
Macrophage Polarization States in the Tumor Microenvironment


Abstract
:1. Introduction
2. Macrophage Markers
2.1. Human Macrophage Markers
2.2. Mouse Macrophage Markers
3. Macrophage Polarization
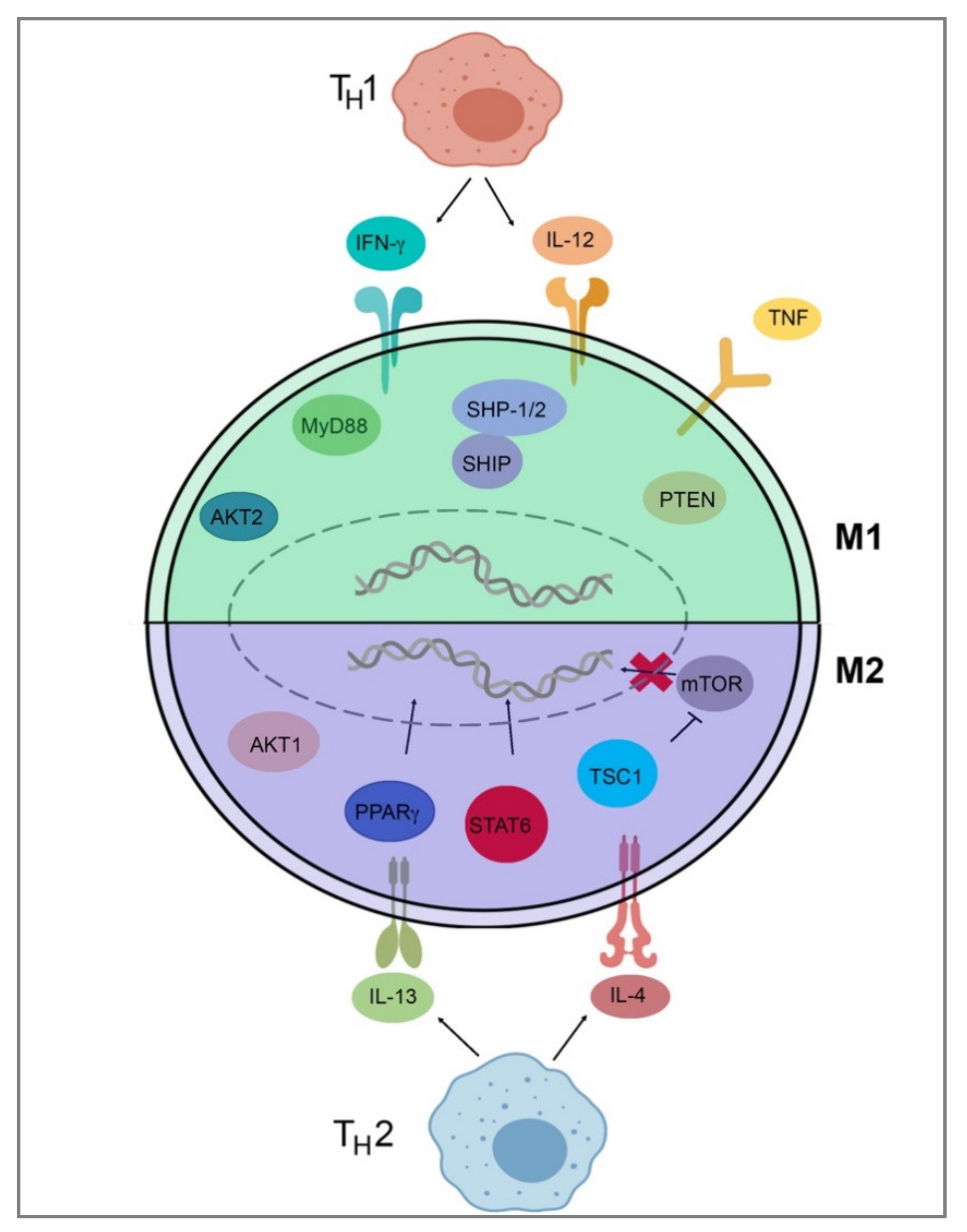
3.1. M1 and M2 Macrophages
3.2. Extrinsic Polarization
3.3. Hypoxia-Induced Polarization
3.4. Intrinsic Polarization
4. Inflammation
4.1. Role of Macrophages in Inflammation
4.2. Role of Inflammation in the Tumor Microenvironment
4.3. Influence of the Tumor Microenvironment on Macrophage Polarization
5. Pro-Tumorigenic Outcomes
5.1. Immune Suppression
5.2. Proliferation
5.3. Lymphangiogenesis, Angiogenesis, and Metastasis
5.4. TIE2-Expressing Macrophages
5.5. Resistance to Therapy
5.6. Proposed Therapies
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Wang, N.; Liang, H.; Zen, K. Molecular Mechanisms That Influence the Macrophage M1–M2 Polarization Balance. Front. Immunol. 2014, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dijkgraaf, E.M.; Heusinkveld, M.; Tummers, B.; Vogelpoel, L.T.C.; Goedemans, R.; Jha, V.; Nortier, J.W.R.; Welters, M.J.P.; Kroep, J.R.; van der Burg, S.H. Chemotherapy Alters Monocyte Differentiation to Favor Generation of Cancer-Supporting M2 Macrophages in the Tumor Microenvironment. Cancer Res. 2013, 73, 2480–2492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujiwara, N.; Kobayashi, K. Macrophages in Inflammation. Curr. Drug Targets Inflamm. Allergy 2005, 4, 281–286. [Google Scholar] [CrossRef] [PubMed]
- Wynn, T.A.; Chawla, A.; Pollard, J.W. Macrophage Biology in Development, Homeostasis and Disease. Nature 2013, 496, 445–455. [Google Scholar] [CrossRef]
- Allavena, P.; Sica, A.; Solinas, G.; Porta, C.; Mantovani, A. The Inflammatory Micro-Environment in Tumor Progression: The Role of Tumor-Associated Macrophages. Crit. Rev Oncol. Hematol. 2008, 66, 1–9. [Google Scholar] [CrossRef]
- Gordy, C.; Pua, H.; Sempowski, G.D.; He, Y.-W. Regulation of Steady-State Neutrophil Homeostasis by Macrophages. Blood 2011, 117, 618–629. [Google Scholar] [CrossRef] [Green Version]
- Goren, I.; Allmann, N.; Nir, Y.; Schürmann, C.; Linke, A.; Holdener, M.; Waisman, A.; Pfeilschifter, J.; Frank, S. A Transgenic Mouse Model of Inducible Macrophage Depletion: Effects of Diphtheria Toxin-Driven Lysozyme M-Specific Cell Lineage Ablation on Wound Inflammatory, Angiogenic, and Contractive Processes. Am. J. Pathol. 2009, 175, 132–147. [Google Scholar] [CrossRef] [Green Version]
- Oishi, Y.; Manabe, I. Macrophages in Inflammation, Repair and Regeneration. Int. Immunol. 2018, 30, 511–528. [Google Scholar] [CrossRef]
- Jaiswal, S.; Jamieson, C.H.M.; Pang, W.W.; Park, C.Y.; Chao, M.P.; Majeti, R.; Traver, D.; van Rooijen, N.; Weissman, I.L. CD47 Is Upregulated on Circulating Hematopoietic Stem Cells and Leukemia Cells to Avoid Phagocytosis. Cell 2009, 138, 271–285. [Google Scholar] [CrossRef] [Green Version]
- Borisenko, G.G.; Matsura, T.; Liu, S.-X.; Tyurin, V.A.; Jianfei, J.; Serinkan, F.B.; Kagan, V.E. Macrophage Recognition of Externalized Phosphatidylserine and Phagocytosis of Apoptotic Jurkat Cells—Existence of a Threshold. Arch. Biochem. Biophys. 2003, 413, 41–52. [Google Scholar] [CrossRef]
- He, H.; Mack, J.J.; Güç, E.; Warren, C.M.; Squadrito, M.L.; Kilarski, W.W.; Baer, C.; Freshman, R.D.; McDonald, A.I.; Ziyad, S.; et al. Perivascular Macrophages Limit Permeability. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 2203–2212. [Google Scholar] [CrossRef] [Green Version]
- Stefanowski, J.; Lang, A.; Rauch, A.; Aulich, L.; Köhler, M.; Fiedler, A.F.; Buttgereit, F.; Schmidt-Bleek, K.; Duda, G.N.; Gaber, T.; et al. Spatial Distribution of Macrophages During Callus Formation and Maturation Reveals Close Crosstalk Between Macrophages and Newly Forming Vessels. Front. Immunol. 2019, 10, 2588. [Google Scholar] [CrossRef] [Green Version]
- Nucera, S.; Biziato, D.; De Palma, M. The Interplay between Macrophages and Angiogenesis in Development, Tissue Injury and Regeneration. Int. J. Dev. Biol. 2011, 55, 495–503. [Google Scholar] [CrossRef] [Green Version]
- Squadrito, M.L.; De Palma, M. Macrophage Regulation of Tumor Angiogenesis: Implications for Cancer Therapy. Mol. Asp. Med. 2011, 32, 123–145. [Google Scholar] [CrossRef]
- Haywood, L.; McWilliams, D.F.; Pearson, C.I.; Gill, S.E.; Ganesan, A.; Wilson, D.; Walsh, D.A. Inflammation and Angiogenesis in Osteoarthritis. Arthritis Rheum. 2003, 48, 2173–2177. [Google Scholar] [CrossRef]
- Rao, S.; Lobov, I.B.; Vallance, J.E.; Tsujikawa, K.; Shiojima, I.; Akunuru, S.; Walsh, K.; Benjamin, L.E.; Lang, R.A. Obligatory Participation of Macrophages in an Angiopoietin 2-Mediated Cell Death Switch. Development 2007, 134, 4449–4458. [Google Scholar] [CrossRef] [Green Version]
- Stefater, J.A.; Lewkowich, I.; Rao, S.; Mariggi, G.; Carpenter, A.C.; Burr, A.R.; Fan, J.; Ajima, R.; Molkentin, J.D.; Williams, B.O.; et al. Regulation of Angiogenesis by a Non-Canonical Wnt-Flt1 Pathway in Myeloid Cells. Nature 2011, 474, 511–515. [Google Scholar] [CrossRef] [Green Version]
