, A.; Tran, H.; Palaniyar, N. Anthracyclines suppress both NADPH oxidase- dependent and -independent NETosis in human neutrophils. Cancers 2019, 11, 1328. [Google Scholar] [CrossRef] [Green Version]
- Yuzhalin, A.E.; Gordon-Weeks, A.N.; Tognoli, M.L.; Jones, K.; Markelc, B.; Konietzny, R.; Fischer, R.; Muth, A.; O’Neill, E.; Thompson, P.R.; et al. Colorectal cancer liver metastatic growth depends on PAD4-driven citrullination of the extracellular matrix. Nat. Commun. 2018, 9, 4783. [Google Scholar] [CrossRef]
- Li, M.; Lin, C.; Deng, H.; Strnad, J.; Bernabei, L.; Vogl, D.T.; Burke, J.J.; Nefedova, Y. A novel peptidylarginine deiminase 4 (PAD4) inhibitor BMS-P5 blocks formation of neutrophil extracellular traps and delays progression of multiple myeloma. Mol. Cancer Ther. 2020, 19, 1530–1538. [Google Scholar] [CrossRef] [PubMed]
- Jiménez-Alcázar, M.; Rangaswamy, C.; Panda, R.; Bitterling, J.; Simsek, Y.J.; Long, A.T.; Bilyy, R.; Krenn, V.; Renné, C.; Renné, T.; et al. Host DNases prevent vascular occlusion by neutrophil extracellular traps. Science 2017, 358, 1202–1206. [Google Scholar] [CrossRef] [Green Version]
- Wong, S.L.; Wagner, D.D. Peptidylarginine deiminase 4, a nuclear button triggering neutrophil extracellular traps in inflammatory diseases and aging. FASEB J. 2018, 32, 6258–6370. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Wysocki, R.W.; Amoozgar, Z.; Maiorino, L.; Fein, M.R.; Jorns, J.; Schott, A.F.; Kinugasa-Katayama, Y.; Lee, Y.; Won, N.H.; et al. Cancer cells induce metastasis-supporting neutrophil extracellular DNA traps. Sci. Transl. Med. 2016, 8, 361ra138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Demers, M.; Wong, S.L.; Martinod, K.; Gallant, M.; Cabral, J.E.; Wang, Y.; Wagner, D.D. Priming of neutrophils toward NETosis promotes tumor growth. Oncoimmunology 2016, 5, e1134073. [Google Scholar] [CrossRef] [Green Version]
- Roberts, A.W. G-CSF: A key regulator of neutrophil production, but that’s not all! Growth Factors 2005, 23, 33–41. [Google Scholar] [CrossRef]
- Lieschke, G.J.; Grail, D.; Hodgson, G.; Metcalf, D.; Stanley, E.; Cheers, C.; Fowler, K.J.; Basu, S.; Zhan, Y.F.; Dunn, A.R. Mice lacking granulocyte colony-stimulating factor have chronic neutropenia, granulocyte and macrophage progenitor cell deficiency, and impaired neutrophil mobilization. Blood 1994, 84, 1737–1746. [Google Scholar] [CrossRef]
- Wang, H.; Aloe, C.; Wilson, N.; Bozinovski, S. G-CSFR antagonism reduces neutrophilic inflammation during pneumococcal and influenza respiratory infections without compromising clearance. Sci. Rep. 2019, 9, 17732. [Google Scholar] [CrossRef]
- Odajima, T.; Onishi, M.; Hayama, E.; Motoji, N.; Momose, Y.; Shigematsu, A. Cytolysis of B-16 melanoma tumor cells mediated by the myeloperoxidase and lactoperoxidase systems. Biol. Chem. 1996, 377, 689–693. [Google Scholar] [PubMed]
- Weel, E.A.; Redekop, W.K.; Weening, R.S. Increased risk of malignancy for patients with chronic granulomatous disease and its possible link to the pathogenesis of cancer. Eur. J. Cancer 1996, 32, 734–735. [Google Scholar] [CrossRef]
- Fuchs, T.A.; Abed, U.; Goosmann, C.; Hurwitz, R.; Schulze, I.; Wahn, V.; Weinrauch, Y.; Brinkmann, V.; Zychlinsky, A. Novel cell death program leads to neutrophil extracellular traps. J. Cell Biol. 2007, 176, 231–241. [Google Scholar] [CrossRef] [PubMed]
