
Molecular Characteristics of RAGE and Advances in Small-Molecule Inhibitors


Abstract
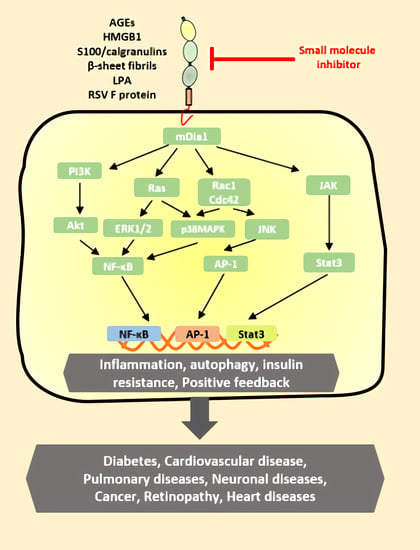
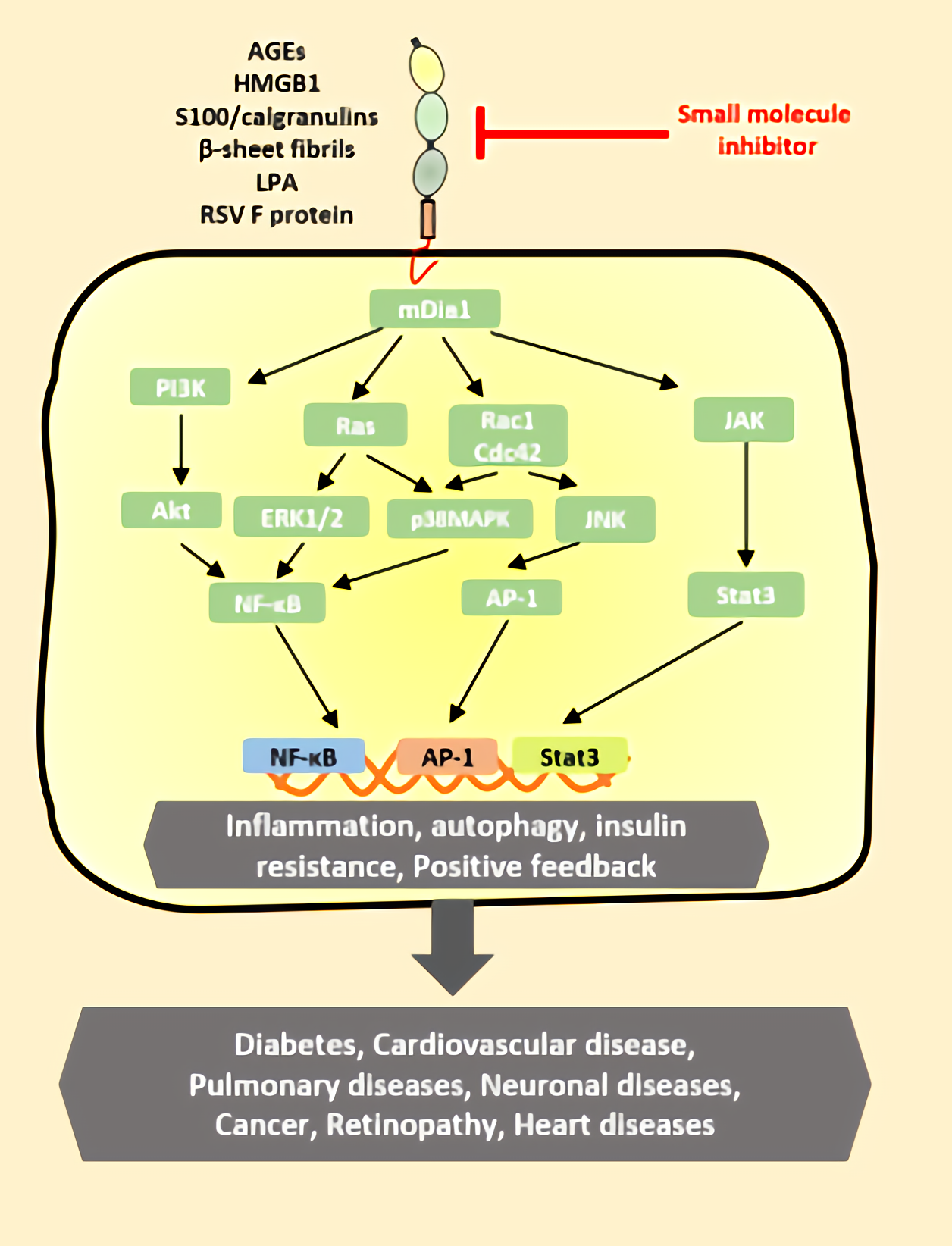
:
1. Introduction
2. Structure and Isoforms of RAGE
3. RAGE as a Multi-Ligand Receptor
3.1. Endogenous RAGE
3.2. Exogenous RAGE Ligands
4. RAGE Ligand Signaling
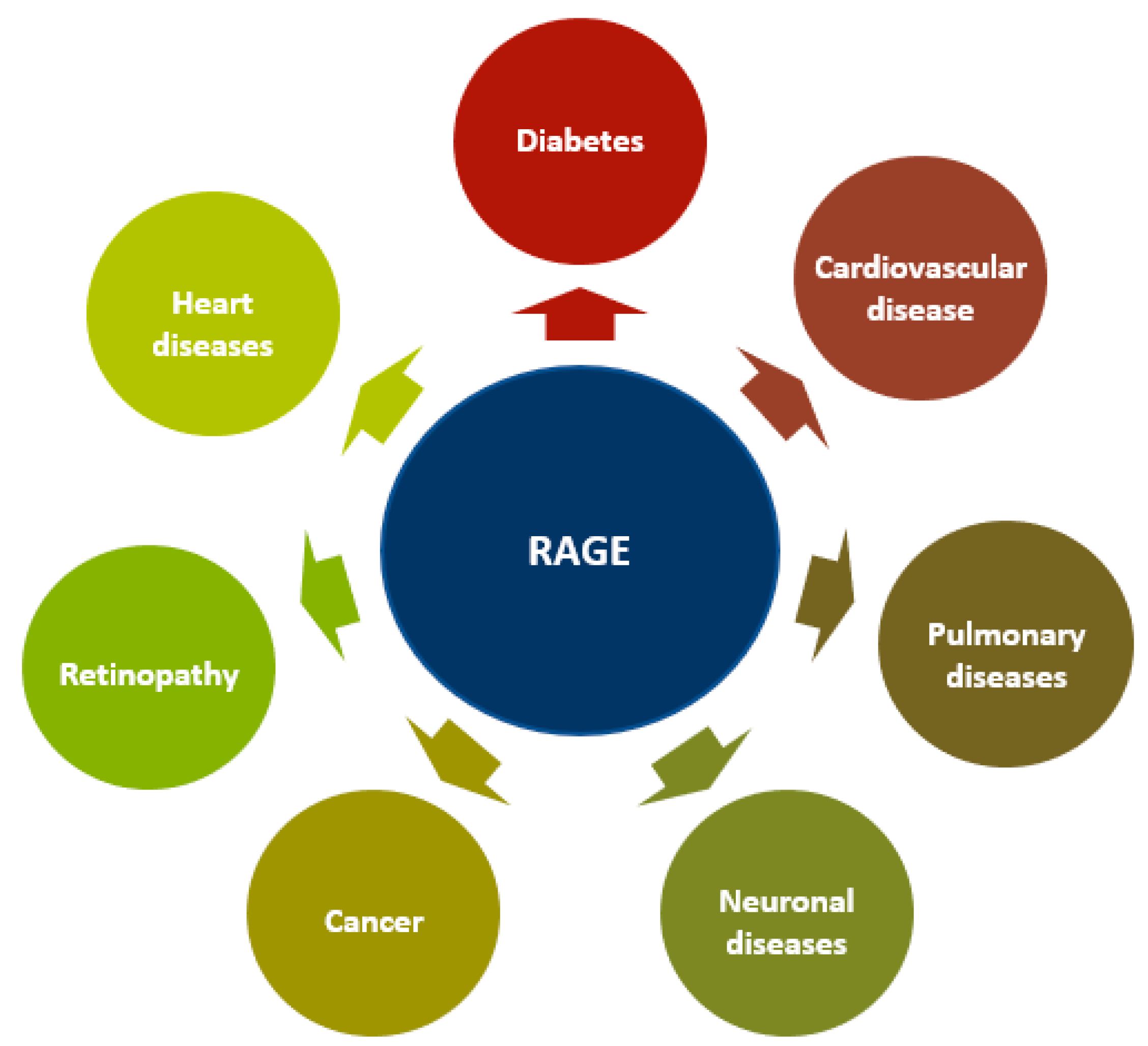
5. RAGE in Diseases
5.1. Diabetes and Cardiovascular Disease
5.2. Neurodegeneration
5.3. Cancer
5.4. Other Diseases
5.5. RAGE Polymorphisms and Inflammatory Disease
6. RAGE Inhibitors
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Neeper, M.; Schmidt, A.; Brett, J.; Yan, S.; Wang, F.; Pan, Y.; Elliston, K.; Stern, D.; Shaw, A. Cloning and expression of a cell surface receptor for advanced glycosylation end products of proteins. J. Biol. Chem. 1992, 267, 14998–15004. [Google Scholar] [CrossRef]
- Leclerc, E.; Fritz, G.; Vetter, S.W.; Heizmann, C.W. Binding of S100 proteins to RAGE: An update. Biochim. et Biophys. Acta (BBA)-Bioenerg. 2009, 1793, 993–1007. [Google Scholar] [CrossRef] [Green Version]
- Hori, O.; Brett, J.; Slattery, T.; Cao, R.; Zhang, J.; Chen, J.X.; Nagashima, M.; Lundh, E.R.; Vijay, S.; Nitecki, D.; et al. The receptor for advanced glycation end products (RAGE) is a cellular binding site for amphoterin: Mediation of neurite outgrowth and co-expression of rage and amphoterin in the developing nervous system. J. Biol. Chem. 1995, 270, 25752–25761. [Google Scholar] [CrossRef] [Green Version]
- Galasko, D.; Bell, J.; Mancuso, J.Y.; Kupiec, J.W.; Sabbagh, M.N.; van Dyck, C.; Thomas, R.G.; Aisen, P.S.; Alzheimer’s Disease Cooperative Study. Clinical trial of an inhibitor of RAGE-Aβ interactions in Alzheimer disease. Neurology 2014, 82, 1536–1542. [Google Scholar] [CrossRef]
- Yan, S.D.; Chen, X.; Fu, J.; Chen, M.; Zhu, H.; Roher, A.; Slattery, T.; Zhao, L.; Nagashima, M.; Morser, J. RAGE and amyloid-β peptide neurotoxicity in Alzheimer’s disease. Nature 1996, 382, 685–691. [Google Scholar] [CrossRef]
- Serratos, I.N.; Castellanos, P.; Pastor, N.; Millán-Pacheco, C.; Rembao, D.; Pérez-Montfort, R.; Cabrera, N.; Reyes-Espinosa, F.; Díaz-Garrido, P.; López-Macay, A. Modeling the interaction between quinolinate and the receptor for advanced glycation end products (RAGE): Relevance for early neuropathological processes. PLoS ONE 2015, 10, e0120221. [Google Scholar] [CrossRef] [PubMed]
- Hudson, B.I.; Lippman, M.E. Targeting RAGE Signaling in Inflammatory Disease. Annu. Rev. Med. 2018, 69, 349–364. [Google Scholar] [CrossRef] [PubMed]
- Sirois, C.M.; Jin, T.; Miller, A.L.; Bertheloot, D.; Nakamura, H.; Horvath, G.L.; Mian, A.; Jiang, J.; Schrum, J.; Bossaller, L.; et al. RAGE is a nucleic acid receptor that promotes inflammatory responses to DNA. J. Exp. Med. 2013, 210, 2447–2463. [Google Scholar] [CrossRef] [Green Version]
- Andersson, U.; Erlandsson-Harris, H.; Yang, H.; Tracey, K.J. HMGB1 as a DNA-binding cytokine. J. Leukoc. Biol. 2002, 72, 1084–1091. [Google Scholar]
- Yamamoto, Y.; Harashima, A.; Saito, H.; Tsuneyama, K.; Munesue, S.; Motoyoshi, S.; Han, D.; Watanabe, T.; Asano, M.; Takasawa, S. Septic shock is associated with receptor for advanced glycation end products ligation of LPS. J. Immunol. 2011, 186, 3248–3257. [Google Scholar] [CrossRef] [Green Version]
- Tian, J.; Huang, K.; Krishnan, S.; Svabek, C.; Rowe, D.C.; Brewah, Y.; Sanjuan, M.; Patera, A.C.; Kolbeck, R.; Herbst, R.; et al. RAGE inhibits human respiratory syncytial virus syncytium formation by interfering with F-protein function. J. Gen. Virol. 2013, 94, 1691–1700. [Google Scholar] [CrossRef]
- Anisuzzaman, A.; Hatta, T.; Miyoshi, T.; Matsubayashi, M.; Islam, M.K.; Alim, M.A.; Abu Anas, M.; Hasan, M.M.; Matsumoto, Y.; Yamamoto, Y.; et al. Longistatin in tick saliva blocks advanced glycation end-product receptor activation. J. Clin. Investig. 2014, 124, 4429–4444. [Google Scholar] [CrossRef] [Green Version]
- Rai, V.; Maldonado, A.Y.; Burz, D.S.; Reverdatto, S.; Schmidt, A.M.; Shekhtman, A. Signal Transduction in Receptor for Advanced Glycation End Products (RAGE). J. Biol. Chem. 2012, 287, 5133–5144. [Google Scholar] [CrossRef] [Green Version]
- Manigrasso, M.B.; Pan, J.; Rai, V.; Zhang, J.; Reverdatto, S.; Quadri, N.; DeVita, R.J.; Ramasamy, R.; Shekhtman, A.; Schmidt, A.M. Small Molecule Inhibition of Ligand-Stimulated RAGE-DIAPH1 Signal Transduction. Sci. Rep. 2016, 6, 22450. [Google Scholar] [CrossRef] [Green Version]
- Hudson, B.I.; Kalea, A.Z.; Arriero, M.d.; Harja, E.; Boulanger, E.; D’Agati, V.; Schmidt, A.M. Interaction of the RAGE cytoplasmic domain with diaphanous-1 is required for ligand-stimulated cellular migration through activation of Rac1 and Cdc. J. Biol. Chem. 2008, 283, 34457–34468. [Google Scholar] [CrossRef] [Green Version]
- Ishihara, K.; Tsutsumi, K.; Kawane, S.; Nakajima, M.; Kasaoka, T. The receptor for advanced glycation end-products (RAGE) directly binds to ERK by a D-domain-like docking site. FEBS Lett. 2003, 550, 107–113. [Google Scholar] [CrossRef] [Green Version]
- Koch, M.; Chitayat, S.; Dattilo, B.M.; Schiefner, A.; Diez, J.; Chazin, W.J.; Fritz, G. Structural Basis for Ligand Recognition and Activation of RAGE. Structures 2010, 18, 1342–1352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lander, H.M.; Tauras, J.M.; Ogiste, J.S.; Hori, O.; Moss, R.A.; Schmidt, A.M. Activation of the receptor for advanced glycation end products triggers a p21 ras-dependent mitogen-activated protein kinase pathway regulated by oxidant stress. J. Biol. Chem. 1997, 272, 17810–17814. [Google Scholar] [CrossRef] [Green Version]
- Huang, J.-S.; Guh, J.-Y.; Chen, H.-C.; Hung, W.-C.; Lai, Y.-H.; Chuang, L.-Y. Role of receptor for advanced glycation end-product (RAGE) and the JAK/STAT-signaling pathway in AGE-induced collagen production in NRK-49F cells. J. Cell. Biochem. 2001, 81, 102–113. [Google Scholar] [CrossRef]
- Goldin, A.; Beckman, J.A.; Schmidt, A.M.; Creager, M.A. Advanced glycation end products: Sparking the development of diabetic vascular injury. Circulation 2006, 114, 597–605. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nenna, A.; Spadaccio, C.; Lusini, M.; Ulianich, L.; Chello, M.; Nappi, F. Basic and Clinical Research Against Advanced Glycation End Products (AGEs): New Compounds to Tackle Cardiovascular Disease and Diabetic Complications. Recent Pat. Cardiovasc. Drug Discov. 2016, 10, 10–33. [Google Scholar] [CrossRef] [PubMed]
- Santilli, F.; Vazzana, N.; Bucciarelli, L.G.; Davì, G. Soluble forms of RAGE in human diseases: Clinical and therapeutical implications. Curr. Med. Chem. 2009, 16, 940–952. [Google Scholar] [CrossRef] [PubMed]
- Bierhaus, A.; Schiekofer, S.; Schwaninger, M.; Andrassy, M.; Humpert, P.M.; Chen, J.; Hong, M.; Luther, T.; Henle, T.; Klöting, I.; et al. Diabetes-associated sustained activation of the transcription factor nuclear factor-κB. Diabetes 2001, 50, 2792–2808. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soro-Paavonen, A.; Watson, A.M.D.; Li, J.; Paavonen, K.; Koitka, A.; Calkin, A.C.; Barit, D.; Coughlan, M.T.; Drew, B.G.; Lancaster, G.I.; et al. Receptor for Advanced Glycation End Products (RAGE) Deficiency Attenuates the Development of Atherosclerosis in Diabetes. Diabetes 2008, 57, 2461–2469. [Google Scholar] [CrossRef] [Green Version]
- Chavakis, T.; Bierhaus, A.; Nawroth, P.P. RAGE (receptor for advanced glycation end products): A central player in the inflammatory response. Microbes Infect. 2004, 6, 1219–1225. [Google Scholar] [CrossRef]
- Turovskaya, O.; Foell, D.; Sinha, P.; Vogl, T.; Newlin, R.; Nayak, J.; Nguyen, M.; Olsson, A.; Nawroth, P.P.; Bierhaus, A.; et al. RAGE, carboxylated glycans and S100A8/A9 play essential roles in colitis-associated carcinogenesis. Carcinogenesis 2008, 29, 2035–2043. [Google Scholar] [CrossRef]
- Bongarzone, S.; Savickas, V.; Luzi, F.; Gee, A.D. Targeting the Receptor for Advanced Glycation Endproducts (RAGE): A Medicinal Chemistry Perspective. J. Med. Chem. 2017, 60, 7213–7232. [Google Scholar] [CrossRef] [Green Version]
- Xue, J.; Rai, V.; Singer, D.; Chabierski, S.; Xie, J.; Reverdatto, S.; Burz, D.S.; Schmidt, A.M.; Hoffmann, R.; Shekhtman, A. Advanced Glycation End Product Recognition by the Receptor for AGEs. Structures 2011, 19, 722–732. [Google Scholar] [CrossRef] [Green Version]