- Gordon, E.J.; Rao, S.; Pollard, J.W.; Nutt, S.L.; Lang, R.A.; Harvey, N.L. Macrophages Define Dermal Lymphatic Vessel Calibre during Development by Regulating Lymphatic Endothelial Cell Proliferation. Development 2010, 137, 3899–3910. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Liu, Z.; Sun, J.; Song, X.; Bian, M.; Wang, F.; Yan, F.; Yu, Z. Inhibition of NADPH Oxidase 4 Attenuates Lymphangiogenesis and Tumor Metastasis in Breast Cancer. FASEB J. 2021, 35. [Google Scholar] [CrossRef]
- Khazen, W.; M’Bika, J.-P.; Tomkiewicz, C.; Benelli, C.; Chany, C.; Achour, A.; Forest, C. Expression of Macrophage-Selective Markers in Human and Rodent Adipocytes. FEBS Lett. 2005, 579, 5631–5634. [Google Scholar] [CrossRef] [Green Version]
- Barros, M.H.M.; Hauck, F.; Dreyer, J.H.; Kempkes, B.; Niedobitek, G. Macrophage Polarisation: An Immunohistochemical Approach for Identifying M1 and M2 Macrophages. PLoS ONE 2013, 8, e80908. [Google Scholar] [CrossRef] [Green Version]
- Holness, C.; Simmons, D. Molecular Cloning of CD68, a Human Macrophage Marker Related to Lysosomal Glycoproteins. Blood 1993, 81, 1607–1613. [Google Scholar] [CrossRef] [Green Version]
- Stöger, J.L.; Gijbels, M.J.J.; van der Velden, S.; Manca, M.; van der Loos, C.M.; Biessen, E.A.L.; Daemen, M.J.A.P.; Lutgens, E.; de Winther, M.P.J. Distribution of Macrophage Polarization Markers in Human Atherosclerosis. Atherosclerosis 2012, 225, 461–468. [Google Scholar] [CrossRef] [Green Version]
- Xu, N.; Tang, X.-H.; Pan, W.; Xie, Z.-M.; Zhang, G.-F.; Ji, M.-H.; Yang, J.-J.; Zhou, M.-T.; Zhou, Z.-Q. Spared Nerve Injury Increases the Expression of Microglia M1 Markers in the Prefrontal Cortex of Rats and Provokes Depression-Like Behaviors. Front. Neurosci. 2017, 11. [Google Scholar] [CrossRef]
- Gensel, J.C.; Kopper, T.J.; Zhang, B.; Orr, M.B.; Bailey, W.M. Predictive Screening of M1 and M2 Macrophages Reveals the Immunomodulatory Effectiveness of Post Spinal Cord Injury Azithromycin Treatment. Sci. Rep. 2017, 7, 40144. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Zhang, Y.; Wang, Z.; Zhang, J.; Qiao, R.R.; Xu, M.; Yang, N.; Gao, L.; Qiao, H.; Gao, M.; et al. Optical/MRI Dual-Modality Imaging of M1 Macrophage Polarization in Atherosclerotic Plaque with MARCO-Targeted Upconversion Luminescence Probe. Biomaterials 2019, 219, 119378. [Google Scholar] [CrossRef]
- Jablonski, K.A.; Amici, S.A.; Webb, L.M.; Ruiz-Rosado, J.d.D.; Popovich, P.G.; Partida-Sanchez, S.; Guerau-de-Arellano, M. Novel Markers to Delineate Murine M1 and M2 Macrophages. PLoS ONE 2015, 10, e0145342. [Google Scholar] [CrossRef] [Green Version]
- Jaguin, M.; Houlbert, N.; Fardel, O.; Lecureur, V. Polarization Profiles of Human M-CSF-Generated Macrophages and Comparison of M1-Markers in Classically Activated Macrophages from GM-CSF and M-CSF Origin. Cell. Immunol. 2013, 281, 51–61. [Google Scholar] [CrossRef]
- Rőszer, T. Understanding the Mysterious M2 Macrophage through Activation Markers and Effector Mechanisms. Available online: https://www.hindawi.com/journals/mi/2015/816460/ (accessed on 6 May 2021). [CrossRef] [Green Version]
- Yeung, O.W.H.; Lo, C.-M.; Ling, C.-C.; Qi, X.; Geng, W.; Li, C.-X.; Ng, K.T.P.; Forbes, S.J.; Guan, X.-Y.; Poon, R.T.P.; et al. Alternatively Activated (M2) Macrophages Promote Tumour Growth and Invasiveness in Hepatocellular Carcinoma. J. Hepatol. 2015, 62, 607–616. [Google Scholar] [CrossRef]
- Sindrilaru, A.; Peters, T.; Wieschalka, S.; Baican, C.; Baican, A.; Peter, H.; Hainzl, A.; Schatz, S.; Qi, Y.; Schlecht, A.; et al. An Unrestrained Proinflammatory M1 Macrophage Population Induced by Iron Impairs Wound Healing in Humans and Mice. J. Clin. Invest. 2011, 121, 985–997. [Google Scholar] [CrossRef] [Green Version]
- Kawane, K.; Fukuyama, H.; Kondoh, G.; Takeda, J.; Ohsawa, Y.; Uchiyama, Y.; Nagata, S. Requirement of DNase II for Definitive Erythropoiesis in the Mouse Fetal Liver. Science 2001, 292, 1546–1549. [Google Scholar] [CrossRef] [PubMed]
- Verreck, F.A.W.; de Boer, T.; Langenberg, D.M.L.; Hoeve, M.A.; Kramer, M.; Vaisberg, E.; Kastelein, R.; Kolk, A.; de Waal-Malefyt, R.; Ottenhoff, T.H.M. Human IL-23-Producing Type 1 Macrophages Promote but IL-10-Producing Type 2 Macrophages Subvert Immunity to (Myco) Bacteria. Proc. Natl. Acad. Sci. USA 2004, 101, 4560–4565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bouhlel, M.A.; Derudas, B.; Rigamonti, E.; Diévart, R.; Brozek, J.; Haulon, S.; Zawadski, C.; Jude, B.; Topier, G.; Marx, N.; et al. PPARγ Activation Primes Human Monocytes into Alternative M2 Macrophages with Anti-Inflammatory Properties. Cell Metabol. 2007, 6, 137–143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orecchioni, M.; Ghosheh, Y.; Pramod, A.B.; Ley, K. Macrophage Polarization: Different Gene Signatures in M1 (LPS+) vs. Classically and M2 (LPS–) vs. Alternatively Activated Macrophages. Front. Immunol. 2019, 10, 1084. [Google Scholar] [CrossRef] [PubMed]