- Schenten, V.; Plançon, S.; Jung, N.; Hann, J.; Bueb, J.-L.; Bréchard, S.; Tschirhart, E.J.; Tolle, F. Secretion of the phosphorylated form of S100A9 from neutrophils is essential for the proinflammatory functions of extracellular S100A8/A9. Front. Immunol. 2018, 9, 447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shabani, F.; Farasat, A.; Mahdavi, M.; Gheibi, N. Calprotectin (S100A8/S100A9): A key protein between inflammation and cancer. Inflamm. Res. 2018, 67, 801–812. [Google Scholar] [CrossRef] [PubMed]
- Passey, R.J.; Xu, K.; Hume, D.A.; Geczy, C. S100A8: Emerging functions and regulation. J. Leukoc. Biol. 1999, 66, 549–556. [Google Scholar] [CrossRef] [Green Version]
- Hiratsuka, S.; Watanabe, A.; Sakurai, Y.; Akashi-Takamura, S.; Ishibashi, S.; Miyake, K.; Shibuya, M.; Akira, S.; Aburatani, H.; Maru, Y. The S100A8-serum amyloid A3-TLR4 paracrine cascade establishes a pre-metastatic phase. Nat. Cell Biol. 2008, 10, 1349–1355. [Google Scholar] [CrossRef]
- Ghavami, S.; Chitayat, S.; Hashemi, M.; Eshraghi, M.; Chazin, W.J.; Halayko, A.J.; Kerkhoff, C. S100A8/A9: A Janus-faced molecule in cancer therapy and tumorgenesis. Eur. J. Pharmacol. 2009, 625, 73–83. [Google Scholar] [CrossRef]
- Ghavami, S.; Kerkhoff, C.; Los, M.; Hashemi, M.; Sorg, C.; Karami-Tehrani, F. Mechanism of apoptosis induced by S100A8/A9 in colon cancer cell lines: The role of ROS and the effect of metal ions. J. Leukoc. Biol. 2004, 76, 169–175. [Google Scholar] [CrossRef] [Green Version]
- Ghavami, S.; Kerkhoff, C.; Chazin, W.J.; Kadkhoda, K.; Xiao, W.; Zuse, A.; Hashemi, M.; Eshraghi, M.; Schulze-Osthoff, K.; Klonisch, T.; et al. S100A8/9 induces cell death via a novel, RAGE-independent pathway that involves selective release of Smac/DIABLO and Omi/HtrA2. Biochim. Biophys. Acta 2008, 1783, 297–311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Condamime, T.; Dominguez, G.A.; Youn, J.-I.; Kossenkov, A.V.; Mony, S.; Alicea-Torres, K.; Tcyganov, E.; Hashimoto, A.; Nefedova, Y.; Lin, C.; et al. Lectin-type oxidized LDL receptor-1 distinguishes population of human polymorphonuclear myeloid-derived suppressor cells in cancer patients. Sci. Immunol. 2016, 1, aaf8943. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, P.; Corzo, C.A.; Luetteke, N.; Yu, B.; Nagaraj, S.; Bui, M.M.; Ortiz, M.; Nacken, W.; Sorg, C.; Vogl, T.; et al. Inhibition of dendritic cell differentiation and accumulation of myeloid-derived suppressor cells in cancer is regulated by S100A9 protein. J. Exp. Med. 2008, 205, 2235–2249. [Google Scholar] [CrossRef]
- Hermani, A.; De Servi, B.; Medunjanin, S.; Tessier, P.A.; Mayer, D. S100A8 and S100A9 activate MAP kinase and NF-kappaB signaling pathways and trigger translocation of RAGE in human prostate cancer cells. Ex. Cell Res. 2006, 312, 184–197. [Google Scholar] [CrossRef]
- Ichikawa, M.; Williams, R.; Wang, L.; Vogl, T.; Srikrishna, G. S100A8/A9 activate key genes and pathways in colon tumor progression. Mol. Cancer Res. 2011, 9, 133–148. [Google Scholar] [CrossRef] [Green Version]