- Hudson, B.I.; Carter, A.M.; Harja, E.; Kalea, A.Z.; Arriero, M.; Yang, H.; Grant, P.J.; Schmidt, A.M. Identification, classification, and expression of RAGE gene splice variants. FASEB J. 2007, 22, 1572–1580. [Google Scholar] [CrossRef]
- Xu, D.; Young, J.H.; Krahn, J.M.; Song, D.; Corbett, K.D.; Chazin, W.J.; Pedersen, L.C.; Esko, J.D. Stable RAGE-Heparan Sulfate Complexes Are Essential for Signal Transduction. ACS Chem. Biol. 2013, 8, 1611–1620. [Google Scholar] [CrossRef] [Green Version]
- Xie, J.; Burz, D.S.; He, W.; Bronstein, I.B.; Lednev, I.; Shekhtman, A. Hexameric Calgranulin C (S100A12) Binds to the Receptor for Advanced Glycated End Products (RAGE) Using Symmetric Hydrophobic Target-binding Patches. J. Biol. Chem. 2007, 282, 4218–4231. [Google Scholar] [CrossRef] [Green Version]
- Wei, W.; Lampe, L.; Park, S.; Vangara, B.S.; Waldo, G.S.; Cabantous, S.; Subaran, S.S.; Yang, N.; Lakatta, E.; Lin, L. Disulfide Bonds within the C2 Domain of RAGE Play Key Roles in Its Dimerization and Biogenesis. PLoS ONE 2012, 7, e50736. [Google Scholar] [CrossRef] [Green Version]
- Russ, W.P.; Engelman, D.M. The GxxxG motif: A framework for transmembrane helix-helix association. J. Mol. Biol. 2000, 296, 911–919. [Google Scholar] [CrossRef]
- Huttunen, H.J.; Fages, C.; Rauvala, H. Receptor for advanced glycation end products (RAGE)-mediated neurite outgrowth and activation of NF-κB require the cytoplasmic domain of the receptor but different downstream signaling pathways. J. Biol. Chem. 1999, 274, 19919–19924. [Google Scholar] [CrossRef] [Green Version]
- Harja, E.; Bu, D.-X.; Hudson, B.; Chang, J.S.; Shen, X.; Hallam, K.; Kalea, A.Z.; Lu, Y.; Rosario, R.H.; Oruganti, S.; et al. Vascular and inflammatory stresses mediate atherosclerosis via RAGE and its ligands in apoE–/– mice. J. Clin. Investig. 2008, 118, 183–194. [Google Scholar] [CrossRef] [Green Version]
- Grossin, N.; Boulanger, E.; Wautier, M.-P.; Wautier, J.-L. The different isoforms of the receptor for advanced glycation end products are modulated by pharmacological agents. Clin. Hemorheol. Microcirc. 2010, 45, 143–153. [Google Scholar] [CrossRef] [PubMed]
- Pan, H.; He, L.; Wang, B.; Niu, W. The relationship between RAGE gene four common polymorphisms and breast cancer risk in northeastern Han Chinese. Sci. Rep. 2015, 4, 4355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, W.; Li, J.; Xu, J.-X.; Liu, Y.; Zhang, W.; Xiao, J.-R.; Zhu, L.-Y.; Liu, J.-Y. Association of 2184AG Polymorphism in the RAGE Gene with Diabetic Nephropathy in Chinese Patients with Type 2 Diabetes. J. Diabetes Res. 2015, 2015, 1–6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yonekura, H.; Yamamoto, Y.; Sakurai, S.; Petrova, R.G.; Abedin, J.; Li, H.; Yasui, K.; Takeuchi, M.; Makita, Z.; Takasawa, S.; et al. Novel splice variants of the receptor for advanced glycation end-products expressed in human vascular endothelial cells and pericytes, and their putative roles in diabetes-induced vascular injury. Biochem. J. 2003, 370, 1097–1109. [Google Scholar] [CrossRef]
- Zong, H.; Ward, M.; Stitt, A.W. AGEs, RAGE, and diabetic retinopathy. Curr. Diabetes Rep. 2011, 11, 244–252. [Google Scholar] [CrossRef] [PubMed]
- Kalea, A.Z.; Schmidt, A.M.; Hudson, B.I. RAGE: A novel biological and genetic marker for vascular disease. Clin. Sci. 2009, 116, 621–637. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prasad, K. Is there any evidence that AGE/sRAGE is a universal biomarker/risk marker for diseases? Mol. Cell. Biochem. 2019, 451, 139–144. [Google Scholar] [CrossRef] [PubMed]
- Hamasaki, M.Y.; Barbeiro, H.V.; De Souza, H.P.; Machado, M.C.C.; Da Silva, F.P. sRAGE in septic shock: A potential biomarker of mortality. Rev. Bras. Ter. Intensiv. 2014, 26, 392–396. [Google Scholar] [CrossRef]
- Hofmann, M.A.; Drury, S.; Fu, C.; Qu, W.; Taguchi, A.; Lu, Y.; Avila, C.; Kambham, N.; Bierhaus, A.; Nawroth, P.; et al. RAGE Mediates a Novel Proinflammatory Axis: A Central Cell Surface Receptor for S100/Calgranulin Polypeptides. Cell 1999, 97, 889–901. [Google Scholar] [CrossRef] [Green Version]
- Chavakis, T.; Bierhaus, A.; Al-Fakhri, N.; Schneider, D.; Witte, S.; Linn, T.; Nagashima, M.; Morser, J.; Arnold, B.; Preissner, K.T. The pattern recognition receptor (RAGE) is a counterreceptor for leukocyte Integrins a novel pathway for inflammatory cell recruitment. J. Exp. Med. 2003, 198, 1507–1515. [Google Scholar] [CrossRef]
- Schmidt, A.M.; Du Yan, S.; Stern, D.M. The biology of the receptor for advanced glycation end products and its ligands. Biochim. et Biophys. Acta (BBA)-Bioenerg. 2000, 1498, 99–111. [Google Scholar] [CrossRef] [Green Version]
- Orlova, V.V.; Choi, E.Y.; Xie, C.; Chavakis, E.; Bierhaus, A.; Ihanus, E.; Ballantyne, C.M.; Gahmberg, C.; Bianchi, M.E.; Nawroth, P.P.; et al. A novel pathway of HMGB1-mediated inflammatory cell recruitment that requires Mac-1-integrin. EMBO J. 2007, 26, 1129–1139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, J.; Reverdatto, S.; Frolov, A.; Hoffmann, R.; Burz, D.S.; Shekhtman, A. Structural Basis for Pattern Recognition by the Receptor for Advanced Glycation End Products (RAGE). J. Biol. Chem. 2008, 283, 27255–27269. [Google Scholar] [CrossRef] [Green Version]
- Ibrahim, Z.A.; Armour, C.; Phipps, S.; Sukkar, M. RAGE and TLRs: Relatives, friends or neighbours? Mol. Immunol. 2013, 56, 739–744. [Google Scholar] [CrossRef] [PubMed]
- Fritz, G. RAGE: A single receptor fits multiple ligands. Trends Biochem. Sci. 2011, 36, 625–632. [Google Scholar] [CrossRef]
- Kierdorf, K.; Fritz, G. RAGE regulation and signaling in inflammation and beyond. J. Leukoc. Biol. 2013, 94, 55–68. [Google Scholar] [CrossRef] [PubMed]
- Han, S.-H.; Kim, Y.H.; Mook-Jung, I. RAGE: The beneficial and deleterious effects by diverse mechanisms of actions. Mol. Cells 2011, 31, 91–97. [Google Scholar] [CrossRef] [Green Version]
- Kaneko, M.; Bucciarelli, L.; Hwang, Y.C.; Lee, L.; Yan, S.F.; Schmidt, A.M.; Ramasamy, R. Aldose Reductase and AGE-RAGE Pathways: Key Players in Myocardial Ischemic Injury. Ann. N. Y. Acad. Sci. 2005, 1043, 702–709. [Google Scholar] [CrossRef]
- Vincent, A.M.; Perrone, L.; Sullivan, K.A.; Backus, C.; Sastry, A.M.; Lastoskie, C.; Feldman, E.L. Receptor for Advanced Glycation End Products Activation Injures Primary Sensory Neurons via Oxidative Stress. Endocrinology 2007, 148, 548–558. [Google Scholar] [CrossRef] [Green Version]
- Wendt, T.; Tanji, N.; Guo, J.; Hudson, B.; Bierhaus, A.; Ramasamy, R.; Arnold, B.; Nawroth, P.P.; Yan, S.F.; D’Agati, V.; et al. Glucose, Glycation, and RAGE: Implications for Amplification of Cellular Dysfunction in Diabetic Nephropathy. J. Am. Soc. Nephrol. 2003, 14, 1383–1395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmidt, A.M.; Yan, S.D.; Wautier, J.-L.; Stern, D. Activation of receptor for advanced glycation end products: A mechanism for chronic vascular dysfunction in diabetic vasculopathy and atherosclerosis. Circ. Res. 1999, 84, 489–497. [Google Scholar] [CrossRef] [Green Version]
- Basta, G. Receptor for advanced glycation endproducts and atherosclerosis: From basic mechanisms to clinical implications. Atherosclerosis 2008, 196, 9–21. [Google Scholar] [CrossRef] [PubMed]
- Tanji, N.; Markowitz, G.S.; Fu, C.; Kislinger, T.; Taguchi, A.; Pischetsrieder, M.; Stern, D.; Schmidt, A.M.; D’Agati, V.D. Expression of Advanced Glycation End Products and Their Cellular Receptor RAGE in Diabetic Nephropathy and Nondiabetic Renal Disease. J. Am. Soc. Nephrol. 2000, 11, 1656–1666. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, M.A.; Drury, S.; Hudson, B.; Gleason, M.R.; Qu, W.; Lu, Y.; Lalla, E.; Chitnis, S.; Monteiro, J.; Stickland, M.H.; et al. RAGE and arthritis: The G82S polymorphism amplifies the inflammatory response. Genes Immun. 2002, 3, 123–135. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, A.M.; Yan, S.D.; Yan, S.F.; Stern, D.M. The multiligand receptor RAGE as a progression factor amplifying immune and inflammatory responses. J. Clin. Investig. 2001, 108, 949–955. [Google Scholar] [CrossRef] [PubMed]