- Murray, P.J. Macrophage Polarization. Annu. Rev. Physiol. 2017, 79, 541–566. [Google Scholar] [CrossRef]
- Cohen, H.B.; Mosser, D.M. Extrinsic and Intrinsic Control of Macrophage Inflammatory Responses. J. Leukoc. Biol. 2013, 94, 913–919. [Google Scholar] [CrossRef] [Green Version]
- Rutschman, R.; Lang, R.; Hesse, M.; Ihle, J.N.; Wynn, T.A.; Murray, P.J. Cutting Edge: Stat6-Dependent Substrate Depletion Regulates Nitric Oxide Production. J. Immunol. 2001, 166, 2173–2177. [Google Scholar] [CrossRef] [Green Version]
- Covarrubias, A.J.; Aksoylar, H.I.; Horng, T. Control of Macrophage Metabolism and Activation by MTOR and Akt Signaling. Semin. Immunol. 2015, 27, 286–296. [Google Scholar] [CrossRef] [Green Version]
- Arranz, A.; Doxaki, C.; Vergadi, E.; de la Torre, Y.M.; Vaporidi, K.; Lagoudaki, E.D.; Ieronymaki, E.; Androulidaki, A.; Venihaki, M.; Margioris, A.N.; et al. Akt1 and Akt2 Protein Kinases Differentially Contribute to Macrophage Polarization. Proc. Natl. Acad. Sci. USA 2012, 109, 9517–9522. [Google Scholar] [CrossRef] [Green Version]
- Fukao, T.; Koyasu, S. PI3K and Negative Regulation of TLR Signaling. Trends Immunol. 2003, 24, 358–363. [Google Scholar] [CrossRef]
- Zheng, C.; Yang, Q.; Xu, C.; Shou, P.; Cao, J.; Jiang, M.; Chen, Q.; Cao, G.; Han, Y.; Li, F.; et al. CD11b Regulates Obesity-Induced Insulin Resistance via Limiting Alternative Activation and Proliferation of Adipose Tissue Macrophages. Proc. Natl. Acad. Sci. USA 2015, 112, E7239–E7248. [Google Scholar] [CrossRef] [Green Version]
- Rauh, M.J.; Ho, V.; Pereira, C.; Sham, A.; Lam, V.; Huxham, L.; Minchinton, A.I.; Mui, A.; Krystal, G. SHIP Represses the Generation of Alternatively Activated Macrophages. Immunity 2005, 23, 361–374. [Google Scholar] [CrossRef] [Green Version]
- Yue, S.; Rao, J.; Zhu, J.; Busuttil, R.W.; Kupiec-Weglinski, J.W.; Lu, L.; Wang, X.; Zhai, Y. Myeloid PTEN Deficiency Protects Livers from Ischemia Reperfusion Injury by Facilitating M2 Macrophage Differentiation. J. Immunol. 2014, 192, 5343–5353. [Google Scholar] [CrossRef] [Green Version]
- Kratochvill, F.; Neale, G.; Haverkamp, J.M.; de Velde, L.A.V.; Smith, A.M.; Kawauchi, D.; McEvoy, J.; Roussel, M.F.; Dyer, M.A.; Qualls, J.E.; et al. TNF Counterbalances the Emergence of M2 Tumor Macrophages. Cell Rep. 2015, 12, 1902–1914. [Google Scholar] [CrossRef] [Green Version]
- Murdoch, C.; Muthana, M.; Lewis, C.E. Hypoxia Regulates Macrophage Functions in Inflammation. J. Immunol. 2005, 175, 6257–6263. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Yan, W.; Tohme, S.; Chen, M.; Fu, Y.; Tian, D.; Lotze, M.; Tang, D.; Tsung, A. Hypoxia Induced HMGB1 and Mitochondrial DNA Interactions Mediate Tumor Growth in Hepatocellular Carcinoma through Toll-like Receptor 9. J. Hepatol. 2015, 63, 114–121. [Google Scholar] [CrossRef] [Green Version]
- Gebhardt, C.; Riehl, A.; Durchdewald, M.; Németh, J.; Fürstenberger, G.; Müller-Decker, K.; Enk, A.; Arnold, B.; Bierhaus, A.; Nawroth, P.P.; et al. RAGE Signaling Sustains Inflammation and Promotes Tumor Development. J. Exp. Med. 2008, 205, 275–285. [Google Scholar] [CrossRef] [Green Version]
- Mittal, D.; Saccheri, F.; Vénéreau, E.; Pusterla, T.; Bianchi, M.E.; Rescigno, M. TLR4-Mediated Skin Carcinogenesis Is Dependent on Immune and Radioresistant Cells. EMBO J. 2010, 29, 2242–2252. [Google Scholar] [CrossRef]
- Huber, R.; Meier, B.; Otsuka, A.; Fenini, G.; Satoh, T.; Gehrke, S.; Widmer, D.; Levesque, M.P.; Mangana, J.; Kerl, K.; et al. Tumour Hypoxia Promotes Melanoma Growth and Metastasis via High Mobility Group Box-1 and M2-like Macrophages. Sci. Rep. 2016, 6, 29914. [Google Scholar] [CrossRef] [Green Version]
- Laviron, M.; Boissonnas, A. Ontogeny of Tumor-Associated Macrophages. Front. Immunol. 2019, 10, 1799. [Google Scholar] [CrossRef] [Green Version]
- Mondini, M.; Loyher, P.-L.; Hamon, P.; Gerbé de Thoré, M.; Laviron, M.; Berthelot, K.; Clémenson, C.; Salomon, B.L.; Combadière, C.; Deutsch, E.; et al. CCR2-Dependent Recruitment of Tregs and Monocytes Following Radiotherapy Is Associated with TNFα-Mediated Resistance. Cancer Immunol. Res. 2019, 7, 376–387. [Google Scholar] [CrossRef] [PubMed]
- Kalbasi, A.; Komar, C.; Tooker, G.M.; Liu, M.; Lee, J.W.; Gladney, W.L.; Ben-Josef, E.; Beatty, G.L. Tumor-Derived CCL2 Mediates Resistance to Radiotherapy in Pancreatic Ductal Adenocarcinoma. Clin. Cancer Res. 2017, 23, 137–148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, Y.; Herndon, J.M.; Sojka, D.K.; Kim, K.-W.; Knolhoff, B.L.; Zuo, C.; Cullinan, D.R.; Luo, J.; Bearden, A.R.; Lavine, K.J.; et al. Tissue-Resident Macrophages in Pancreatic Ductal Adenocarcinoma Originate from Embryonic Hematopoiesis and Promote Tumor Progression. Immunity 2017, 47, 323–338.e6. [Google Scholar] [CrossRef] [PubMed]