- Voss, A.; Bode, G.; Sopalla, C.; Benedyk, M.; Varga, G.; Böhm, M.; Nacken, W.; Kerkhoff, C. Expression of S100A8/A9 in HaCaT keratinocytes alters the rate of cell proliferation and differentiation. FEBS Lett. 2011, 585, 440–446. [Google Scholar] [CrossRef] [Green Version]
- Hiratsuka, S.; Watanabe, A.; Aburatani, H.; Maru, Y. Tumour-mediated upregulation of chemoattractants and recruitment of myeloid cells predetermines lung metastasis. Nat. Cell Biol. 2006, 8, 1369–1375. [Google Scholar] [CrossRef] [PubMed]
- Bronte, V.; Brandau, S.; Chen, S.H.; Colombo, M.P.; Frey, A.B.; Greten, T.F.; Mandruzzato, S.; Murray, P.J.; Ochoa, A.; Ostrand-Rosenberg, S.; et al. Recommendations for myeloid-derived suppressor cell nomenclature and characterization standards. Nat. Commun. 2016, 7, 12150. [Google Scholar] [CrossRef] [Green Version]
- Veglia, F.; Sanseviero, E.; Gabrilovich, D.I. Myeloid-derived suppressor cells in the era of increasing myeloid cell diversity. Nat. Rev. Immunol. 2021, 1–14. [Google Scholar] [CrossRef]
- Ostrand-Rosenberg, S.; Fenselau, C. Myeloid-derived suppressor cells: Immune-suppressive cells that impair antitumor immunity and are sculpted by their environment. J. Immunol. 2018, 200, 422–431. [Google Scholar] [CrossRef] [Green Version]
- Groth, C.; Hu, X.; Weber, R.; Fleming, V.; Altevogt, P.; Utikal, J.; Umansky, V. Immunosuppression mediated by myeloid-derived suppressor cells (MDSCs) during tumour progression. Br. J. Cancer 2019, 120, 16–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Solito, S.; Marigo, I.; Pinton, L.; Damuzzo, V.; Mandruzzato, S.; Bronte, V. Myeloid-derived suppressor cell heterogeneity in human cancers. Ann. N. Y. Acad. Sci. 2014, 1319, 47–65. [Google Scholar] [CrossRef]
- Cassetta, L.; Bruderek, K.; Skrzeczynska-Moncznik, J.; Osiecka, O.; Hu, X.; Rundgren, I.M.; Lin, A.; Santegoets, K.; Horzum, U.; Godinho-Santos, A.; et al. Differential expansion of circulating human MDSC subsets in patients with cancer, infection and inflammation. J. Immunother. Cancer 2020, 8, e001223. [Google Scholar] [CrossRef]
- Zhou, J.; Nefedova, Y.; Lei, A.; Gabrilovich, D. Neutrophils and PMN-MDSCs: Their biological role and interaction with stromal cells. Semin. Immunol. 2018, 35, 19–28. [Google Scholar] [CrossRef] [PubMed]
- Si, Y.; Merz, S.F.; Jansen, P.; Wang, B.; Bruderek, K.; Altenhoff, P.; Mattheis, S.; Lang, S.; Gunzer, M.; Klode, J.; et al. Multidimensional imaging provides evidence for down-regulation of T cell effector function by MDSC in human cancer tissue. Sci. Immunol. 2019, 4, eaaw9159. [Google Scholar] [CrossRef]
- Groth, C.; Arpinati, L.; Shaul, M.E.; Winkler, N.; Diester, K.; Gengenbacher, N.; Weber, R.; Arkhypov, I.; Lasser, S.; Petrova, V.; et al. Blocking Migration of Polymorphonuclear Myeloid-Derived Suppressor Cells Inhibits Mouse Melanoma Progression. Cancers 2021, 13, 726. [Google Scholar] [CrossRef]