- Rong, L.L.; Gooch, C.; Szabolcs, M.; Herold, K.C.; Lalla, E.; Hays, A.P.; Yan, S.F.; Du Yan, S.S.; Schmidt, A.M. RAGE: A journey from the complications of diabetes to disorders of the nervous system-striking a fine balance between injury and repair. Restor. Neurol. Neurosci. 2005, 23, 355–365. [Google Scholar] [PubMed]
- Taguchi, A.; Blood, D.C.; Del Toro, G.; Canet, A.; Lee, D.C.; Qu, W.; Tanji, N.; Lu, Y.; Lalla, E.; Fu, C.; et al. Blockade of RAGE–amphoterin signalling suppresses tumour growth and metastases. Nat. Cell Biol. 2000, 405, 354–360. [Google Scholar] [CrossRef]
- Ramasamy, R.; Vannucci, S.J.; Du Yan, S.S.; Herold, K.; Yan, S.F.; Schmidt, A.M. Advanced glycation end products and RAGE: A common thread in aging, diabetes, neurodegeneration, and inflammation. Glycobiology 2005, 15, 16R–28R. [Google Scholar] [CrossRef] [PubMed]
- Pinto, S.S.; Gottfried, C.; Mendez, A.; Gonçalves, D.; Karl, J.; Gonçalves, C.A.; Wofchuk, S.; Rodnight, R. Immunocontent and secretion of S100B in astrocyte cultures from different brain regions in relation to morphology. FEBS Lett. 2000, 486, 203–207. [Google Scholar] [CrossRef] [Green Version]
- Dong, X.D.; Ito, N.; Lotze, M.T.; DeMarco, R.A.; Popovic, P.; Shand, S.H.; Watkins, S.; Winikoff, S.; Brown, C.K.; Bartlett, D.L.; et al. High Mobility Group Box I (HMGB1) Release From Tumor Cells After Treatment: Implications for Development of Targeted Chemoimmunotherapy. J. Immunother. 2007, 30, 596–606. [Google Scholar] [CrossRef] [PubMed]
- Deane, R.; Du Yan, S.; Submamaryan, R.K.; LaRue, B.; Jovanovic, S.; Hogg, E.; Welch, D.; Manness, L.; Lin, C.; Yu, J.; et al. RAGE mediates amyloid-β peptide transport across the blood-brain barrier and accumulation in brain. Nat. Med. 2003, 9, 907–913. [Google Scholar] [CrossRef] [PubMed]
- Lugo-Huitrón, R.; Muñiz, P.U.; Pineda, B.; Pedraza-Chaverrí, J.; Ríos, C.; La Cruz, V.P.-D. Quinolinic Acid: An Endogenous Neurotoxin with Multiple Targets. Oxidative Med. Cell. Longev. 2013, 2013, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rai, V.; Touré, F.; Chitayat, S.; Pei, R.; Song, F.; Li, Q.; Zhang, J.; Rosario, R.; Ramasamy, R.; Chazin, W.J. Lysophosphatidic acid targets vascular and oncogenic pathways via RAGE signaling. J. Exp. Med. 2012, 209, 2339–2350. [Google Scholar] [CrossRef] [Green Version]
- He, M.; Kubo, H.; Morimoto, K.; Fujino, N.; Suzuki, T.; Takahashi, T.; Yamada, M.; Yamaya, M.; Maekawa, T.; Yamamoto, Y.; et al. Receptor for advanced glycation end products binds to phosphatidylserine and assists in the clearance of apoptotic cells. EMBO Rep. 2011, 12, 358–364. [Google Scholar] [CrossRef] [Green Version]
- Ma, W.; Rai, V.; Hudson, B.I.; Song, F.; Schmidt, A.M.; Barile, G.R. RAGE binds C1q and enhances C1q-mediated phagocytosis. Cell. Immunol. 2012, 274, 72–82. [Google Scholar] [CrossRef]
- Semba, R.D.; Najjar, S.S.; Sun, K.; Lakatta, E.G.; Ferrucci, L. Serum carboxymethyl–lysine, an advanced glycation end product, is associated with increased aortic pulse wave velocity in adults. Am. J. Hypertens. 2009, 22, 74–79. [Google Scholar] [CrossRef] [Green Version]
- Kellow, N.; Coughlan, M.T. Effect of diet-derived advanced glycation end products on inflammation. Nutr. Rev. 2015, 73, 737–759. [Google Scholar] [CrossRef] [PubMed]
- Senatus, L.M.; Schmidt, A.M. The AGE-RAGE Axis: Implications for Age-Associated Arterial Diseases. Front. Genet. 2017, 8, 187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xue, J.; Ray, R.; Singer, D.; Böhme, D.; Burz, D.S.; Rai, V.; Hoffmann, R.; Shekhtman, A. The Receptor for Advanced Glycation End Products (RAGE) Specifically Recognizes Methylglyoxal-Derived AGEs. Biochemistry 2014, 53, 3327–3335. [Google Scholar] [CrossRef] [PubMed]
- Schäfer, B.W.; Wicki, R.; Engelkamp, D.; Mattei, M.-G.; Heizmann, C.W. Isolation of a YAC clone covering a cluster of nine S100 genes on human chromosome 1q21: Rationale for a new nomenclature of the S100 calcium-binding protein family. Genomics 1995, 25, 638–643. [Google Scholar] [CrossRef]
- Steiner, J.; Bernstein, H.-G.; Bogerts, B.; Gos, T.; Richter-Landsberg, C.; Wunderlich, M.; Keilhoff, G. S100B is expressed in, and released from, OLN-93 oligodendrocytes: Influence of serum and glucose deprivation. Neuroscience 2008, 154, 496–503. [Google Scholar] [CrossRef]
- Baudier, J.; Glasser, N.; Gerard, D. Ions binding to S100 proteins. I. Calcium- and zinc-binding properties of bovine brain S100 alpha alpha, S100a (alpha beta), and S100b (beta beta) protein: Zn2+ regulates Ca2+ binding on S100b protein. J. Biol. Chem. 1986, 261, 8192–8203. [Google Scholar] [CrossRef]
- Leclerc, E.; Fritz, G.; Weibel, M.; Heizmann, C.W.; Galichet, A. S100B and S100A6 Differentially Modulate Cell Survival by Interacting with Distinct RAGE (Receptor for Advanced Glycation End Products) Immunoglobulin Domains. J. Biol. Chem. 2007, 282, 31317–31331. [Google Scholar] [CrossRef] [Green Version]
- Rothermundt, M.; Peters, M.; Prehn, J.H.; Arolt, V. S100B in brain damage and neurodegeneration. Microsc. Res. Tech. 2003, 60, 614–632. [Google Scholar] [CrossRef] [PubMed]
- Sinha, P.; Okoro, C.; Foell, D.; Freeze, H.; Ostrand-Rosenberg, S.; Srikrishna, G. Proinflammatory S100 Proteins Regulate the Accumulation of Myeloid-Derived Suppressor Cells. J. Immunol. 2008, 181, 4666–4675. [Google Scholar] [CrossRef] [Green Version]
- Zimmer, D.B.; Chaplin, J.; Baldwin, A.; Rast, M. S100-mediated signal transduction in the nervous system and neurological diseases. Cell. Mol. Boil. 2005, 51, 201–214. [Google Scholar]
- Huttunen, H.J.; Kuja-Panula, J.; Sorci, G.; Agneletti, A.L.; Donato, R.; Rauvala, H. Coregulation of Neurite Outgrowth and Cell Survival by Amphoterin and S100 Proteins through Receptor for Advanced Glycation End Products (RAGE) Activation. J. Biol. Chem. 2000, 275, 40096–40105. [Google Scholar] [CrossRef] [Green Version]
- Mueller, A.; Schäfer, B.W.; Ferrari, S.; Weibel, M.; Makek, M.; Höchli, M.; Heizmann, C.W. The Calcium-binding Protein S100A2 Interacts with p53 and Modulates Its Transcriptional Activity. J. Biol. Chem. 2005, 280, 29186–29193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maelandsmo, G.M.; Flørenes, V.A.; Mellingsaeter, T.; Hovig, E.; Kerbel, R.S.; Fodstad, Ø. Differential expression patterns of S100A2, S100A4 and S100A6 during progression of human malignant melanoma. Int. J. Cancer 1997, 74, 464–469. [Google Scholar] [CrossRef]
- Gupta, S.; Hussain, T.; MacLennan, G.T.; Fu, P.; Patel, J.; Mukhtar, H. Differential Expression of S100A2 and S100A4 During Progression of Human Prostate Adenocarcinoma. J. Clin. Oncol. 2003, 21, 106–112. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, F.; Oridate, N.; Homma, A.; Nakamaru, Y.; Nagahashi, T.; Yagi, K.; Yamaguchi, S.; Furuta, Y.; Fukuda, S. S100A2 expression as a predictive marker for late cervical metastasis in stage I and II invasive squamous cell carcinoma of the oral cavity. Oncol. Rep. 2005, 14, 1493–1498. [Google Scholar] [CrossRef]
- Feng, G.; Xu, X.; Youssef, E.M.; Lotan, R. Diminished expression of S100A2, a putative tumor suppressor, at early stage of human lung carcinogenesis. Cancer Res. 2001, 61, 7999–8004. [Google Scholar]
- Lee, S.W.; Tomasetto, C.; Swisshelm, K.; Keyomarsi, K.; Sager, R. Down-regulation of a member of the S100 gene family in mammary carcinoma cells and reexpression by azadeoxycytidine treatment. Proc. Natl. Acad. Sci. USA 1992, 89, 2504–2508. [Google Scholar] [CrossRef] [Green Version]
- Imazawa, M.; Hibi, K.; Fujitake, S.-I.; Kodera, Y.; Ito, K.; Akiyama, S.; Nakao, A. S100A2 overexpression is frequently observed in esophageal squamous cell carcinoma. Anticancer. Res. 2005, 25, 1247–1250. [Google Scholar]
- Smith, S.L.; Gugger, M.; Hoban, P.; Ratschiller, D.; Watson, S.G.; Field, J.K.; Betticher, D.C.; Heighway, J. S100A2 is strongly expressed in airway basal cells, preneoplastic bronchial lesions and primary non-small cell lung carcinomas. Br. J. Cancer 2004, 91, 1515–1524. [Google Scholar] [CrossRef] [Green Version]
- El-Rifai, W.; Moskaluk, C.A.; Abdrabbo, M.K.; Harper, J.; Yoshida, C.; Riggins, G.J.; Frierson, H.F.; Powell, S.M. Gastric cancers overexpress S100A calcium-binding proteins. Cancer Res. 2002, 62, 6823–6826. [Google Scholar]