- Bain, C.C.; Hawley, C.A.; Garner, H.; Scott, C.L.; Schridde, A.; Steers, N.J.; Mack, M.; Joshi, A.; Guilliams, M.; Mowat, A.M.I.; et al. Long-Lived Self-Renewing Bone Marrow-Derived Macrophages Displace Embryo-Derived Cells to Inhabit Adult Serous Cavities. Nat. Commun. 2016, 7, ncomms11852. [Google Scholar] [CrossRef] [Green Version]
- Gibbings, S.L.; Goyal, R.; Desch, A.N.; Leach, S.M.; Prabagar, M.; Atif, S.M.; Bratton, D.L.; Janssen, W.; Jakubzick, C.V. Transcriptome Analysis Highlights the Conserved Difference between Embryonic and Postnatal-Derived Alveolar Macrophages. Blood 2015, 126, 1357–1366. [Google Scholar] [CrossRef]
- Van de Laar, L.; Saelens, W.; De Prijck, S.; Martens, L.; Scott, C.; Van Isterdael, G.; Hoffmann, E.; Beyaert, R.; Saeys, Y.; Lambrecht, B.; et al. Yolk Sac Macrophages, Fetal Liver, and Adult Monocytes Can Colonize an Empty Niche and Develop into Functional Tissue-Resident Macrophages. Immunity 2016, 44, 755–768. [Google Scholar] [CrossRef] [Green Version]
- Lavin, Y.; Winter, D.; Blecher-Gonen, R.; David, E.; Keren-Shaul, H.; Merad, M.; Jung, S.; Amit, I. Tissue-Resident Macrophage Enhancer Landscapes Are Shaped by the Local Microenvironment. Cell 2014, 159, 1312–1326. [Google Scholar] [CrossRef] [Green Version]
- Medzhitov, R. Origin and Physiological Roles of Inflammation. Nature 2008, 454, 428–435. [Google Scholar] [CrossRef]
- Kim, J.; Bae, J.-S. Tumor-Associated Macrophages and Neutrophils in Tumor Microenvironment. Mediat. Inflamm. 2016, 2016, 6058147. [Google Scholar] [CrossRef] [Green Version]
- Prame Kumar, K.; Nicholls, A.J.; Wong, C.H.Y. Partners in Crime: Neutrophils and Monocytes/Macrophages in Inflammation and Disease. Cell Tissue Res. 2018, 371, 551–565. [Google Scholar] [CrossRef] [Green Version]
- Nathan, C.; Ding, A. Nonresolving Inflammation. Cell 2010, 140, 871–882. [Google Scholar] [CrossRef] [Green Version]
- Krausgruber, T.; Blazek, K.; Smallie, T.; Alzabin, S.; Lockstone, H.; Sahgal, N.; Hussell, T.; Feldmann, M.; Udalova, I.A. IRF5 Promotes Inflammatory Macrophage Polarization and TH1-TH17 Responses. Nat. Immunol. 2011, 12, 231–238. [Google Scholar] [CrossRef]
- Murray, P.J.; Wynn, T.A. Protective and Pathogenic Functions of Macrophage Subsets. Nat. Rev. Immunol. 2011, 11, 723–737. [Google Scholar] [CrossRef]
- Wynn, T.A.; Barron, L. Macrophages: Master Regulators of Inflammation and Fibrosis. Semin. Liver Dis. 2010, 30, 245–257. [Google Scholar] [CrossRef] [Green Version]
- Mantovani, A.; Sozzani, S.; Locati, M.; Allavena, P.; Sica, A. Macrophage Polarization: Tumor-Associated Macrophages as a Paradigm for Polarized M2 Mononuclear Phagocytes. Trends Immunol. 2002, 23, 549–555. [Google Scholar] [CrossRef]
- Han, W.; Jackson, D.A.; Matissek, S.J.; Misurelli, J.A.; Neil, M.S.; Sklavanitis, B.; Amarsaikhan, N.; Elsawa, S.F. Novel Molecular Mechanism of Regulation of CD40 Ligand by the Transcription Factor GLI2. J. Immunol. 2017, 198, 4481–4489. [Google Scholar] [CrossRef] [Green Version]
- Jackson, D.A.; Smith, T.D.; Amarsaikhan, N.; Han, W.; Neil, M.S.; Boi, S.K.; Vrabel, A.M.; Tolosa, E.J.; Almada, L.L.; Fernandez-Zapico, M.E.; et al. Modulation of the IL-6 Receptor α Underlies GLI2-Mediated Regulation of Ig Secretion in Waldenström Macroglobulinemia Cells. J. Immunol. 2015, 195, 2908–2916. [Google Scholar] [CrossRef] [Green Version]
- Elsawa, S.F.; Almada, L.L.; Ziesmer, S.C.; Novak, A.J.; Witzig, T.E.; Ansell, S.M.; Fernandez-Zapico, M.E. GLI2 Transcription Factor Mediates Cytokine Cross-Talk in the Tumor Microenvironment. J. Biol. Chem. 2011, 286, 21524–21534. [Google Scholar] [CrossRef] [Green Version]
- Elsawa, S.F.; Novak, A.J.; Ziesmer, S.C.; Almada, L.L.; Hodge, L.S.; Grote, D.M.; Witzig, T.E.; Fernandez-Zapico, M.E.; Ansell, S.M. Comprehensive Analysis of Tumor Microenvironment Cytokines in Waldenstrom Macroglobulinemia Identifies CCL5 as a Novel Modulator of IL-6 Activity. Blood 2011, 118, 5540–5549. [Google Scholar] [CrossRef] [Green Version]
- Coussens, L.M.; Werb, Z. Inflammation and Cancer. Nature 2002, 420, 860–867. [Google Scholar] [CrossRef]
- Torisu, H.; Ono, M.; Kiryu, H.; Furue, M.; Ohmoto, Y.; Nakayama, J.; Nishioka, Y.; Sone, S.; Kuwano, M. Macrophage Infiltration Correlates with Tumor Stage and Angiogenesis in Human Malignant Melanoma: Possible Involvement of TNFalpha and IL-1alpha. Int. J. Cancer 2000, 85, 182–188. [Google Scholar] [CrossRef]
- Mosser, D.M.; Edwards, J.P. Exploring the Full Spectrum of Macrophage Activation. Nat. Rev. Immunol. 2008, 8, 958–969. [Google Scholar] [CrossRef]
- Fan, Z.; Yu, P.; Wang, Y.; Wang, Y.; Fu, M.L.; Liu, W.; Sun, Y.; Fu, Y.-X. NK-Cell Activation by LIGHT Triggers Tumor-Specific CD8+ T-Cell Immunity to Reject Established Tumors. Blood 2006, 107, 1342–1351. [Google Scholar] [CrossRef]
- Hadrup, S.; Donia, M.; thor Straten, P. Effector CD4 and CD8 T Cells and Their Role in the Tumor Microenvironment. Cancer Microenviron. 2013, 6, 123–133. [Google Scholar] [CrossRef] [Green Version]
- Susek, K.H.; Karvouni, M.; Alici, E.; Lundqvist, A. The Role of CXC Chemokine Receptors 1–4 on Immune Cells in the Tumor Microenvironment. Front. Immunol. 2018, 9, 2159. [Google Scholar] [CrossRef]