- Lang, S.; Bruderek, K.; Kaspar, C.; Höing, B.; Kanaan, O.; Dominas, N.; Hussain, T.; Droege, F.; Eyth, C.; Hadaschik, B.; et al. Clinical Relevance and Suppressive Capacity of Human Myeloid-Derived Suppressor Cell Subsets. Clin. Cancer Res. 2018, 24, 4834–4844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diaz-Montero, C.M.; Salem, M.L.; Nishimura, M.I.; Garrett-Mayer, E.; Cole, D.J.; Montero, A.J. Increased circulating myeloid-derived suppressor cells correlate with clinical cancer stage, metastatic tumor burden, and doxorubicin-cyclophosphamide chemotherapy. Cancer Immunol. Immunother. 2009, 58, 49–59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ben-Meir, K.; Twaik, N.; Baniyash, M. Plasticity and biological diversity of myeloid derived suppressor cells. Curr. Opin. Immunol. 2018, 51, 154–161. [Google Scholar] [CrossRef] [PubMed]
- Ugolini, A.; Tyurin, V.A.; Tyurina, Y.Y.; Tcyganov, E.N.; Donthireddy, L.; Kagan, V.E.; Gabrilovich, D.I.; Veglia, F. Polymorphonuclear myeloid-derived suppressor cells limit antigen cross-presentation by dendritic cells in cancer. JCI Insight 2020, 5, e138581. [Google Scholar] [CrossRef]
- Serafini, P.; Mgebroff, S.; Noonan, K.; Borrello, I. Myeloid-derived suppressor cells promote cross-tolerance in B-cell lymphoma by expanding regulatory T cells. Cancer Res. 2008, 68, 5439–5449. [Google Scholar] [CrossRef] [Green Version]
- Pan, P.Y.; Ma, G.; Weber, K.J.; Ozao-Choy, J.; Wang, G.; Yin, B.; Divino, C.M.; Chen, S.H. Immune stimulatory receptor CD40 is required for T-cell suppression and T regulatory cell activation mediated by myeloid-derived suppressor cells in cancer. Cancer Res. 2010, 70, 99–108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Christiansson, L.; Söderlund, S.; Svensson, E.; Mustjoki, S.; Bengtsson, M.; Simonsson, B.; Olsson-Strömberg, U.; Loskog, A.S.I. Increased level of myeloid-derived suppressor cells, programmed death receptor ligand 1/programmed death receptor 1, and soluble CD25 in Sokal high risk chronic myeloid leukemia. PLoS ONE 2013, 8, e55818. [Google Scholar] [CrossRef] [Green Version]
- Ramachandran, I.; Martner, A.; Pisklakova, A.; Condamine, T.; Chase, T.; Vogl, T.; Roth, J.; Gabrilovich, D.; Nefedova, Y. Myeloid derived suppressor cells regulate growth of multiple myeloma by inhibiting T cells in bone marrow. J. Immunol. 2013, 190, 3815–3823. [Google Scholar] [CrossRef] [Green Version]
- Giallongo, C.; Parrinello, N.; Tibullo, D.; La Cava, P.; Romano, A.; Chiarenza, A.; Barbagallo, I.; Palumbo, G.A.; Stagno, F.; Vigneri, P.; et al. Myeloid derived suppressor cells (MDSCs) are increased and exert immunosuppressive activity together with Polymorphonuclear Leukocytes (PMNs) in chronic myeloid leukemia patients. PLoS ONE. 2014, 9, e101848. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kittang, A.O.; Kordasti, S.; Sand, K.E.; Costantini, B.; Kramer, A.M.; Perezabellan, P.; Seidl, T.; Rye, K.P.; Hagen, K.M.; Kulasekararaj, A.; et al. Expansion of myeloid derived suppressor cells correlates with number of T regulatory cells and disease progression in myelodysplastic syndrome. Oncoimmunology 2016, 5, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Marini, O.; Spina, C.; Mimiola, E.; Cassaro, A.; Malerba, G.; Todeschini, G.; Perbellini, O.; Scupoli, M.; Carli, G.; Facchinelli, D.; et al. Identification of granulocytic myeloid-derived suppressor cells (G-MDSCs) in the peripheral blood of Hodgkin and non-Hodgkin lymphoma patients. Oncotarget 2016, 7, 27676–27688. [Google Scholar] [CrossRef] [Green Version]