- Hough, C.D.; Cho, K.R.; Zonderman, A.B.; Schwartz, D.R.; Morin, P.J. Coordinately up-regulated genes in ovarian cancer. Cancer Res. 2001, 61, 3869–3876. [Google Scholar] [PubMed]
- Yammani, R.R.; Carlson, C.S.; Bresnick, A.R.; Loeser, R.F. Increase in production of matrix metalloproteinase 13 by human articular chondrocytes due to stimulation with S100A4: Role of the receptor for advanced glycation end products. Arthritis Rheum. 2006, 54, 2901–2911. [Google Scholar] [CrossRef] [PubMed]
- Camby, I.; Lefranc, F.; Titeca, G.; Neuci, S.; Fastrez, M.; Dedecken, L.; Schäfer, B.W.; Brotchi, J.; Heizmann, C.W.; Pochet, R.; et al. Differential expression of S100 calcium-binding proteins characterizes distinct clinical entities in both WHO grade II and III astrocytic tumours. Neuropathol. Appl. Neurobiol. 2000, 26, 76–90. [Google Scholar] [CrossRef] [PubMed]
- Kuźnicki, J.; Filipek, A.; Hunziker, P.E.; Huber, S.; Heizmann, C.W. Calcium-binding protein from mouse Ehrlich ascites-tumour cells is homologous to human calcyclin. Biochem. J. 1989, 263, 951–956. [Google Scholar] [CrossRef] [Green Version]
- Kuźnicki, J.; Filipek, A.; Heimann, P.; Kaczmarek, L.; Kamińska, B. Tissue specific distribution of calcyclin—10.5 kDa Ca2+-binding protein. FEBS Lett. 1989, 254, 141–144. [Google Scholar] [CrossRef] [Green Version]
- Stradal, T.; Gimona, M. Ca2+-dependent Association of S100A6 (Calcyclin) with the Plasma Membrane and the Nuclear Envelope. J. Biol. Chem. 1999, 274, 31593–31596. [Google Scholar] [CrossRef] [Green Version]
- Komatsu, K.; Andoh, A.; Ishiguro, S.; Suzuki, N.; Hunai, H.; Kobune-Fujiwara, Y.; Kameyama, M.; Miyoshi, J.; Akedo, H.; Nakamura, H. Increased expression of S100A6 (Calcyclin), a calcium-binding protein of the S100 family, in human colorectal adenocarcinomas. Clin. Cancer Res. 2000, 6, 172–177. [Google Scholar]
- De Petris, L.; Orre, L.M.; Kanter, L.; Pernemalm, M.; Koyi, H.; Lewensohn, R.; Lehtiö, J. Tumor expression of S100A6 correlates with survival of patients with stage I non-small-cell lung cancer. Lung Cancer 2009, 63, 410–417. [Google Scholar] [CrossRef] [PubMed]
- Ohuchida, K.; Mizumoto, K.; Yu, J.; Yamaguchi, H.; Konomi, H.; Nagai, E.; Yamaguchi, K.; Tsuneyoshi, M.; Tanaka, M. S100A6 Is Increased in a Stepwise Manner during Pancreatic Carcinogenesis: Clinical Value of Expression Analysis in 98 Pancreatic Juice Samples. Cancer Epidemiol. Biomark. Prev. 2007, 16, 649–654. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.Q.; Zhang, L.J.; Dong, H.; Jiang, C.L.; Zhu, Z.G.; Wu, J.X.; Wu, Y.L.; Han, J.S.; Xiao, H.S.; Gao, H.J.; et al. Upregulated expression of S100A6 in human gastric cancer. J. Dig. Dis. 2007, 8, 186–193. [Google Scholar] [CrossRef]
- Wolf, R.; Howard, O.M.Z.; Dong, H.-F.; Voscopoulos, C.; Boeshans, K.; Winston, J.; Divi, R.; Gunsior, M.; Goldsmith, P.; Ahvazi, B.; et al. Chemotactic Activity of S100A7 (Psoriasin) Is Mediated by the Receptor for Advanced Glycation End Products and Potentiates Inflammation with Highly Homologous but Functionally Distinct S100A. J. Immunol. 2008, 181, 1499–1506. [Google Scholar] [CrossRef] [Green Version]
- Brandtzaeg, P.; Gabrielsen, T.-Ø.; Dale, I.; Műller, F.; Steinbakk, M.; Fagerhol, M.K. The Leucocyte Protein L1 (Calprotectin): A Putative Nonspecific Defence Factor at Epithelial Surfaces. In Chemistry and Biology of Pteridines and Folates; Springer Science and Business Media LLC: Berlin/Heidelberg, Germany, 1995; Volume 371, pp. 201–206. [Google Scholar]
- Murao, S.; Collart, F.; Huberman, E. A protein complex expressed during terminal differentiation of monomyelocytic cells is an inhibitor of cell growth. Cell Growth Differ. Mol. Boil. J. Am. Assoc. Cancer Res. 1990, 1, 447–454. [Google Scholar]
- Kelly, S.E.; Jones, D.B.; Fleming, S. Calgranulin expression in inflammatory dermatoses. J. Pathol. 1989, 159, 17–21. [Google Scholar] [CrossRef] [PubMed]
- Korndörfer, I.P.; Brueckner, F.; Skerra, A. The Crystal Structure of the Human (S100A8/S100A9)2 Heterotetramer, Calprotectin, Illustrates how Conformational Changes of Interacting α-Helices Can Determine Specific Association of Two EF-hand Proteins. J. Mol. Biol. 2007, 370, 887–898. [Google Scholar] [CrossRef] [PubMed]
- Lügering, N.; Stoll, R.; Kucharzik, T.; Schmid, K.W.; Rohlmann, G.; Burmeister, G.; Sorg, C.; Domschke, W. Immunohistochemical distribution and serum levels of the Ca2+-binding proteins MRP8, MRP14 and their heterodimeric form MRP8/14 in Crohn’s disease. Digestion 1995, 56, 406–414. [Google Scholar] [CrossRef] [PubMed]
- Ehlermann, P.; Eggers, K.; Bierhaus, A.; Most, P.; Weichenhan, D.; Greten, J.; Nawroth, P.P.; A. Katus, H.; Remppis, A. Increased proinflammatory endothelial response to S100A8/A9 after preactivation through advanced glycation end products. Cardiovasc. Diabetol. 2006, 5, 6. [Google Scholar] [CrossRef] [Green Version]
- Yong, H.-Y.; Moon, A. Roles of calcium-binding proteins, S100A8 and S100A9, in invasive phenotype of human gastric cancer cells. Arch. Pharmacal Res. 2007, 30, 75–81. [Google Scholar] [CrossRef]
- Kerkhoff, C.; Klempt, M.; Sorg, C. Novel insights into structure and function of MRP8 (S100A8) and MRP14 (S100A9). Biochim. et Biophys. Acta (BBA)-Bioenerg. 1998, 1448, 200–211. [Google Scholar] [CrossRef] [Green Version]
- Hermani, A.; de Servi, B.; Medunjanin, S.; Tessier, P.A.; Mayer, D. S100A8 and S100A9 activate MAP kinase and NF-κB signaling pathways and trigger translocation of RAGE in human prostate cancer cells. Exp. Cell Res. 2006, 312, 184–197. [Google Scholar] [CrossRef]
- Stulík, J.; Österreicher, J.; Koupilová, K.; Knížek, J.; Macela, A.; Bureš, J.; Jandík, P.; Langr, F.; Dědič, K.; Jungblut, P.R. The analysis of S100A9 and S100A8 expression in matched sets of macroscopically normal colon mucosa and colorectal carcinoma: The S100A9 and S100A8 positive cells underlie and invade tumor mass. Electrophoresis 1999, 20, 1047–1054. [Google Scholar] [CrossRef]
- Hermani, A.; Hess, J.; de Servi, B.; Medunjanin, S.; Grobholz, R.; Trojan, L.; Angel, P.; Mayer, D. Calcium-binding proteins S100A8 and S100A9 as novel diagnostic markers in human prostate cancer. Clin. Cancer Res. 2005, 11, 5146–5152. [Google Scholar] [CrossRef] [Green Version]
- Ghavami, S.; Rashedi, I.; Dattilo, B.M.; Eshraghi, M.; Chazin, W.J.; Hashemi, M.; Wesselborg, S.; Kerkhoff, C.; Los, M. S100A8/A9 at low concentration promotes tumor cell growth via RAGE ligation and MAP kinase-dependent pathway. J. Leukoc. Biol. 2008, 83, 1484–1492. [Google Scholar] [CrossRef] [PubMed]
- Sunahori, K.; Yamamura, M.; Yamana, J.; Takasugi, K.; Kawashima, M.; Yamamoto, H.; Chazin, W.J.; Nakatani, Y.; Yui, S.; Makino, H. The S100A8/A9 heterodimer amplifies proinflammatory cytokine production by macrophages via activation of nuclear factor kappa B and p38 mitogen-activated protein kinase in rheumatoid arthritis. Arthritis Res. 2006, 8, R69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rehman, I.; Azzouzi, A.R.; Cross, S.S.; Deloulme, J.C.; Catto, J.W.; Wylde, N.; Larre, S.; Champigneuille, J.; Hamdy, F.C. Dysregulated expression of S100A11 (calgizzarin) in prostate cancer and precursor lesions. Hum. Pathol. 2004, 35, 1385–1391. [Google Scholar] [CrossRef]
- Cross, S.S.; Hamdy, F.C.; Deloulme, J.C.; Rehman, I. Expression of S100 proteins in normal human tissues and common cancers using tissue microarrays: S100A6, S100A8, S100A9 and S100A11 are all overexpressed in common cancers. Histopathology 2005, 46, 256–269. [Google Scholar] [CrossRef] [PubMed]
- Ohuchida, K.; Mizumoto, K.; Ohhashi, S.; Yamaguchi, H.; Konomi, H.; Nagai, E.; Yamaguchi, K.; Tsuneyoshi, M.; Tanaka, M. S100A11, A Putative Tumor Suppressor Gene, Is Overexpressed in Pancreatic Carcinogenesis. Clin. Cancer Res. 2006, 12, 5417–5422. [Google Scholar] [CrossRef] [Green Version]
- Memon, A.; Sorensen, B.; Meldgaard, P.; Fokdal, L.; Thykjaer, T.; Nexo, E. Down-regulation of S100C is associated with bladder cancer progression and poor survival. Clin. Cancer Res. 2005, 11, 606–611. [Google Scholar]