- Zhang, M.; He, Y.; Sun, X.; Li, Q.; Wang, W.; Zhao, A.; Di, W. A High M1/M2 Ratio of Tumor-Associated Macrophages Is Associated with Extended Survival in Ovarian Cancer Patients. J. Ovarian Res. 2014, 7, 19. [Google Scholar] [CrossRef] [Green Version]
- Lopez-Gonzalez, J.S.; Avila-Moreno, F.; Prado-Garcia, H.; Aguilar-Cazares, D.; Mandoki, J.J.; Meneses-Flores, M. Lung Carcinomas Decrease the Number of Monocytes/Macrophages (CD14+ Cells) That Produce TNF-Alpha. Clin. Immunol. 2007, 122, 323–329. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Liu, L.; Che, G.; Yu, N.; Dai, F.; You, Z. The M1 Form of Tumor-Associated Macrophages in Non-Small Cell Lung Cancer Is Positively Associated with Survival Time. BMC Cancer 2010, 10, 112. [Google Scholar] [CrossRef] [Green Version]
- Jackute, J.; Zemaitis, M.; Pranys, D.; Sitkauskiene, B.; Miliauskas, S.; Vaitkiene, S.; Sakalauskas, R. Distribution of M1 and M2 Macrophages in Tumor Islets and Stroma in Relation to Prognosis of Non-Small Cell Lung Cancer. BMC Immunol. 2018, 19, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edin, S.; Wikberg, M.L.; Oldenborg, P.-A.; Palmqvist, R. Macrophages: Good Guys in Colorectal Cancer. OncoImmunology 2013, 2, e23038. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, L.; Cheng, X.; Chen, H.; Chen, C.; Xie, S.; Zhao, M.; Liu, D.; Deng, Q.; Liu, Y.; Wang, X.; et al. Induction of Breast Cancer Stem Cells by M1 Macrophages through Lin-28B-Let-7-HMGA2 Axis. Cancer Lett. 2019, 452, 213–225. [Google Scholar] [CrossRef]
- Alves, A.M.; Diel, L.F.; Lamers, M.L. Macrophages and Prognosis of Oral Squamous Cell Carcinoma: A Systematic Review. J. Oral Pathol. Med. 2018, 47, 460–467. [Google Scholar] [CrossRef]
- Kovaleva, O.V.; Samoilova, D.V.; Shitova, M.S.; Gratchev, A. Tumor Associated Macrophages in Kidney Cancer. Analyt. Cell. Pathol. 2016, 2016, 9307549. [Google Scholar] [CrossRef] [Green Version]
- Falleni, M.; Savi, F.; Tosi, D.; Agape, E.; Cerri, A.; Moneghini, L.; Bulfamante, G.P. M1 and M2 Macrophages’ Clinicopathological Significance in Cutaneous Melanoma. Melanoma Res. 2017, 27, 200–210. [Google Scholar] [CrossRef]
- Talmadge, J.E.; Donkor, M.; Scholar, E. Inflammatory Cell Infiltration of Tumors: Jekyll or Hyde. Cancer Metastasis Rev. 2007, 26, 373–400. [Google Scholar] [CrossRef]
- Sica, A.; Mantovani, A. Macrophage Plasticity and Polarization: In Vivo Veritas. J. Clin. Invest. 2012, 122, 787–795. [Google Scholar] [CrossRef] [PubMed]
- Xu, F.; Wei, Y.; Tang, Z.; Liu, B.; Dong, J. Tumor associated Macrophages in Lung Cancer: Friend or Foe? (Review). Mol. Med. Rep. 2020, 22, 4107–4115. [Google Scholar] [CrossRef]
- Allavena, P.; Mantovani, A. Immunology in the Clinic Review Series; Focus on Cancer: Tumour-Associated Macrophages: Undisputed Stars of the Inflammatory Tumour Microenvironment. Clin. Exp. Immunol. 2012, 167, 195–205. [Google Scholar] [CrossRef]
- Krishnan, V.; Schaar, B.; Tallapragada, S.; Dorigo, O. Tumor Associated Macrophages in Gynecologic Cancers. Gynecol. Oncol. 2018, 149, 205–213. [Google Scholar] [CrossRef]
- Gupta, V.; Yull, F.; Khabele, D. Bipolar Tumor-Associated Macrophages in Ovarian Cancer as Targets for Therapy. Cancers 2018, 10, 366. [Google Scholar] [CrossRef] [Green Version]
- Raggi, C.; Mousa, H.S.; Correnti, M.; Sica, A.; Invernizzi, P. Cancer Stem Cells and Tumor-Associated Macrophages: A Roadmap for Multitargeting Strategies. Oncogene 2016, 35, 671–683. [Google Scholar] [CrossRef]
- Komohara, Y.; Fujiwara, Y.; Ohnishi, K.; Takeya, M. Tumor-Associated Macrophages: Potential Therapeutic Targets for Anti-Cancer Therapy. Adv. Drug Deliv. Rev. 2016, 99, 180–185. [Google Scholar] [CrossRef] [PubMed]
- Parker, K.H.; Sinha, P.; Horn, L.A.; Clements, V.K.; Yang, H.; Li, J.; Tracey, K.J.; Ostrand-Rosenberg, S. HMGB1 Enhances Immune Suppression by Facilitating the Differentiation and Suppressive Activity of Myeloid-Derived Suppressor Cells. Cancer Res. 2014, 74, 5723–5733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gabrilovich, D.I.; Nagaraj, S. Myeloid-Derived Suppressor Cells as Regulators of the Immune System. Nat. Rev. Immunol. 2009, 9, 162–174. [Google Scholar] [CrossRef] [PubMed]
- Columba-Cabezas, S.; Serafini, B.; Ambrosini, E.; Sanchez, M.; Penna, G.; Adorini, L.; Aloisi, F. Induction of Macrophage-Derived Chemokine/CCL22 Expression in Experimental Autoimmune Encephalomyelitis and Cultured Microglia: Implications for Disease Regulation. J. Neuroimmunol. 2002, 130, 10–21. [Google Scholar] [CrossRef]
- Bloch, O.; Crane, C.A.; Kaur, R.; Safaee, M.; Rutkowski, M.J.; Parsa, A.T. Gliomas Promote Immunosuppression through Induction of B7-H1 Expression in Tumor-Associated Macrophages. Clin. Cancer Res. 2013, 19, 3165–3175. [Google Scholar] [CrossRef] [Green Version]
- Matsunaga, T.; Saito, H.; Ikeguchi, M. Increased B7-H1 and B7-H4 Expressions on Circulating Monocytes and Tumor-Associated Macrophages Are Involved in Immune Evasion in Patients with Gastric Cancer. Yonago Acta Med. 2011, 54, 1–10. [Google Scholar]