- Romano, A.; Parrinello, N.L.; La Cava, P.; Tibullo, D.; Giallongo, C.; Camiolo, G.; Puglisi, F.; Parisi, M.; Pirosa, M.C.; Martino, E.; et al. PMN-MDSC and arginase are increased in myeloma and may contribute to resistance to therapy. Expert. Rev. Mol. Diagn. 2018, 18, 675–683. [Google Scholar] [CrossRef] [PubMed]
- Umansky, V.; Adema, G.J.; Baran, J.; Brandau, S.; Van Ginderachter, J.A.; Hu, X.; Jablonska, J.; Mojsilovic, S.; Papadaki, H.A.; Pico de Coaña, Y.; et al. Interactions among myeloid regulatory cells in cancer. Cancer Immunol. Immunother. 2019, 68, 645–660. [Google Scholar] [CrossRef] [PubMed]
- Veglia, F.; Hashimoto, A.; Dweep, H.; Sanseviero, E.; De Leo, A.; Tcyganov, E.; Kossenkov, A.; Mulligan, C.; Nam, B.; Masters, G.; et al. Analysis of classical neutrophils and polymorphonuclear myeloid-derived suppressor cells in cancer patients and tumor-bearing mice. J. Exp. Med. 2012, 218, e20201803. [Google Scholar] [CrossRef] [PubMed]
- Sinha, P.; Okoro, C.; Foell, D.; Freeze, H.H.; Ostrand-Rosenberg, S.; Srikrishna, G. Proinflammatory S100 proteins regulate the accumulation of myeloid-derived suppressor cells. J. Immunol. 2008, 181, 4666–4675. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Srikrishna, G.; Toomre, D.K.; Manzi, A.; Panneerselvam, K.; Freeze, H.H.; Varki, A.; Varki, N.M. A novel anionic modification of N-glycans on mammalian endothelial cells is recognized by activated neutrophils and modulates acute inflammatory responses. J. Immunol. 2001, 166, 624–632. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turovskaya, O.; Foell, D.; Sinha, P.; Vogl, T.; Newlin, R.; Nayak, J.; Nguyen, M.; Nawroth, P.; Bierhaus, A.; Varki, N.; et al. RAGE, carboxylated glycans and S100A8/A9 play essential roles in colitis associated carcinogenesis. Carcinogenesis 2008, 29, 2035–2043. [Google Scholar] [CrossRef] [PubMed]
- Leclerc, E.; Vetter, S.W. The role of S100 proteins and their receptor RAGE in pancreatic cancer. Biochim. Biophys. Acta 2015, 1852, 2706–2711. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Azizan, N.; Suter, M.A.; Liu, Y.; Logsdon, C.D. RAGE maintains high levels of NFκB and oncogenic Kras activity in pancreatic cancer. Biochem. Biophys. Res. Commun. 2017, 493, 592–597. [Google Scholar] [CrossRef]
- Shahab, U.; Ahmad, M.K.; Mahdi, A.A.; Waseem, M.; Arif, B.; Moinuddin, A.B.; Ahmad, S. The receptor for advanced glycation end products: A fuel to pancreatic cancer. Semin. Cancer Biol. 2018, 49, 37–43. [Google Scholar] [CrossRef]
- Zhao, M.; Mishra, L.; Deng, C.-X. The role of TGF-β/SMAD4 signaling in cancer. Int. J. Biol. Sci. 2018, 14, 111–123. [Google Scholar] [CrossRef] [Green Version]
- Papageorgis, P.; Cheng, K.H.; Ozturk, S.; Gong, Y.; Lambert, A.W.; Abdolmaleky, H.M.; Zhou, J.-R.; Thiagalingam, S. Smad4 inactivation promotes malignancy and drug resistance of colon cancer. Cancer Res. 2011, 71, 998–1008. [Google Scholar] [CrossRef] [Green Version]
- Takaku, K.; Oshima, M.; Miyoshi, H.; Matsui, M.; Seldin, M.F.; Taketo, M.M. Intestinal tumorigenesis in compound mutant mice of both Dpc4 (Smad4) and Apc genes. Cell 1998, 92, 645–656. [Google Scholar] [CrossRef] [Green Version]
- Kojima, K.; Vickers, S.M.; Adsay, N.V.; Jhala, N.C.; Kim, H.-G.; Schoeb, T.R.; Grizzle, W.E.; Klug, C.A. Inactivation of Smad4 accelerates Kras(G12D)-mediated pancreatic neoplasia. Cancer Res. 2007, 67, 8121–8130. [Google Scholar] [CrossRef] [Green Version]