- Kondo, A.; Sakaguchi, M.; Makino, E.; Namba, M.; Okada, S.; Huh, N.-H. Localization of S100C immunoreactivity in various human tissues. Acta Med. Okayama 2002, 56, 31–34. [Google Scholar]
- Cecil, D.L.; Johnson, K.; Rediske, J.; Lotz, M.; Schmidt, A.M.; Terkeltaub, R. Inflammation-Induced Chondrocyte Hypertrophy Is Driven by Receptor for Advanced Glycation End Products. J. Immunol. 2005, 175, 8296–8302. [Google Scholar] [CrossRef] [Green Version]
- Foell, D.; Ichida, F.; Vogl, T.; Yu, X.; Chen, R.; Miyawaki, T.; Sorg, C.; Roth, J. S100A12 (EN-RAGE) in monitoring Kawasaki disease. Lancet 2003, 361, 1270–1272. [Google Scholar] [CrossRef]
- Foell, D.; Kane, D.; Bresnihan, B.; Vogl, T.; Nacken, W.; Sorg, C.; FitzGerald, O.; Roth, J. Expression of the pro-inflammatory protein S100A12 (EN-RAGE) in rheumatoid and psoriatic arthritis. Rheumatology 2003, 42, 1383–1389. [Google Scholar] [CrossRef] [Green Version]
- Foell, D.; Kucharzik, T.; Kraft, M.; Vogl, T.; Sorg, C.; Domschke, W.; Roth, J. Neutrophil derived human S100A12 (EN-RAGE) is strongly expressed during chronic active inflammatory bowel disease. Gut 2003, 52, 847–853. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foell, D.; Seeliger, S.; Vogl, T.; Koch, H.-G.; Maschek, H.; Harms, E.; Sorg, C.; Roth, J. Expression of S100A12 (EN-RAGE) in cystic fibrosis. Thorax 2003, 58, 613–617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mori, Y.; Kosaki, A.; Kishimoto, N.; Kimura, T.; Iida, K.; Fukui, M.; Nakajima, F.; Nagahara, M.; Urakami, M.; Iwasaka, T.; et al. Increased Plasma S100A12 (EN-RAGE) Levels in Hemodialysis Patients with Atherosclerosis. Am. J. Nephrol. 2009, 29, 18–24. [Google Scholar] [CrossRef] [PubMed]
- Pietzsch, J.; Hoppmann, S. Human S100A12: A novel key player in inflammation? Amino Acids 2008, 36, 381–389. [Google Scholar] [CrossRef]
- Mikkelsen, S.E.; Novitskaya, V.; Kriajevska, M.; Berezin, V.; Bock, E.; Norrild, B.; Lukanidin, E. S100A12 protein is a strong inducer of neurite outgrowth from primary hippocampal neurons. J. Neurochem. 2008, 79, 767–776. [Google Scholar] [CrossRef] [Green Version]
- Hsieh, H.-L.; Schäfer, B.W.; Weigle, B.; Heizmann, C.W. S100 protein translocation in response to extracellular S100 is mediated by receptor for advanced glycation endproducts in human endothelial cells. Biochem. Biophys. Res. Commun. 2004, 316, 949–959. [Google Scholar] [CrossRef]
- Wicki, R.; Schäfer, B.W.; Erne, P.; Heizmann, C.W. Characterization of the human and mouse cDNAs coding for S100A13, a new member of the S100 protein family. Biochem. Biophys Res. Commun. 1996, 227, 594–599. [Google Scholar] [CrossRef]
- Ridinger, K.; Schäfer, B.W.; Durussel, I.; Cox, J.A.; Heizmann, C.W. S100ABiochemical characterization and subcellular localization in different cell lines. J. Biol. Chem. 2000, 275, 8686–8694. [Google Scholar] [CrossRef] [Green Version]
- Landriscina, M.; Schinzari, G.; Di Leonardo, G.; Quirino, M.; Cassano, A.; D’Argento, E.; Lauriola, L.; Scerrati, M.; Prudovsky, I.; Barone, C. S100A13, a new marker of angiogenesis in human astrocytic gliomas. J. Neuro-Oncol. 2006, 80, 251–259. [Google Scholar] [CrossRef]
- Pierce, A.; Barron, N.; Linehan, R.; Ryan, E.; O’Driscoll, L.; Daly, C.; Clynes, M. Identification of a novel, functional role for S100A13 in invasive lung cancer cell lines. Eur. J. Cancer 2008, 44, 151–159. [Google Scholar] [CrossRef]
- Parkkila, S.; Pan, P.-W.; Ward, A.; Gibadulinova, A.; Oveckova, I.; Pastorekova, S.; Pastorek, J.; Martinez, A.R.; Helin, H.O.; Isola, J. The calcium-binding protein S100P in normal and malignant human tissues. BMC Clin. Pathol. 2008, 8, 2. [Google Scholar] [CrossRef] [Green Version]
- Arumugam, T.; Simeone, D.M.; Van Golen, K.; Logsdon, C.D. S100P Promotes Pancreatic Cancer Growth, Survival, and Invasion. Clin. Cancer Res. 2005, 11, 5356–5364. [Google Scholar] [CrossRef] [Green Version]
- Da Silva, I.D.G.; Hu, Y.F.; Russo, I.H.; Ao, X.; Salicioni, A.M.; Yang, X.; Russo, J. S100P calcium-binding protein overexpression is associated with immortalization of human breast epithelial cells in vitro and early stages of breast cancer development in vivo. Int. J. Oncol. 2000, 16, 231–240. [Google Scholar] [CrossRef]
- Arumugam, T.; Simeone, D.M.; Schmidt, A.M.; Logsdon, C.D. S100P Stimulates Cell Proliferation and Survival via Receptor for Activated Glycation End Products (RAGE). J. Biol. Chem. 2004, 279, 5059–5065. [Google Scholar] [CrossRef] [Green Version]
- Müller-Knapp, S.; Scaffidi, P.; Degryse, B.; Bonaldi, T.; Ronfani, L.; Agresti, A.; Beltrame, M.; Bianchi, M.E. NEW EMBO MEMBERS’ REVIEW: The double life of HMGB1 chromatin protein: Architectural factor and extracellular signal. EMBO J. 2001, 20, 4337–4340. [Google Scholar] [CrossRef]
- Tian, J.; Avalos, A.M.; Mao, S.-Y.; Chen, B.; Senthil, K.; Wu, H.; Parroche, P.; Drabic, S.; Golenbock, D.T.; Sirois, C.M.; et al. Toll-like receptor 9–dependent activation by DNA-containing immune complexes is mediated by HMGB1 and RAGE. Nat. Immunol. 2007, 8, 487–496. [Google Scholar] [CrossRef]
- Tang, D.; Kang, R.; Iii, H.J.Z.; Lotze, M.T. High-mobility group box 1 and cancer. Biochim. et Biophys. Acta (BBA)-Bioenerg. 2010, 1799, 131–140. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Yang, H.; Tracey, K.J. Extracellular role of HMGB1 in inflammation and sepsis. J. Intern. Med. 2004, 255, 320–331. [Google Scholar] [CrossRef]
- Andersson, U.; Tracey, K.J. HMGB1 as a mediator of necrosis-induced inflammation and a therapeutic target in arthritis. Rheum. Dis. Clin. N. Am. 2004, 30, 627–637. [Google Scholar] [CrossRef] [PubMed]
- Enokido, Y.; Yoshitake, A.; Ito, H.; Okazawa, H. Age-dependent change of HMGB1 and DNA double-strand break accumulation in mouse brain. Biochem. Biophys. Res. Commun. 2008, 376, 128–133. [Google Scholar] [CrossRef] [PubMed]
- Sparvero, L.J.; Asafu-Adjei, D.; Kang, R.; Tang, D.; Amin, N.; Im, J.; Rutledge, R.; Lin, B.; Amoscato, A.A.; Zeh, H.J.; et al. RAGE (Receptor for Advanced Glycation Endproducts), RAGE Ligands, and their role in Cancer and Inflammation. J. Transl. Med. 2009, 7, 17–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lotze, M.T.; Demarco, R.A. Dealing with death: HMGB1 as a novel target for cancer therapy. Curr. Opin. Investig. Drugs (Lond. Engl. 2000) 2003, 4, 1405–1409. [Google Scholar]
- Sims, G.P.; Rowe, D.C.; Rietdijk, S.T.; Herbst, R.; Coyle, A.J. HMGB1 and RAGE in Inflammation and Cancer. Annu. Rev. Immunol. 2010, 28, 367–388. [Google Scholar] [CrossRef]
- Kuniyasu, H.; Chihara, Y.; Takahashi, T. Co-expression of receptor for advanced glycation end products and the ligand amphoterin associates closely with metastasis of colorectal cancer. Oncol. Rep. 2003, 10, 445–448. [Google Scholar] [CrossRef]
- Huang, C.-Y.; Chiang, S.-F.; Chen, W.T.-L.; Ke, T.-W.; Chen, T.-W.; You, Y.-S.; Lin, C.-Y.; Chao, K.S.C. HMGB1 promotes ERK-mediated mitochondrial Drp1 phosphorylation for chemoresistance through RAGE in colorectal cancer. Cell Death Dis. 2018, 9, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Mills, G.B.; Moolenaar, W.H. The emerging role of lysophosphatidic acid in cancer. Nat. Rev. Cancer 2003, 3, 582–591. [Google Scholar] [CrossRef]
- Glade, M.J.; Smith, K. Phosphatidylserine and the human brain. Nutrition 2015, 31, 781–786. [Google Scholar] [CrossRef]
- Sakaguchi, M.; Murata, H.; Yamamoto, K.-I.; Ono, T.; Sakaguchi, Y.; Motoyama, A.; Hibino, T.; Kataoka, K.; Huh, N.-H. TIRAP, an Adaptor Protein for TLR2/4, Transduces a Signal from RAGE Phosphorylated upon Ligand Binding. PLoS ONE 2011, 6, e23132. [Google Scholar] [CrossRef] [Green Version]
- Gąsiorowski, K.; Brokos, B.; Echeverria, V.; Barreto, G.E.; Leszek, J. RAGE-TLR Crosstalk Sustains Chronic Inflammation in Neurodegeneration. Mol. Neurobiol. 2018, 55, 1463–1476. [Google Scholar] [CrossRef]
- Wang, L.; Wu, J.; Guo, X.; Huang, X.; Huang, Q. RAGE plays a role in LPS-induced NF-κB activation and endothelial hyperpermeability. Sensors 2017, 17, 722. [Google Scholar] [CrossRef]
- Rouhiainen, A.; Kuja-Panula, J.; Tumova, S.; Rauvala, H. RAGE-mediated cell signaling. Calcium-Bind. Proteins RAGE, 2013; 239–263. [Google Scholar] [CrossRef]