- Anderson, K.M.; Czinn, S.J.; Redline, R.W.; Blanchard, T.G. Induction of CTLA-4-Mediated Anergy Contributes to Persistent Colonization in the Murine Model of Gastric Helicobacter Pylori Infection. J. Immunol. 2006, 176, 5306–5313. [Google Scholar] [CrossRef] [Green Version]
- Jeong, H.; Kim, S.; Hong, B.-J.; Lee, C.-J.; Kim, Y.-E.; Bok, S.; Oh, J.-M.; Gwak, S.-H.; Yoo, M.Y.; Lee, M.S.; et al. Tumor-Associated Macrophages Enhance Tumor Hypoxia and Aerobic Glycolysis. Cancer Res. 2019, 79, 795–806. [Google Scholar] [CrossRef] [Green Version]
- Obermajer, N.; Muthuswamy, R.; Odunsi, K.; Edwards, R.P.; Kalinski, P. PGE2-Induced CXCL12 Production and CXCR4 Expression Controls the Accumulation of Human MDSCs in Ovarian Cancer Environment. Cancer Res. 2011, 71, 7463–7470. [Google Scholar] [CrossRef] [Green Version]
- Kzhyshkowska, J.; Riabov, V.; Gudima, A.; Wang, N.; Orekhov, A.; Mickley, A. Role of Tumor Associated Macrophages in Tumor Angiogenesis and Lymphangiogenesis. Front. Physiol. 2014, 5. [Google Scholar] [CrossRef] [Green Version]
- Mitsudomi, T.; Yatabe, Y. Epidermal Growth Factor Receptor in Relation to Tumor Development: EGFR Gene and Cancer. FEBS J. 2010, 277, 301–308. [Google Scholar] [CrossRef]
- Turner, N.; Grose, R. Fibroblast Growth Factor Signalling: From Development to Cancer. Nat. Rev. Cancer 2010, 10, 116–129. [Google Scholar] [CrossRef]
- Gold, L.I. The role for transforming growth factor-beta (TGF-beta) in human cancer. Critical Rev. Oncog. 1999, 10, 303–360. [Google Scholar]
- Zhang, J.; Yan, Y.; Yang, Y.; Wang, L.; Li, M.; Wang, J.; Liu, X.; Duan, X.; Wang, J. High Infiltration of Tumor-Associated Macrophages Influences Poor Prognosis in Human Gastric Cancer Patients, Associates with the Phenomenon of EMT. Medicine 2016, 95, e2636. [Google Scholar] [CrossRef]
- Diepenbruck, M.; Christofori, G. Epithelial–Mesenchymal Transition (EMT) and Metastasis: Yes, No, Maybe? Curr. Opin. Cell Biol. 2016, 43, 7–13. [Google Scholar] [CrossRef] [Green Version]
- Cortés, M.; Sanchez-Moral, L.; de Barrios, O.; Fernández-Aceñero, M.J.; Martínez-Campanario, M.C.; Esteve-Codina, A.; Darling, D.S.; Győrffy, B.; Lawrence, T.; Dean, D.C.; et al. Tumor-Associated Macrophages (TAMs) Depend on ZEB1 for Their Cancer-Promoting Roles. EMBO J. 2017, 36, 3336–3355. [Google Scholar] [CrossRef]
- Zhu, L.; Fu, X.; Chen, X.; Han, X.; Dong, P. M2 Macrophages Induce EMT through the TGF-β/Smad2 Signaling Pathway. Cell Biol. Int. 2017, 41, 960–968. [Google Scholar] [CrossRef]
- Zhang, Q.; Mao, Z.; Sun, J. NF-ΚB Inhibitor, BAY11-7082, Suppresses M2 Tumor-Associated Macrophage Induced EMT Potential via MiR-30a/NF-ΚB/Snail Signaling in Bladder Cancer Cells. Gene 2019, 710, 91–97. [Google Scholar] [CrossRef]
- Jetten, N.; Verbruggen, S.; Gijbels, M.J.; Post, M.J.; De Winther, M.P.; Donners, M.M. Anti-Inflammatory M2, but not pro-Inflammatory M1 Macrophages Promote Angiogenesis In Vivo. Angiogenesis 2014, 17, 109–118. [Google Scholar] [CrossRef]
- Fabregat, I.; Fernando, J.; Mainez, J.; Sancho, P. TGF-Beta Signaling in Cancer Treatment. Curr. Pharm. Des. 2014, 20, 2934–2947. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, E.S.; Koizumi, K.; Kobayashi, M.; Saiki, I. Inhibition of Lymphangiogenesis-Related Properties of Murine Lymphatic Endothelial Cells and Lymph Node Metastasis of Lung Cancer by the Matrix Metalloproteinase Inhibitor MMI270. Cancer Sci. 2004, 95, 25–31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, D. Parallels of Resistance between Angiogenesis and Lymphangiogenesis Inhibition in Cancer Therapy. Cells 2020, 9, 762. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turrini, R.; Pabois, A.; Xenarios, I.; Coukos, G.; Delaloye, J.-F.; Doucey, M.-A. TIE-2 Expressing Monocytes in Human Cancers. OncoImmunology 2017, 6, e1303585. [Google Scholar] [CrossRef] [Green Version]
- Guex, N.; Crespo, I.; Bron, S.; Ifticene-Treboux, A.; Faes-van’t Hull, E.; Kharoubi, S.; Liechti, R.; Werffeli, P.; Ibberson, M.; Majo, F.; et al. Angiogenic Activity of Breast Cancer Patients’ Monocytes Reverted by Combined Use of Systems Modeling and Experimental Approaches. PLoS Comput. Biol. 2015, 11, e1004050. [Google Scholar] [CrossRef] [Green Version]
- De Palma, M.; Venneri, M.A.; Galli, R.; Sergi, L.S.; Politi, L.S.; Sampaolesi, M.; Naldini, L. Tie2 Identifies a Hematopoietic Lineage of Proangiogenic Monocytes Required for Tumor Vessel Formation and a Mesenchymal Population of Pericyte Progenitors. Cancer Cell 2005, 8, 211–226. [Google Scholar] [CrossRef] [Green Version]
- Bron, S.; Henry, L.; Faes-van’t Hull, E.; Turrini, R.; Vanhecke, D.; Guex, N.; Ifticene-Treboux, A.; Marina Iancu, E.; Semilietof, A.; Rufer, N.; et al. TIE-2-Expressing Monocytes Are Lymphangiogenic and Associate Specifically with Lymphatics of Human Breast Cancer. OncoImmunology 2016, 5, e1073882. [Google Scholar] [CrossRef]