- Bera, A.; Zhao, S.; Cao, L.; Chiao, P.J.; Freeman, J.W. Oncogenic K-Ras and loss of Smad4 mediate invasion by activating an EGFR/NF-κB axis that induces expression of MMP9 and uPA in human pancreas progenitor cells. PLoS ONE 2013, 8, e82282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ziemke, M.; Patil, T.; Nolan, K.; Tippimanchai, D.; Malkoski, S.P. Reduced Smad4 expression and DNA topoisomerase inhibitor chemosensitivity in non-small cell lung cancer. Lung Cancer 2017, 109, 28–35. [Google Scholar] [CrossRef]
- Hernandez, A.L.; Young, C.D.; Wang, J.H.; Wang, X.-J. Lessons learned from SMAD4 loss in squamous cell carcinomas. Mol. Carcinog. 2019, 58, 1648–1655. [Google Scholar] [CrossRef] [Green Version]
- Ang, C.W.; Nedjadi, T.; Sheikh, A.A.; Tweedle, E.M.; Tonack, S.; Honap, S.; Jenkins, R.E.; Park, B.K.; Schwarte-Waldhoff, I.; Khattak, I.; et al. Smad4 loss is associated with fewer S100A8-positive monocytes in colorectal tumors and attenuated response to S100A8 in colorectal and pancreatic cancer cells. Carcinogenesis 2010, 31, 1541–1551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qin, H.; Lerman, B.; Sakamaki, I.; Wei, G.; Cha, S.; Rao, S.S.; Qian, J.; Hailemichael, Y.; Nurieva, R.; Dwyer, K.C.; et al. Generation of a novel therapeutic peptide that depletes MDSC in tumor-bearing mice. Nat. Med. 2014, 20, 676–681. [Google Scholar] [CrossRef] [PubMed]
- Eruslanov, E.B.; Singhal, S.; Albelda, S.M. Mouse versus human neutrophils in cancer—A major knowledge gap. Trends Cancer 2017, 3, 149–160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mestas, J.; Hughes, C.C.W. Of mice and not men: Differences between mouse and human immunology. J. Immunol. 2004, 172, 2731–2738. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Guoqiang, L.; Sun, M.; Lu, X. Targeting and exploitation of tumor-associated neutrophils to enhance immunotherapy and drug delivery for cancer treatment. Cancer Biol. Med. 2020, 17, 32–43. [Google Scholar] [CrossRef]
- Orillion, A.; Hashimoto, A.; Damayanti, N.; Shen, L.; Adelaiye-Ogala, R.; Arisa, S.; Chintala, S.; Ordentlich, P.; Kao, C.; Elzey, B.; et al. Entinostat neutralizes myeloid derived suppressor cells and enhances the antitumor effect of PD-1 inhibition in murine models of lung and renal cell carcinoma. Clin. Cancer Res. 2017, 23, 5187–5201. [Google Scholar] [CrossRef] [Green Version]
- Xue, J.; Zhao, Z.; Zhang, L.; Xue, L.; Shen, S.; Wen, Y.; Wei, Z.; Wang, L.; Kong, L.; Sun, H.; et al. Neutrophil-mediated anticancer drug delivery for suppression of postoperative malignant glioma recurrence. Nat. Nanotechnol. 2017, 12, 692–700. [Google Scholar] [CrossRef]
- Chu, D.; Dong, X.; Shi, X.; Zhang, C.; Wang, Z. Neutrophil-based drug delivery systems. Adv. Mater. 2018, 30, e1706245. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| TAN1 = “Classical Neutrophils”? (Anti-Tumoral) | TAN2 = PMN-MDSCs? (Pro-Tumoral) | ||||
|---|---|---|---|---|---|
| Mature/Segmented Nucleus/High-Density Neutrophils | Immature/Ring-Shaped Nucleus/Low-Density Neutrophils | ||||
| CD66b+/CD11b+/CD14−/HLA-DR+/CD177+/CD15hig | CD15+/HLA-DR/CD11b+/CD14−/CD33+/Lox-1+ | ||||