- Yamamoto, K.-I.; Murata, H.; Putranto, E.W.; Kataoka, K.; Motoyama, A.; Hibino, T.; Inoue, Y.; Sakaguchi, M.; Huh, N.-H. DOCK7 is a critical regulator of the RAGE-Cdc42 signaling axis that induces formation of dendritic pseudopodia in human cancer cells. Oncol. Rep. 2012, 29, 1073–1079. [Google Scholar] [CrossRef] [Green Version]
- Touré, F.; Fritz, G.; Li, Q.; Rai, V.; Daffu, G.; Zou, Y.S.; Rosario, R.; Ramasamy, R.; Alberts, A.S.; Yan, S.F.; et al. Formin mDia1 Mediates Vascular Remodeling via Integration of Oxidative and Signal Transduction Pathways. Circ. Res. 2012, 110, 1279–1293. [Google Scholar] [CrossRef]
- Bianchi, R.; Kastrisianaki, E.; Giambanco, I.; Donato, R. S100B Protein Stimulates Microglia Migration via RAGE-dependent Up-regulation of Chemokine Expression and Release. J. Biol. Chem. 2011, 286, 7214–7226. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.; Toure, F.; Qu, W.; Lin, L.; Song, F.; Shen, X.; Rosario, R.; Garcia, J.; Schmidt, A.M.; Yan, S.-F. Advanced Glycation End Product (AGE)-Receptor for AGE (RAGE) Signaling and Up-regulation of Egr-1 in Hypoxic Macrophages. J. Biol. Chem. 2010, 285, 23233–23240. [Google Scholar] [CrossRef] [Green Version]
- Wendt, T.; Harja, E.; Bucciarelli, L.; Qu, W.; Lu, Y.; Rong, L.L.; Jenkins, D.G.; Stein, G.; Schmidt, A.M.; Yan, S.F. RAGE modulates vascular inflammation and atherosclerosis in a murine model of type 2 diabetes. Atherosclerosis 2006, 185, 70–77. [Google Scholar] [CrossRef]
- Ahmed, N. Advanced glycation endproducts—Role in pathology of diabetic complications. Diabetes Res. Clin. Pract. 2005, 67, 3–21. [Google Scholar] [CrossRef]
- Chuah, Y.K.; Basir, R.; Talib, H.; Tie, T.H.; Nordin, N. Receptor for Advanced Glycation End Products and Its Involvement in Inflammatory Diseases. Int. J. Inflamm. 2013, 2013, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eleazu, C.; Omar, N.; Lim, O.Z.; Yeoh, B.S.; Hussain, N.H.N.; Mohamed, M. Obesity and comorbidity: Could simultaneous targeting of esRAGE and sRAGE be the panacea? Front. Physiol. 2019, 10, 787. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Yu, M.; Zhang, Z.; Yu, Y.; Chen, Q.; Zhang, W.; Zhao, X. Blockade of receptor for advanced glycation end products protects against systolic overload-induced heart failure after transverse aortic constriction in mice. Eur. J. Pharmacol. 2016, 791, 535–543. [Google Scholar] [CrossRef]
- Warboys, C.M.; Toh, H.-B.; Fraser, P.A. Role of NADPH Oxidase in Retinal Microvascular Permeability Increase by RAGE Activation. Investig. Opthalmol. Vis. Sci. 2009, 50, 1319–1328. [Google Scholar] [CrossRef]
- Jensen, L.J.N.; Flyvbjerg, A.; Bjerre, M. Soluble Receptor for Advanced Glycation End Product: A Biomarker for Acute Coronary Syndrome. BioMed Res. Int. 2015, 2015, 815942. [Google Scholar] [CrossRef] [Green Version]
- Ray, R.; Juranek, J.K.; Rai, V. RAGE axis in neuroinflammation, neurodegeneration and its emerging role in the pathogenesis of amyotrophic lateral sclerosis. Neurosci. Biobehav. Rev. 2016, 62, 48–55. [Google Scholar] [CrossRef]
- Bettiga, A.; Fiorio, F.; Di Marco, F.; Trevisani, F.; Romani, A.; Porrini, E.; Salonia, A.; Montorsi, F.; Vago, R. The Modern Western Diet Rich in Advanced Glycation End-Products (AGEs): An Overview of Its Impact on Obesity and Early Progression of Renal Pathology. Nutrition 2019, 11, 1748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oczypok, E.A.; Perkins, T.N.; Oury, T.D. All the “RAGE” in lung disease: The receptor for advanced glycation endproducts (RAGE) is a major mediator of pulmonary inflammatory responses. Paediatr. Respir. Rev. 2017, 23, 40–49. [Google Scholar] [CrossRef]
- Liu, Y.; Liang, C.; Liu, X.; Liao, B.; Pan, X.; Ren, Y.; Fan, M.; Li, M.; He, Z.; Wu, J.; et al. AGEs increased migration and inflammatory responses of adventitial fibroblasts via RAGE, MAPK and NF-κB pathways. Atherosclerosis 2010, 208, 34–42. [Google Scholar] [CrossRef]
- Yeh, C.-H.; Sturgis, L.; Haidacher, J.; Zhang, X.-N.; Sherwood, S.J.; Bjercke, R.J.; Juhasz, O.; Crow, M.T.; Tilton, R.G.; Denner, L. Requirement for p38 and p44/p42 mitogen-activated protein kinases in RAGE-mediated nuclear factor-κB transcriptional activation and cytokine secretion. Diabetes 2001, 50, 1495–1504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kislinger, T.; Tanji, N.; Wendt, T.; Qu, W.; Lu, Y.; Ferran, L.J.; Taguchi, A.; Olson, K.; Bucciarelli, L.; Goova, M.; et al. Receptor for Advanced Glycation End Products Mediates Inflammation and Enhanced Expression of Tissue Factor in Vasculature of Diabetic Apolipoprotein E–Null Mice. Arter. Thromb. Vasc. Biol. 2001, 21, 905–910. [Google Scholar] [CrossRef] [Green Version]
- Bangert, A.; Andrassy, M.; Müller, A.-M.; Bockstahler, M.; Fischer, A.; Volz, C.H.; Leib, C.; Göser, S.; Korkmaz-Icöz, S.; Zittrich, S. Critical role of RAGE and HMGB1 in inflammatory heart disease. Proc. Natl. Acad. Sci. USA 2016, 113, E155–E164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kay, A.M.; Simpson, C.L.; Stewart, J.A. The Role of AGE/RAGE Signaling in Diabetes-Mediated Vascular Calcification. J. Diabetes Res. 2016, 2016, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Tanikawa, T.; Okada, Y.; Tanikawa, R.; Tanaka, Y. Advanced Glycation End Products Induce Calcification of Vascular Smooth Muscle Cells through RAGE/p38 MAPK. J. Vasc. Res. 2009, 46, 572–580. [Google Scholar] [CrossRef]
- Coughlan, M.T.; Thorburn, D.; Penfold, S.A.; Laskowski, A.; Harcourt, B.E.; Sourris, K.C.; Tan, A.L.; Fukami, K.; Thallas-Bonke, V.; Nawroth, P.P.; et al. RAGE-Induced Cytosolic ROS Promote Mitochondrial Superoxide Generation in Diabetes. J. Am. Soc. Nephrol. 2009, 20, 742–752. [Google Scholar] [CrossRef] [Green Version]
- Bu, D.-X.; Rai, V.; Shen, X.; Rosario, R.; Lu, Y.; Dagati, V.D.; Yan, S.F.; Friedman, R.A.; Nuglozeh, E.; Schmidt, A.M. Activation of the ROCK1 Branch of the Transforming Growth Factor-β Pathway Contributes to RAGE-Dependent Acceleration of Atherosclerosis in Diabetic ApoE-Null Mice. Circ. Res. 2010, 106, 1040–1051. [Google Scholar] [CrossRef] [Green Version]
- Cepas, V.; Collino, M.; Mayo, J.C.; Sainz, R.M. Redox Signaling and Advanced Glycation Endproducts (AGEs) in Diet-Related Diseases. Antioxidants 2020, 9, 142. [Google Scholar] [CrossRef] [Green Version]
- Juranek, J.K.; Kothary, P.; Mehra, A.; Hays, A.; Iii, T.H.B.; Schmidt, A.M. Increased expression of the receptor for advanced glycation end-products in human peripheral neuropathies. Brain Behav. 2013, 3, 701–709. [Google Scholar] [CrossRef]
- Williams, N.; Okun, M. Deep brain stimulation (DBS) at the interface of neurology and psychiatry. J. Clin. Investig. 2013, 123, 4546–4556. [Google Scholar] [CrossRef] [Green Version]
- Kouidrat, Y.; Amad, A.; Arai, M.; Miyashita, M.; Lalau, J.-D.; Loas, G.; Itokawa, M. Advanced glycation end products and schizophrenia: A systematic review. J. Psychiatr. Res. 2015, 66–67, 112–117. [Google Scholar] [CrossRef]
- MacLean, M.; Derk, J.; Ruiz, H.H.; Juranek, J.; Ramasamy, R.; Schmidt, A.M. The Receptor for Advanced Glycation End Products (RAGE) and DIAPH1: Implications for vascular and neuroinflammatory dysfunction in disorders of the central nervous system. Neurochem. Int. 2019, 126, 154–164. [Google Scholar] [CrossRef]
- Richard, S.A.; Sackey, M.; Su, Z.; Xu, H. Pivotal neuroinflammatory and therapeutic role of high mobility group box 1 in ischemic stroke. Biosci. Rep. 2017, 37. [Google Scholar] [CrossRef] [Green Version]
- Sasaki, N.; Toki, S.; Chowei, H.; Saito, T.; Nakano, N.; Hayashi, Y.; Takeuchi, M.; Makita, Z. Immunohistochemical distribution of the receptor for advanced glycation end products in neurons and astrocytes in Alzheimer’s disease. Brain Res. 2001, 888, 256–262. [Google Scholar] [CrossRef]
- Ding, Q.; Keller, J.N. Evaluation of rage isoforms, ligands, and signaling in the brain. Biochim. et Biophys. Acta (BBA)-Bioenerg. 2005, 1746, 18–27. [Google Scholar] [CrossRef] [Green Version]
- Deane, R.J. Is RAGE still a therapeutic target for Alzheimer’s disease? Futur. Med. Chem. 2012, 4, 915–925. [Google Scholar] [CrossRef] [Green Version]
- Deane, R.; Singh, I.; Sagare, A.P.; Bell, R.D.; Ross, N.T.; LaRue, B.; Love, R.; Perry, S.; Paquette, N.; Deane, R.J.; et al. A multimodal RAGE-specific inhibitor reduces amyloid β–mediated brain disorder in a mouse model of Alzheimer disease. J. Clin. Investig. 2012, 122, 1377–1392. [Google Scholar] [CrossRef] [Green Version]