- Quandt, D.; Fiedler, E.; Boettcher, D.; Marsch, W.C.; Seliger, B. B7-H4 Expression in Human Melanoma: Its Association with Patients’ Survival and Antitumor Immune Response. Clin. Cancer Res. 2011, 17, 3100–3111. [Google Scholar] [CrossRef] [Green Version]
- Thurston, G.; Daly, C. The Complex Role of Angiopoietin-2 in the Angiopoietin-Tie Signaling Pathway. Cold Spring Harb. Perspect. Med. 2012, 2, a006650. [Google Scholar] [CrossRef] [Green Version]
- Akwii, R.G.; Sajib, M.S.; Zahra, F.T.; Mikelis, C.M. Role of Angiopoietin-2 in Vascular Physiology and Pathophysiology. Cells 2019, 8, 471. [Google Scholar] [CrossRef] [Green Version]
- Kim, I.; Kim, J.-H.; Ryu, Y.S.; Liu, M.; Koh, G.Y. Tumor Necrosis Factor-α Upregulates Angiopoietin-2 in Human Umbilical Vein Endothelial Cells. Biochem. Biophys. Res. Commun. 2000, 269, 361–365. [Google Scholar] [CrossRef]
- Leblond, M.M.; Pérès, E.A.; Helaine, C.; Gérault, A.N.; Moulin, D.; Anfray, C.; Divoux, D.; Petit, E.; Bernaudin, M.; Valable, S. M2 Macrophages Are More Resistant than M1 Macrophages Following Radiation Therapy in the Context of Glioblastoma. Oncotarget 2017, 8, 72597–72612. [Google Scholar] [CrossRef] [Green Version]
- Castro, B.A.; Flanigan, P.; Jahangiri, A.; Hoffman, D.; Chen, W.; Kuang, R.; De Lay, M.; Yagnik, G.; Wagner, J.R.; Mascharak, S.; et al. Macrophage Migration Inhibitory Factor Downregulation: A Novel Mechanism of Resistance to Anti-Angiogenic Therapy. Oncogene 2017, 36, 3749–3759. [Google Scholar] [CrossRef] [Green Version]
- DeNardo, D.G.; Brennan, D.J.; Rexhepaj, E.; Ruffell, B.; Shiao, S.L.; Madden, S.F.; Gallagher, W.M.; Wadhwani, N.; Keil, S.D.; Junaid, S.A.; et al. Leukocyte Complexity Predicts Breast Cancer Survival and Functionally Regulates Response to Chemotherapy. Cancer Dis. 2011, 1, 54–67. [Google Scholar] [CrossRef] [Green Version]
- Nakasone, E.S.; Askautrud, H.A.; Kees, T.; Park, J.-H.; Plaks, V.; Ewald, A.J.; Fein, M.; Rasch, M.G.; Tan, Y.-X.; Qiu, J.; et al. Imaging Tumor-Stroma Interactions during Chemotherapy Reveals Contributions of the Microenvironment to Resistance. Cancer Cell 2012, 21, 488–503. [Google Scholar] [CrossRef] [Green Version]
- Salvagno, C.; Ciampricotti, M.; Tuit, S.; Hau, C.-S.; van Weverwijk, A.; Coffelt, S.B.; Kersten, K.; Vrijland, K.; Kos, K.; Ulas, T.; et al. Therapeutic Targeting of Macrophages Enhances Chemotherapy Efficacy by Unleashing Type I Interferon Response. Nat. Cell Biol. 2019, 21, 511–521. [Google Scholar] [CrossRef]
- Ruffell, B.; Coussens, L.M. Macrophages and Therapeutic Resistance in Cancer. Cancer Cell 2015, 27, 462–472. [Google Scholar] [CrossRef] [Green Version]
- Shojaei, F.; Wu, X.; Malik, A.K.; Zhong, C.; Baldwin, M.E.; Schanz, S.; Fuh, G.; Gerber, H.-P.; Ferrara, N. Tumor Refractoriness to Anti-VEGF Treatment Is Mediated by CD11b+Gr1+ Myeloid Cells. Nat. Biotechnol. 2007, 25, 911–920. [Google Scholar] [CrossRef]
- Dalton, H.J.; Pradeep, S.; McGuire, M.; Hailemichael, Y.; Ma, S.; Lyons, Y.; Armaiz-Pena, G.N.; Previs, R.A.; Hansen, J.M.; Rupaimoole, R.; et al. Macrophages Facilitate Resistance to Anti-VEGF Therapy by Altered VEGFR Expression. Clin. Cancer Res. 2017, 23, 7034–7046. [Google Scholar] [CrossRef] [Green Version]
- Mazzieri, R.; Pucci, F.; Moi, D.; Zonari, E.; Ranghetti, A.; Berti, A.; Politi, L.S.; Gentner, B.; Brown, J.L.; Naldini, L.; et al. Targeting the ANG2/TIE2 Axis Inhibits Tumor Growth and Metastasis by Impairing Angiogenesis and Disabling Rebounds of Proangiogenic Myeloid Cells. Cancer Cell 2011, 19, 512–526. [Google Scholar] [CrossRef] [Green Version]
- Klug, F.; Prakash, H.; Huber, P.E.; Seibel, T.; Bender, N.; Halama, N.; Pfirschke, C.; Voss, R.H.; Timke, C.; Umansky, L.; et al. Low-Dose Irradiation Programs Macrophage Differentiation to an INOS+/M1 Phenotype That Orchestrates Effective T Cell Immunotherapy. Cancer Cell 2013, 24, 589–602. [Google Scholar] [CrossRef] [Green Version]
- Canè, S.; Ugel, S.; Trovato, R.; Marigo, I.; De Sanctis, F.; Sartoris, S.; Bronte, V. The Endless Saga of Monocyte Diversity. Front. Immunol. 2019, 10, 1786. [Google Scholar] [CrossRef] [Green Version]
- Argyle, D.; Kitamura, T. Targeting Macrophage-Recruiting Chemokines as a Novel Therapeutic Strategy to Prevent the Progression of Solid Tumors. Front. Immunol. 2018, 9, 2629. [Google Scholar] [CrossRef]
- Brana, I.; Calles, A.; LoRusso, P.M.; Yee, L.K.; Puchalski, T.A.; Seetharam, S.; Zhong, B.; de Boer, C.J.; Tabernero, J.; Calvo, E. Carlumab, an Anti-C-C Chemokine Ligand 2 Monoclonal Antibody, in Combination with Four Chemotherapy Regimens for the Treatment of Patients with Solid Tumors: An Open-Label, Multicenter Phase 1b Study. Targ. Oncol. 2015, 10, 111–123. [Google Scholar] [CrossRef]
- Bonapace, L.; Coissieux, M.-M.; Wyckoff, J.; Mertz, K.D.; Varga, Z.; Junt, T.; Bentires-Alj, M. Cessation of CCL2 Inhibition Accelerates Breast Cancer Metastasis by Promoting Angiogenesis. Nature 2014, 515, 130–133. [Google Scholar] [CrossRef]