| Role | Mode of Action | Tumoral Effect | Role | Mode of Action | Tumoral Effect |
| ROS production | • TRPM2 activation → lethal Ca2+ entry [21] | Tumor growth inhibition | ROS production | • DNA mutations [22] | Tumor promotion /progression |
| Chemokine/cytokine secretion | • Leukocyte recruitment • Proliferation of T-cells [23] | Immune anti-tumor response | Chemokine/cytokine secretion | • ↑ CCL17 expression secretion → Tregs recruitment to the TME [23] | Tumor progression |
| Fas signaling | • Activation of caspase cascade [24] | Apoptosis of cancer cells | Neutrophil elastase secretion | • Activation of EGFR, TLR4→ ERK-dependent gene transcription [25] • Degradation of insulin receptor substrate 1 → PI3K-Akt activation [26] • Inactivation of thrombospondin-1 • Cleavage of EMILIN1 [27] | Tumor proliferation |
| MMP-8 release | • ↓ β1-integrin activity [28] • Cleavage of cytokines [29] • Cleavage of decorin → ↓ active TGF-β → ↓ miR-21 expression → ↓ PDCD4 [30] | Tumor suppression | NET formation | • TME remodeling [31] • Activation of dormant cancer cells [31] | Metastasis |
| MMP-9 and VEGF secretion | • Remodeling of ECM membrane → TGF-β activation [32,33] • ↑ vascular permeability [32,33] • ↑ endothelial cell growth [32,33] | Tumor angiogenesis | |||
| Arginase secretion | • ↓ cytotoxic CD8+T cell effects [34] | Immuno-suppression | |||
| Apoptotic/cytotoxic effects | • Cleavage of pro-caspase by zinc sequestration | [110] |
| • Pertubation of the mitochondrial pathway | [111] | |
| - Absence of cytochrome c release | [111] | |
| - Induction of caspase activity | [111] | |
| - Alteration of the mitochondrial membrane potential | ||
| Cell proliferation | • Recruitment of MDSCs | [112] |
| • Inhibition of dendritic cell differentiation | [113] | |
| • MAPK phosphorylation and NF-κB activation via RAGE | [114] | |
| Cell differentiation | • Increase of NF-κB activation by epithelial NADPH oxidases | [115] |
| • Increase of involucrin and filaggrin expression | [116] | |
| Adhesion and invasion | • Attraction of Mac-1+ myeloid cells | [117] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tolle, F.; Umansky, V.; Utikal, J.; Kreis, S.; Bréchard, S. Neutrophils in Tumorigenesis: Missing Targets for Successful Next Generation Cancer Therapies? Int. J. Mol. Sci. 2021, 22, 6744. https://doi.org/10.3390/ijms22136744
Tolle F, Umansky V, Utikal J, Kreis S, Bréchard S. Neutrophils in Tumorigenesis: Missing Targets for Successful Next Generation Cancer Therapies? International Journal of Molecular Sciences. 2021; 22(13):6744. https://doi.org/10.3390/ijms22136744
Chicago/Turabian StyleTolle, Fabrice, Viktor Umansky, Jochen Utikal, Stephanie Kreis, and Sabrina Bréchard. 2021. "Neutrophils in Tumorigenesis: Missing Targets for Successful Next Generation Cancer Therapies?" International Journal of Molecular Sciences 22, no. 13: 6744. https://doi.org/10.3390/ijms22136744
APA StyleTolle, F., Umansky, V., Utikal, J., Kreis, S., & Bréchard, S. (2021). Neutrophils in Tumorigenesis: Missing Targets for Successful Next Generation Cancer Therapies? International Journal of Molecular Sciences, 22(13), 6744. https://doi.org/10.3390/ijms22136744