- Bianchi, R.; Giambanco, I.; Donato, R. S100B/RAGE-dependent activation of microglia via NF-κB and AP-1: Co-regulation of COX-2 expression by S100B, IL-1β and TNF-α. Neurobiol. Aging 2010, 31, 665–677. [Google Scholar] [CrossRef]
- Kang, R.; Tang, D.; Schapiro, N.E.; Livesey, K.M.; Farkas, A.; Loughran, P.; Bierhaus, A.; Lotze, M.T.; Zeh, H.J. The receptor for advanced glycation end products (RAGE) sustains autophagy and limits apoptosis, promoting pancreatic tumor cell survival. Cell Death Differ. 2010, 17, 666–676. [Google Scholar] [CrossRef] [Green Version]
- Logsdon, C.D.; Fuentes, M.K.; Huang, E.H.; Arumugam, T. RAGE and RAGE ligands in cancer. Curr. Mol. Med. 2007, 7, 777–789. [Google Scholar] [CrossRef]
- Kwak, T.; Drews-Elger, K.; Ergonul, A.; Miller, P.C.; Braley, A.; Hwang, G.H.; Zhao, D.; Besser, A.; Yamamoto, Y.; El-Ashry, D.; et al. Targeting of RAGE-ligand signaling impairs breast cancer cell invasion and metastasis. Oncogene 2017, 36, 1559–1572. [Google Scholar] [CrossRef]
- Kalea, A.Z.; See, F.; Harja, E.; Arriero, M.; Schmidt, A.M.; Hudson, B.I. Alternatively Spliced RAGEv1 Inhibits Tumorigenesis through Suppression of JNK Signaling. Cancer Res. 2010, 70, 5628–5638. [Google Scholar] [CrossRef] [Green Version]
- Sorci, G.; Riuzzi, F.; Arcuri, C.; Tubaro, C.; Bianchi, R.; Giambanco, I.; Donato, R. S100B protein in tissue development, repair and regeneration. World J. Biol. Chem. 2013, 4, 1–12. [Google Scholar] [CrossRef]
- Matou-Nasri, S.; Sharaf, H.; Wang, Q.; Almobadel, N.; Rabhan, Z.; Al-Eidi, H.; Bin Yahya, W.; Trivilegio, T.; Ali, R.; Al-Shanti, N.; et al. Biological impact of advanced glycation endproducts on estrogen receptor-positive MCF-7 breast cancer cells. Biochim. et Biophys. Acta (BBA)-Mol. Basis Dis. 2017, 1863, 2808–2820. [Google Scholar] [CrossRef]
- Azizan, N.; Suter, M.A.; Liu, Y.; Logsdon, C.D. RAGE maintains high levels of NFκB and oncogenic Kras activity in pancreatic cancer. Biochem. Biophys. Res. Commun. 2017, 493, 592–597. [Google Scholar] [CrossRef]
- Syed, D.N.; Aljohani, A.; Waseem, D.; Mukhtar, H. Ousting RAGE in melanoma: A viable therapeutic target? Semin. Cancer Biol. 2018, 49, 20–28. [Google Scholar] [CrossRef] [PubMed]
- Deng, R.; Mo, F.; Chang, B.; Zhang, Q.; Ran, H.; Yang, S.; Zhu, Z.; Hu, L.; Su, Q. Glucose-derived AGEs enhance human gastric cancer metastasis through RAGE/ERK/Sp1/MMP2 cascade. Oncotarget 2017, 8, 104216–104226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Somensi, N.; Brum, P.O.; Ramos, V.d.; Gasparotto, J.; Zanotto-Filho, A.; Rostirolla, D.C.; Morrone, M.d.; Moreira, J.C.F.; Gelain, D.P. Extracellular HSP70 Activates ERK1/2, NF-kB and Pro-Inflammatory Gene Transcription Through Binding with RAGE in A549 Human Lung Cancer Cells. Cell Physiol. Biochem. 2017, 42, 2507–2522. [Google Scholar] [CrossRef]
- Ahmad, S.; Khan, H.; Siddiqui, Z.; Khan, M.Y.; Rehman, S.; Shahab, U.; Godovikova, T.; Silnikov, V.; Moinuddin. AGEs, RAGEs and s-RAGE; friend or foe for cancer. Semin. Cancer Biol. 2018, 49, 44–55. [Google Scholar] [CrossRef]
- Kolonin, M.G.; Sergeeva, A.; Staquicini, D.I.; Smith, T.L.; Tarleton, C.A.; Molldrem, J.J.; Sidman, R.L.; Marchiò, S.; Pasqualini, R.; Arap, W. Interaction between Tumor Cell Surface Receptor RAGE and Proteinase 3 Mediates Prostate Cancer Metastasis to Bone. Cancer Res. 2017, 77, 3144–3150. [Google Scholar] [CrossRef] [Green Version]
- Aboushousha, T.; Mamdouh, S.; Hamdy, H.; Helal, N.; Khorshed, F.; Safwat, G.; Seleem, M. Immunohistochemical and Biochemical Expression Patterns of TTF-1, RAGE, GLUT-1 and SOX2 in HCV-Associated Hepatocellular Carcinomas. Asian Pac. J. Cancer Prev. 2018, 19, 219–227. [Google Scholar] [CrossRef]
- Chen, W.-W.; Guo, Q.; Zhang, Z.-D.; Hu, W.-M. Effects of RAGE on Cell Proliferation and Tumor Growth in Pancreatic Cancer. J. Sichuan Univ. Med Sci. Ed. 2017, 48, 46–51. [Google Scholar]
- Yu, Y.X.; Pan, W.C.; Cheng, Y.F. Silencing of advanced glycosylation and glycosylation and product-specific receptor (RAGE) inhibits the metastasis and growth of non-small cell lung cancer. Am. J. Transl. Res. 2017, 9, 2760–2774. [Google Scholar] [PubMed]
- Adamopoulos, C.; Piperi, C.; Gargalionis, A.N.; Dalagiorgou, G.; Spilioti, E.; Korkolopoulou, P.; Diamanti-Kandarakis, E.; Papavassiliou, A.G. Advanced glycation end products upregulate lysyl oxidase and endothelin-1 in human aortic endothelial cells via parallel activation of ERK1/2-NF-κB and JNK-AP-1 signaling pathways. Cell Mol. Life Sci. 2016, 73, 1685–1698. [Google Scholar] [CrossRef] [PubMed]
- Cai, W.; Duan, X.-M.; Liu, Y.; Yu, J.; Tang, Y.-L.; Liu, Z.-L.; Jiang, S.; Zhang, C.-P.; Liu, J.-Y.; Xu, J.-X. Uric Acid Induces Endothelial Dysfunction by Activating the HMGB1/RAGE Signaling Pathway. BioMed Res. Int. 2017, 2017, 1–11. [Google Scholar] [CrossRef]
- Gardner, T.W.; Antonetti, D.A.; Barber, A.J.; LaNoue, K.F.; Levison, S.W. Diabetic retinopathy: More than meets the eye. Surv. Ophthalmol. 2002, 47 (Suppl. 2), S253–S262. [Google Scholar] [CrossRef]
- McVicar, C.M.; Ward, M.; Colhoun, L.M.; Guduric-Fuchs, J.; Bierhaus, A.; Fleming, T.; Schlotterer, A.; Kolibabka, M.; Hammes, H.-P.; Chen, M.; et al. Role of the receptor for advanced glycation endproducts (RAGE) in retinal vasodegenerative pathology during diabetes in mice. Diabetologia 2015, 58, 1129–1137. [Google Scholar] [CrossRef] [PubMed]
- Kaji, Y.; Usui, T.; Ishida, S.; Yamashiro, K.; Moore, T.C.B.; Moore, J.; Yamamoto, Y.; Yamamoto, H.; Adamis, A.P. Inhibition of Diabetic Leukostasis and Blood-Retinal Barrier Breakdown with a Soluble Form of a Receptor for Advanced Glycation End Products. Investig. Opthalmol. Vis. Sci. 2007, 48, 858–865. [Google Scholar] [CrossRef] [Green Version]
- Barile, G.R.; Pachydaki, S.I.; Tari, S.R.; Lee, S.E.; Donmoyer, C.M.; Ma, W.; Rong, L.L.; Buciarelli, L.G.; Wendt, T.; Ho¨rig, H.; et al. The RAGE Axis in Early Diabetic Retinopathy. Investig. Opthalmol. Vis. Sci. 2005, 46, 2916–2924. [Google Scholar] [CrossRef] [Green Version]
- Riuzzi, F.; Chiappalupi, S.; Arcuri, C.; Giambanco, I.; Sorci, G.; Donato, R. S100 proteins in obesity: Liaisons dangereuses. Cell. Mol. Life Sci. 2019, 77, 129–147. [Google Scholar] [CrossRef]
- Song, F.; Del Pozo, C.H.; Rosario, R.; Zou, Y.S.; Ananthakrishnan, R.; Xu, X.; Patel, P.R.; Benoit, V.M.; Yan, S.F.; Li, H.; et al. RAGE Regulates the Metabolic and Inflammatory Response to High-Fat Feeding in Mice. Diabetes 2014, 63, 1948–1965. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mukherjee, T.K.; Mukhopadhyay, S.; Hoidal, J.R. Implication of receptor for advanced glycation end product (RAGE) in pulmonary health and pathophysiology. Respir. Physiol. Neurobiol. 2008, 162, 210–215. [Google Scholar] [CrossRef] [PubMed]
- Bierhaus, A.; Nawroth, P. Multiple levels of regulation determine the role of the receptor for AGE (RAGE) as common soil in inflammation, immune responses and diabetes mellitus and its complications. Diabetologia 2009, 52, 2251–2263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Catalano, M.; Cortelazzo, A.; Santi, R.; Contino, L.; Demicheli, M.; Yilmaz, Y.; Zorzetto, M.; Campo, I.; Lanati, N.; Emanuele, E. The Pro12Ala polymorphism of peroxisome proliferator-activated receptor-γ2 gene is associated with plasma levels of soluble RAGE (Receptor for Advanced Glycation Endproducts) and the presence of peripheral arterial disease. Clin. Biochem. 2008, 41, 981–985. [Google Scholar] [CrossRef] [PubMed]
- Jang, Y.; Kim, J.Y.; Kang, S.-M.; Kim, J.-S.; Chae, J.S.; Kim, O.Y.; Koh, S.J.; Lee, H.C.; Ahn, C.W.; Song, Y.D.; et al. Association of the Gly82Ser polymorphism in the receptor for advanced glycation end products (RAGE) gene with circulating levels of soluble RAGE and inflammatory markers in nondiabetic and nonobese Koreans. Metabolism 2007, 56, 199–205. [Google Scholar] [CrossRef] [PubMed]
- Weykamp, C. HbA1c: A review of analytical and clinical aspects. AnnLab. Med. 2013, 33, 393. [Google Scholar] [CrossRef] [Green Version]