- Hughes, R.; Qian, B.-Z.; Rowan, C.; Muthana, M.; Keklikoglou, I.; Olson, O.C.; Tazzyman, S.; Danson, S.; Addison, C.; Clemons, M.; et al. Perivascular M2 Macrophages Stimulate Tumor Relapse after Chemotherapy. Cancer Res. 2015, 75, 3479–3491. [Google Scholar] [CrossRef] [Green Version]
- Boimel, P.J.; Smirnova, T.; Zhou, Z.N.; Wyckoff, J.; Park, H.; Coniglio, S.J.; Qian, B.-Z.; Stanley, E.R.; Cox, D.; Pollard, J.W.; et al. Contribution of CXCL12 Secretion to Invasion of Breast Cancer Cells. Breast Cancer Res. 2012, 14, R23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ries, C.H.; Cannarile, M.A.; Hoves, S.; Benz, J.; Wartha, K.; Runza, V.; Rey-Giraud, F.; Pradel, L.P.; Feuerhake, F.; Klaman, I.; et al. Targeting Tumor-Associated Macrophages with Anti-CSF-1R Antibody Reveals a Strategy for Cancer Therapy. Cancer Cell 2014, 25, 846–859. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, J.; Escamilla, J.; Mok, S.; David, J.; Priceman, S.; West, B.; Bollag, G.; McBride, W.; Wu, L. CSF1R Signaling Blockade Stanches Tumor-Infiltrating Myeloid Cells and Improves the Efficacy of Radiotherapy in Prostate Cancer. Cancer Res. 2013, 73, 2782–2794. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quail, D.F.; Bowman, R.L.; Akkari, L.; Quick, M.L.; Schuhmacher, A.J.; Huse, J.T.; Holland, E.C.; Sutton, J.C.; Joyce, J.A. The Tumor Microenvironment Underlies Acquired Resistance to CSF-1R Inhibition in Gliomas. Science 2016, 352, aad3018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papadavid, E.; Stratigos, A.J.; Falagas, M.E. Imiquimod: An Immune Response Modifier in the Treatment of Precancerous Skin Lesions and Skin Cancer. Exp. Opin. Pharmacother. 2007, 8, 1743–1755. [Google Scholar] [CrossRef]
- Maeda, A.; Digifico, E.; Andon, F.T.; Mantovani, A.; Allavena, P. Poly(I:C) Stimulation Is Superior than Imiquimod to Induce the Antitumoral Functional Profile of Tumor-conditioned Macrophages. Eur. J. Immunol. 2019, 49, 801–811. [Google Scholar] [CrossRef]
- Zanganeh, S.; Hutter, G.; Spitler, R.; Lenkov, O.; Mahmoudi, M.; Shaw, A.; Pajarinen, J.S.; Nejadnik, H.; Goodman, S.; Moseley, M.; et al. Iron Oxide Nanoparticles Inhibit Tumour Growth by Inducing Pro-Inflammatory Macrophage Polarization in Tumour Tissues. Nat. Nanotech. 2016, 11, 986–994. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Species | M0 | M1 | M2 |
|---|---|---|---|
| Mouse | Csf1r, F4/80, CD11b | Marco, Cxcl9, Cxcl10, Cxcl11, Nos2, Socs1 | Cd206, Tgm2, Fizz1, Chil3, Arg1, Ccl22, Cd163 |
| Human | CSF1R, CD14, CD68, CD11B | CD86, MARCO, CXCL9, CXCL10, CXCL11, NOS2, SOCS1, CD64 | TGM2, CD23, ARG1, CCL22, CD163, CD206 |
| Protein/Gene | Normal Function | Effect on Polarization |
|---|---|---|
| Interleukin-4 and Interleukin-13 | Cytokines | M2-favored |
| Interleukin-4 receptor alpha | IL-4 and IL-13 signaling | M2-favored |
| Signal transducer and activator of transcription 6 | Transcription factor | M2-favored |
| Peroxisome proliferator activated receptor gamma | Transcription factor | M2-favored |
| Tubular sclerosis 1 | Inhibitor of mTOR | M2-favored |
| AKT Serine/Threonine Kinase 1 | Signaling | M2-favored |
| AKT Serine/Threonine Kinase 2 | Signaling | M1-favored |
| Src homology region 2 domain-containing phosphatase-1/2 | Phosphatases | M1-favored |
| SH2-containing Inositol 5′-Phosphatase | Phosphatase | M1-favored |
| Phosphatase and tensin homolog | Lipid phosphatase | M1-favored |
| Myeloid differentiation primary response 88 | Signaling adapter | M1-favored |
| Tumor necrosis factor | Cytokine | M1-favored |
| Tumor necrosis factor receptor 1 | Cytokine receptor | M1-favored |
| Interferon-gamma, Interleukin-12 | Cytokines | M1-favored |
| Factors Secreted by M2 TAMs | Pro-Tumorigenic Outcome |
|---|---|
| IL-6, EGF, TNF-α IL-8, IL-10, CCL2 | Tumor growth |
| IL-10, TGF-β, MMP-7, PD-1, PDE-2, arginase | Immune suppression |
| CCL18, CCL22, MMPs, TGF-β, EGF, CCL20, IGF-1 | Tumor invasion and metastasis |
| VEGFA, PDGF, COX2, HIF, MMPs, IL-10, adrenomedullin | Tumor angiogenesis and lymphangiogenesis |
| TGF-β, MMPs, IL-6, IL-10 | Anti-cancer therapy resistance |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Boutilier, A.J.; Elsawa, S.F. Macrophage Polarization States in the Tumor Microenvironment. Int. J. Mol. Sci. 2021, 22, 6995. https://doi.org/10.3390/ijms22136995
Boutilier AJ, Elsawa SF. Macrophage Polarization States in the Tumor Microenvironment. International Journal of Molecular Sciences. 2021; 22(13):6995. https://doi.org/10.3390/ijms22136995
Chicago/Turabian StyleBoutilier, Ava J., and Sherine F. Elsawa. 2021. "Macrophage Polarization States in the Tumor Microenvironment" International Journal of Molecular Sciences 22, no. 13: 6995. https://doi.org/10.3390/ijms22136995
APA StyleBoutilier, A. J., & Elsawa, S. F. (2021). Macrophage Polarization States in the Tumor Microenvironment. International Journal of Molecular Sciences, 22(13), 6995. https://doi.org/10.3390/ijms22136995