- Constien, R.; Forde, A.; Liliensiek, B.; Gröne, H.J.; Nawroth, P.; Hämmerling, G.; Arnold, B. Characterization of a novel EGFP reporter mouse to monitor Cre recombination as demonstrated by a Tie2 Cre mouse line. Genesis 2001, 30, 36–44. [Google Scholar] [CrossRef]
- Al-Robaiy, S.; Weber, B.; Simm, A.; Diez, C.; Rolewska, P.; Silber, R.-E.; Bartling, B. The receptor for advanced glycation end-products supports lung tissue biomechanics. Am. J. Physiol. Cell. Mol. Physiol. 2013, 305, L491–L500. [Google Scholar] [CrossRef] [Green Version]
- Park, L.; Raman, K.G.; Lee, K.J.; Lu, Y.; Ferran, L.J., Jr.; Chow, W.S.; Stern, D.; Schmidt, A.M. Suppression of accelerated diabetic atherosclerosis by the soluble receptor for advanced glycation endproducts. Nat. Med. 1998, 4, 1025–1031. [Google Scholar] [CrossRef]
- Chen, Y.; Yan, S.S.; Colgan, J.; Zhang, H.-P.; Luban, J.; Schmidt, A.M.; Stern, D.; Herold, K.C. Blockade of late stages of autoimmune diabetes by inhibition of the receptor for advanced glycation end products. J. Immunol. 2004, 173, 1399–1405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sabbagh, M.N.; Agro, A.; Bell, J.; Aisen, P.S.; Schweizer, E.; Galasko, D. PF-04494700, an Oral Inhibitor of Receptor for Advanced Glycation End Products (RAGE), in Alzheimer Disease. Alzheimer Dis. Assoc. Disord. 2011, 25, 206–212. [Google Scholar] [CrossRef]
- Burstein, A.H.; Grimes, I.; Galasko, D.R.; Aisen, P.S.; Sabbagh, M.; Mjalli, A.M.M. Effect of TTP488 in patients with mild to moderate Alzheimer’s disease. BMC Neurol. 2014, 14, 12. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.S.; Kim, H.; Kim, Y.-H.; Roh, E.J.; Han, H.; Shin, K.J. Synthesis and structure–activity relationships of tri-substituted thiazoles as RAGE antagonists for the treatment of Alzheimer’s disease. Bioorg. Med. Chem. Lett. 2012, 22, 7555–7561. [Google Scholar] [CrossRef]
- Han, Y.T.; Choi, G.-I.; Son, D.; Kim, N.-J.; Yun, H.; Lee, S.; Chang, D.J.; Hong, H.-S.; Kim, H.; Ha, H.-J.; et al. Ligand-Based Design, Synthesis, and Biological Evaluation of 2-Aminopyrimidines, a Novel Series of Receptor for Advanced Glycation End Products (RAGE) Inhibitors. J. Med. Chem. 2012, 55, 9120–9135. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.T.; Kim, K.; Choi, G.-I.; An, H.; Son, D.; Kim, H.; Ha, H.-J.; Son, J.-H.; Chung, S.-J.; Park, H.-J.; et al. Pyrazole-5-carboxamides, novel inhibitors of receptor for advanced glycation end products (RAGE). Eur. J. Med. Chem. 2014, 79, 128–142. [Google Scholar] [CrossRef] [PubMed]
- Choi, K.; Lim, K.S.; Shin, J.; Kim, S.H.; Suh, Y.-G.; Hong, H.-S.; Kim, H.; Ha, H.-J.; Kim, Y.-H.; Lee, J.; et al. 6-Phenoxy-2-phenylbenzoxazoles, novel inhibitors of receptor for advanced glycation end products (RAGE). Bioorg. Med. Chem. 2015, 23, 4919–4935. [Google Scholar] [CrossRef]
- Zhang, J.; Xu, X.; Rao, N.V.; Argyle, B.; McCoard, L.; Rusho, W.J.; Kennedy, T.P.; Prestwich, G.D.; Krueger, G. Novel Sulfated Polysaccharides Disrupt Cathelicidins, Inhibit RAGE and Reduce Cutaneous Inflammation in a Mouse Model of Rosacea. PLoS ONE 2011, 6, e16658. [Google Scholar] [CrossRef] [Green Version]
- Arumugam, T.; Ramachandran, V.; Gomez, S.B.; Schmidt, A.M.; Logsdon, C.D. S100P-Derived RAGE Antagonistic Peptide Reduces Tumor Growth and Metastasis. Clin. Cancer Res. 2012, 18, 4356–4364. [Google Scholar] [CrossRef] [Green Version]
- Huttunen, H.J.; Fages, C.; Kuja-Panula, J.; Ridley, A.J.; Rauvala, H. Receptor for advanced glycation end products-binding COOH-terminal motif of amphoterin inhibits invasive migration and metastasis. Cancer Res. 2002, 62, 4805–4811. [Google Scholar]
- Yamagishi, S.-I.; Matsui, T. Therapeutic potential of DNA-aptamers raised against AGE-RAGE axis in diabetes-related complications. Curr. Pharm. Design 2018, 24, 2802–2809. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.; Zhu, W.; He, F.; Li, Z.; Cai, N.; Wang, H.-H. An Aptamer-Based Antagonist against the Receptor for Advanced Glycation End-Products (RAGE) Blocks Development of Colorectal Cancer. Mediat. Inflamm. 2021, 2021, 1–8. [Google Scholar] [CrossRef]
- Yao, L.; Zhao, H.; Tang, H.; Liang, J.; Liu, L.; Dong, H.; Zou, F.; Cai, S. The receptor for advanced glycation end products is required for β-catenin stabilization in a chemical-induced asthma model. Br. J. Pharmacol. 2016, 173, 2600–2613. [Google Scholar] [CrossRef]
- Mizumoto, S.; Takahashi, J.; Sugahara, K. Receptor for Advanced Glycation End Products (RAGE) Functions as Receptor for Specific Sulfated Glycosaminoglycans, and Anti-RAGE Antibody or Sulfated Glycosaminoglycans Delivered in Vivo Inhibit Pulmonary Metastasis of Tumor Cells. J. Biol. Chem. 2012, 287, 18985–18994. [Google Scholar] [CrossRef] [Green Version]
- Yan, Z.; Luo, H.; Xie, B.; Tian, T.; Li, S.; Chen, Z.; Liu, J.; Zhao, X.; Zhang, L.; Deng, Y.; et al. Targeting adaptor protein SLP76 of RAGE as a therapeutic approach for lethal sepsis. Nat. Commun. 2021, 12, 1–14. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| RAGE Ligands | RAGE Binding Domain | Clinical Significance | Ref. |
|---|---|---|---|
| Endogenous RAGE Ligands | |||
| AGEs | V | Diabetes, chronic inflammation and cancer | [28] |
| S100/calgranulins | V or VC1 or V2 | Inflammatory response and cancer differentiation and progression | [64] |
| HMGB1 | VC1C2 | Cancer development and metastasis and drug resistance | [65] |
| β-sheet fibrils | V | Neuronal disease: Alzheimer’s disease | [66] |
| Mac1 | RAGE-mediated leukocyte recruitment | [45] | |
| Quinolinic acids | VC1 | Neuronal disease: Huntington’s disease | [67] |
| LPA | V | Cell proliferation and migration in C6 glioma and smooth muscle cells | [68] |
| PS | Rac1 activation in alveolar macrophages | [69] | |
| C1q | Recruitment of leukocytes and phagocytosis | [70] | |
| mDia1 | cytoplasmic | Initiation and activation of RAGE-mediated signaling | [13] |
| Exogenous RAGE Ligands | |||
| RNA or DNA | VC1 | RAGE-mediated augmentation of inflammation | [8] |
| RSV F protein | VC1 | Promote the survival of RSV-infected cells | [11] |
| Longistatin | V | Longistatin acts as an antagonist to RAGE and suppresses inflammation | [12] |
| Inhibitors | Targeting of RAGE Domain | Effects | Ref. |
|---|---|---|---|
| TTP488 | V | AGEs, HMGB1, CML, S100B, and Aβ-RAGE binding inhibition | [4,220,221] |
| 4,6-disubstituted 2-amino pyrimidines | V | Aβ-RAGE binding inhibition | [223] |
| 4-fluorophenoxy analog | V | Inhibition of amyloid plaques inside the brain | [224] |
| FPS-ZM1 | V | Aβ-RAGE binding inhibition and low cytotoxicity in vitro and in vivo | [186] |
| GM-1111 | VC1C2 | CML, GMGB1, and S100B-RAGE binding inhibition | [226] |
| S100-derived peptide | VC1C2 | Reduced RAGE-mediated activation of NF-κB, inflammation, tumor growth, and metastasis in various cancer cells | [227] |
| HMGB1-derived Peptide | VC1C2 | Suppressed the formation of pulmonary metastasis and invasion in tumor cells | [228] |
| Alagebrium | AGE cross-link breaker | Reduced AGE accumulation and atherosclerotic plaque formation and lesions | [73] |
| DNA-aptamers | against the AGE-RAGE axis in diabetes-associated complications | [229] | |
| Group of 13 compounds | cytoplasmic | Inhibition of ctRAGE interaction with mDia1 | [14] |
| Aptamer-based antagonist | V | inhibit interaction between RAGE and S100B | [230] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, H.J.; Jeong, M.S.; Jang, S.B. Molecular Characteristics of RAGE and Advances in Small-Molecule Inhibitors. Int. J. Mol. Sci. 2021, 22, 6904. https://doi.org/10.3390/ijms22136904
Kim HJ, Jeong MS, Jang SB. Molecular Characteristics of RAGE and Advances in Small-Molecule Inhibitors. International Journal of Molecular Sciences. 2021; 22(13):6904. https://doi.org/10.3390/ijms22136904
Chicago/Turabian StyleKim, Hyeon Jin, Mi Suk Jeong, and Se Bok Jang. 2021. "Molecular Characteristics of RAGE and Advances in Small-Molecule Inhibitors" International Journal of Molecular Sciences 22, no. 13: 6904. https://doi.org/10.3390/ijms22136904
APA StyleKim, H. J., Jeong, M. S., & Jang, S. B. (2021). Molecular Characteristics of RAGE and Advances in Small-Molecule Inhibitors. International Journal of Molecular Sciences, 22(13), 6904. https://doi.org/10.3390/ijms22136904